
Photophysical Properties of Anthracene Derivatives




Abstract
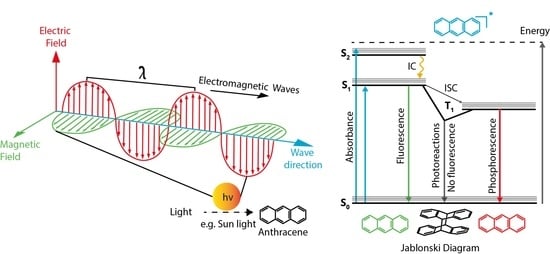
:
1. Introduction
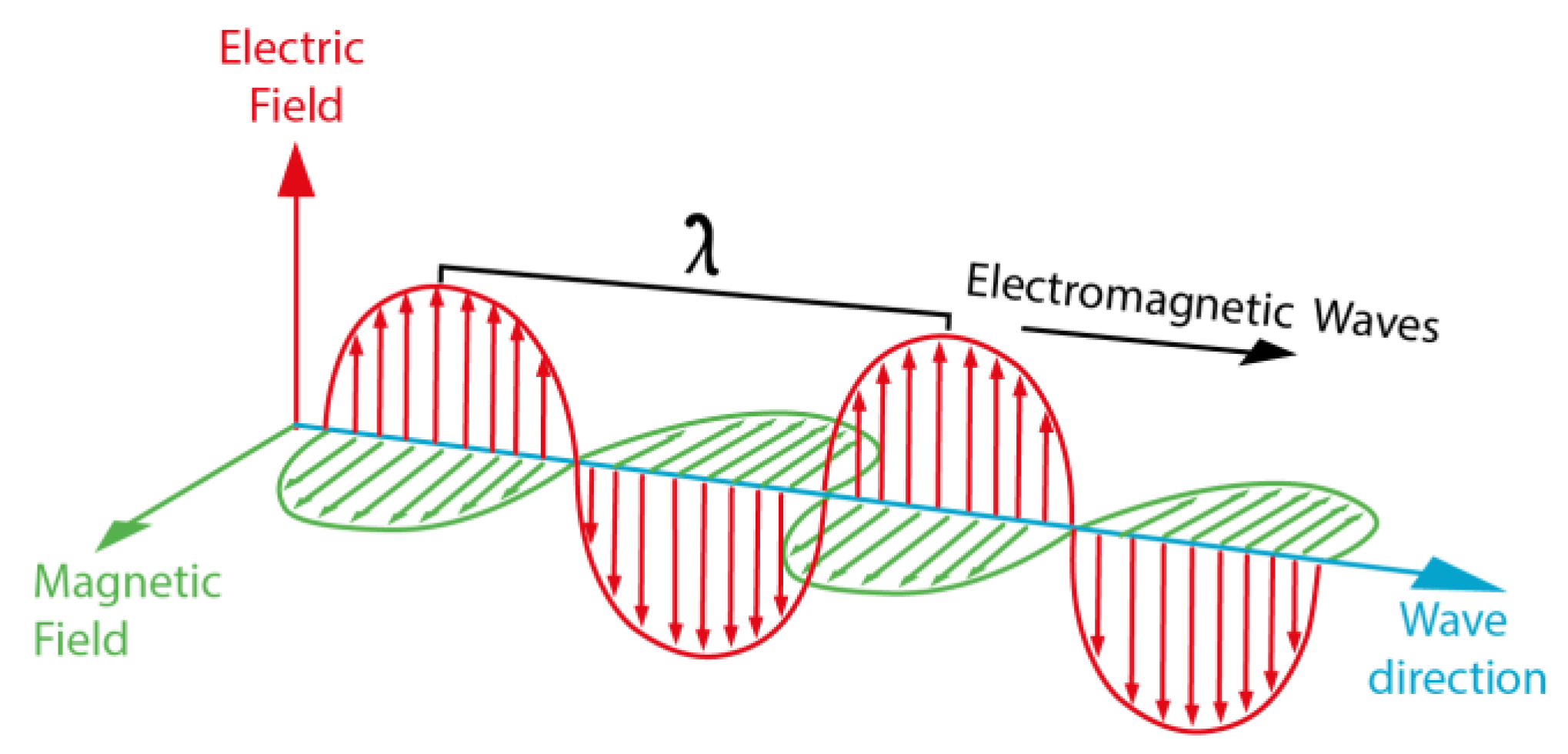
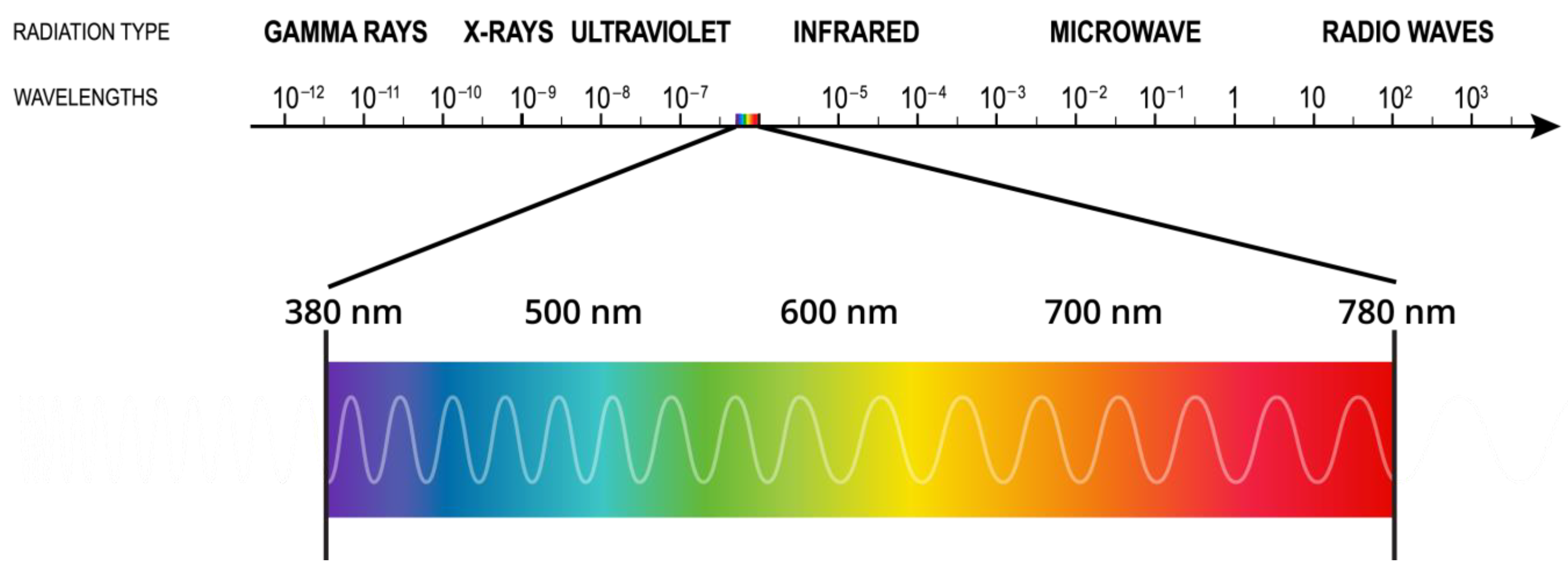
1.1. Light
Light Sources
1.2. Interaction of Light with Matter
1.2.1. Absorption
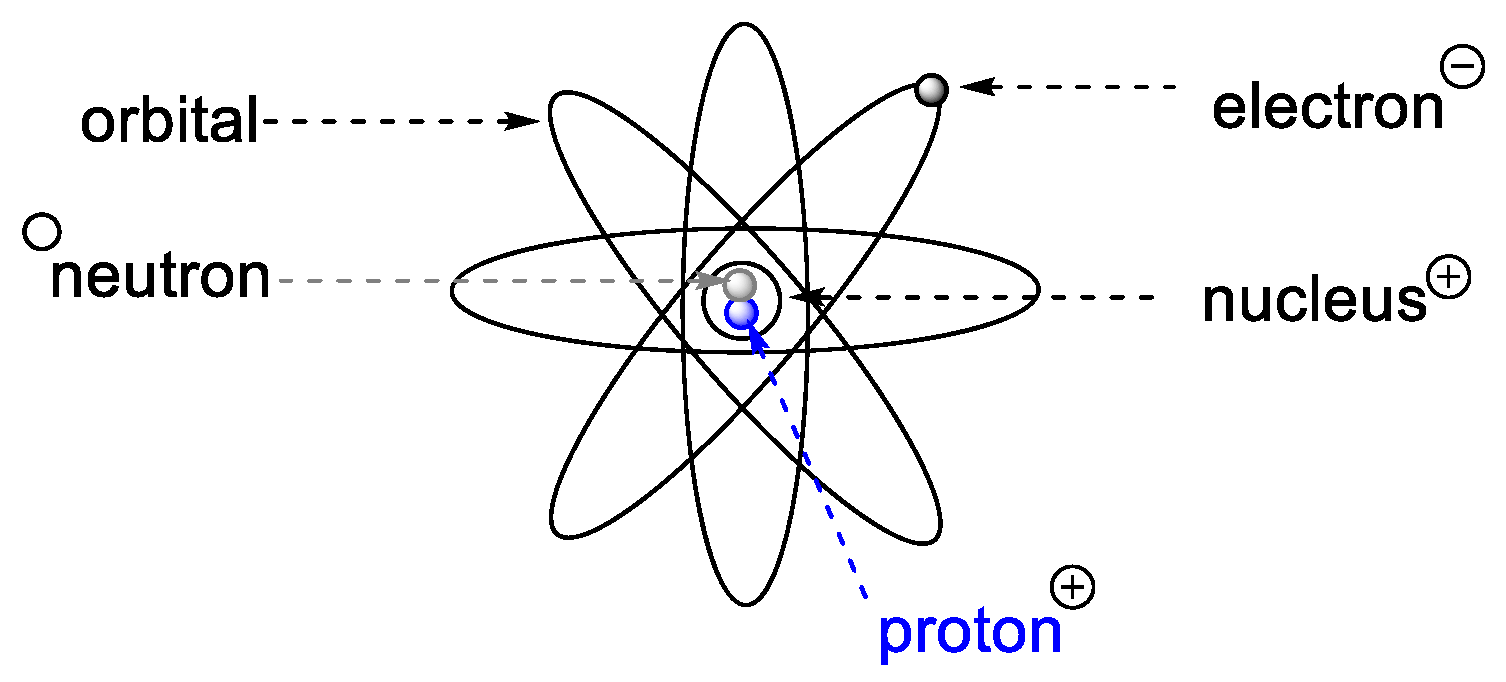
1.2.2. Matter
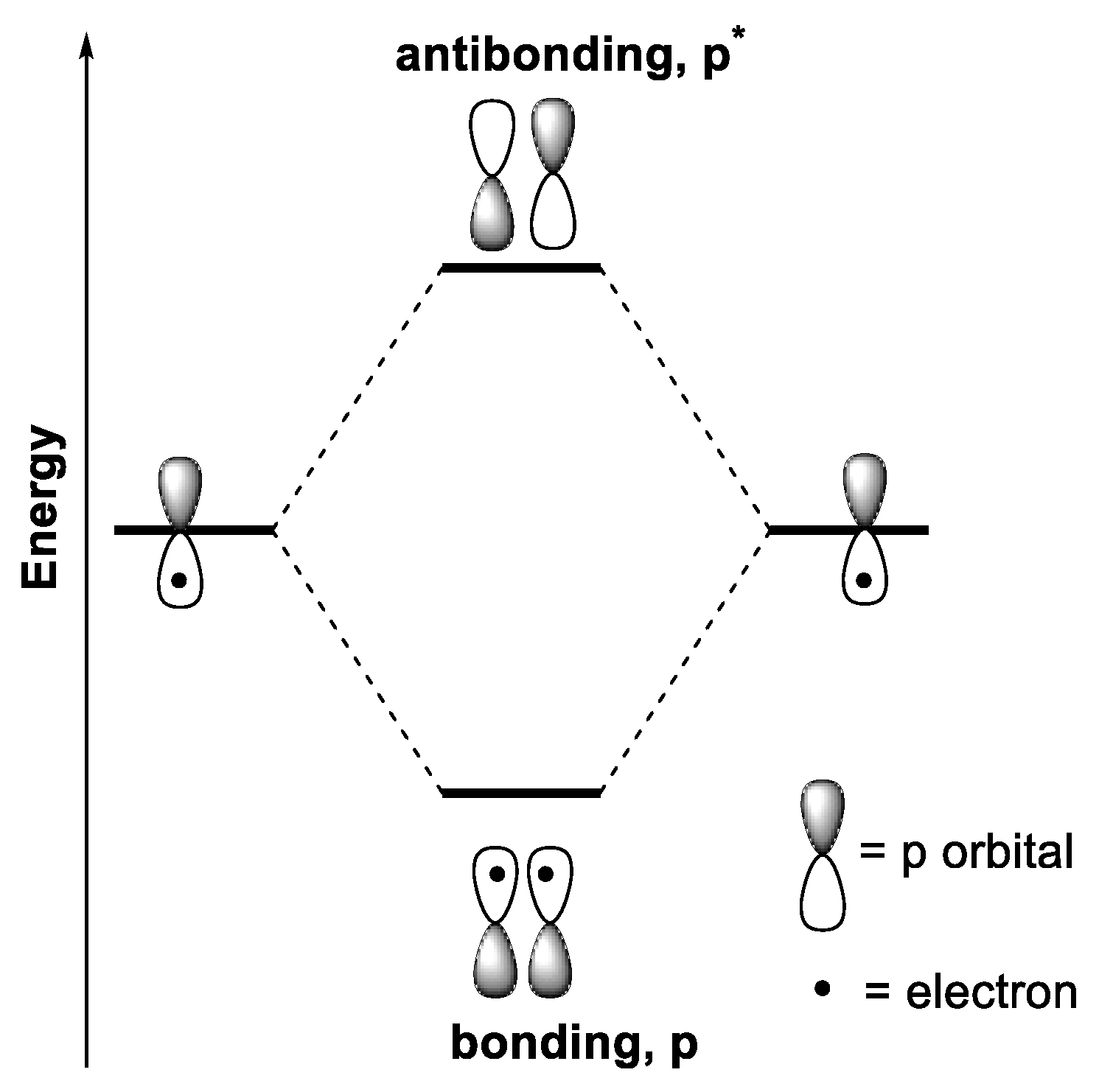
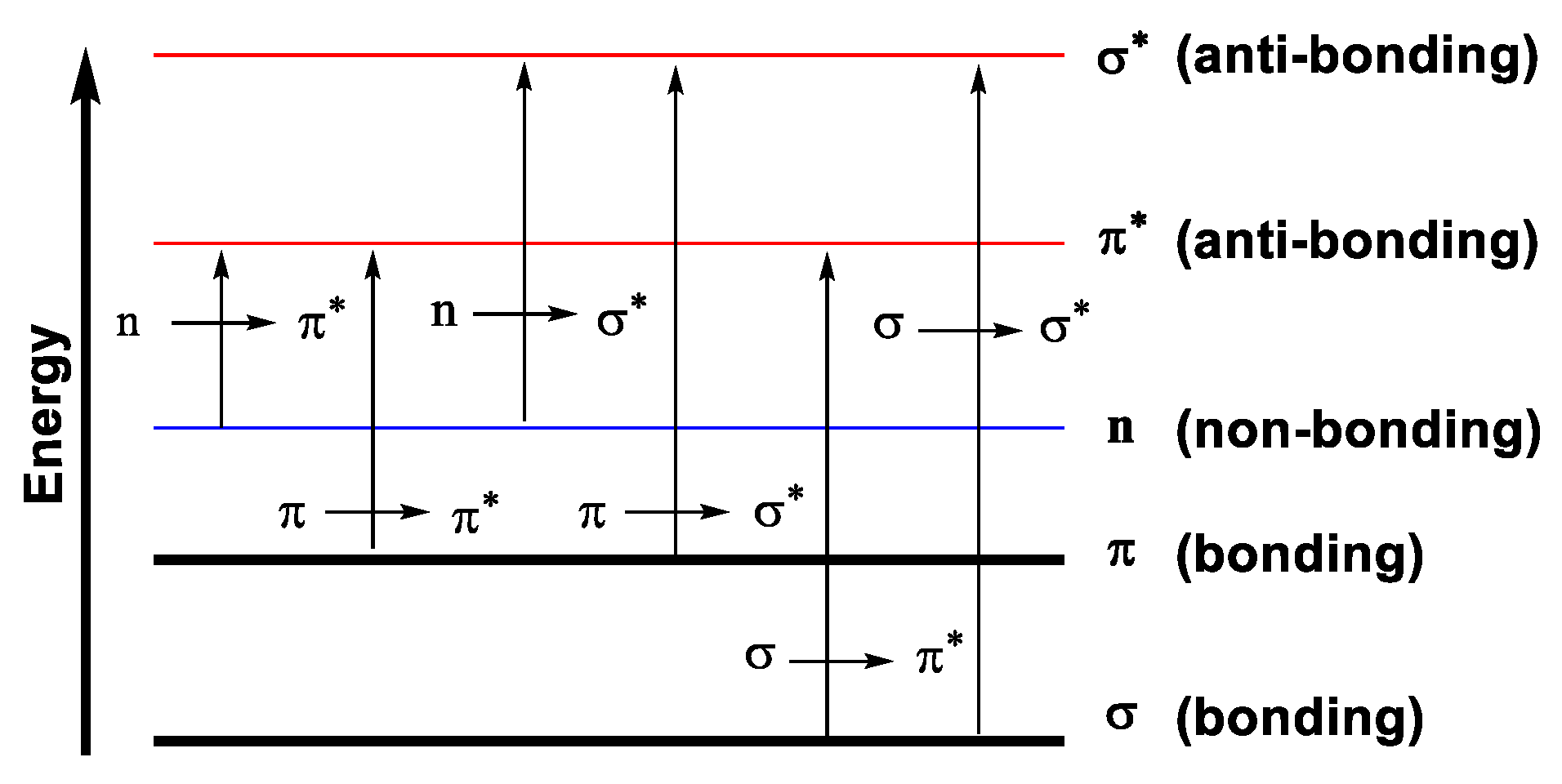
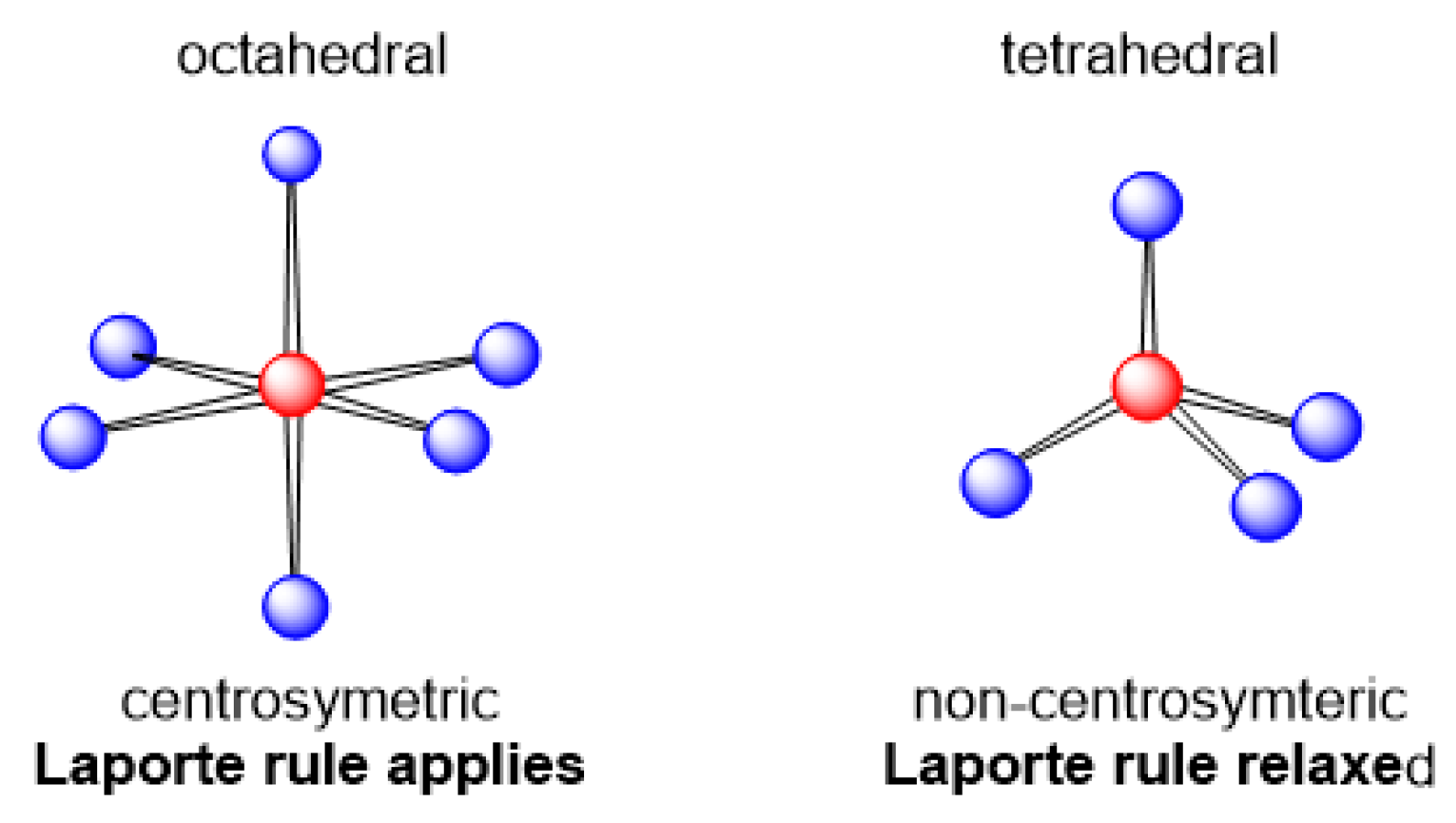
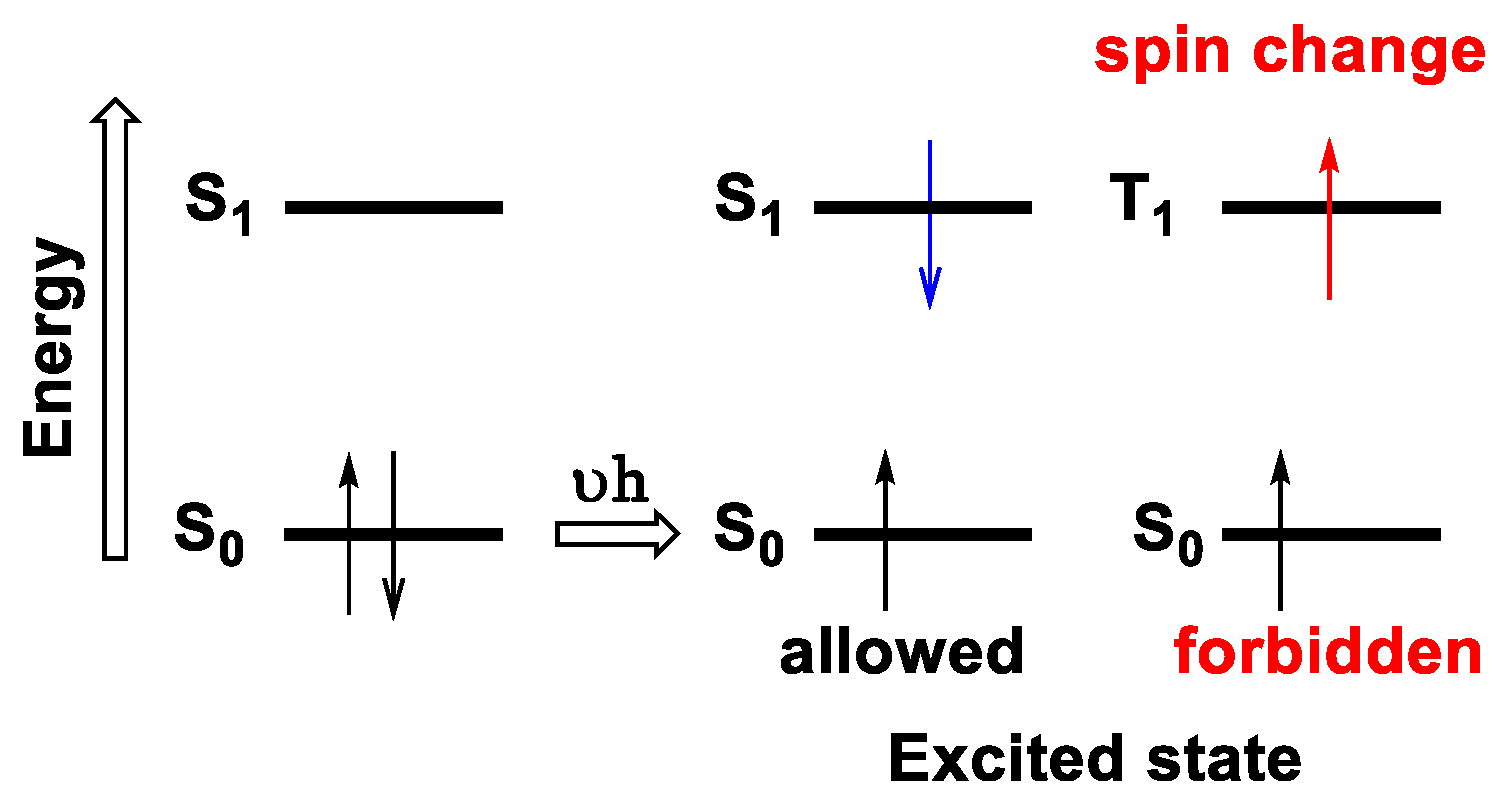
1.2.3. Electronic Transitions
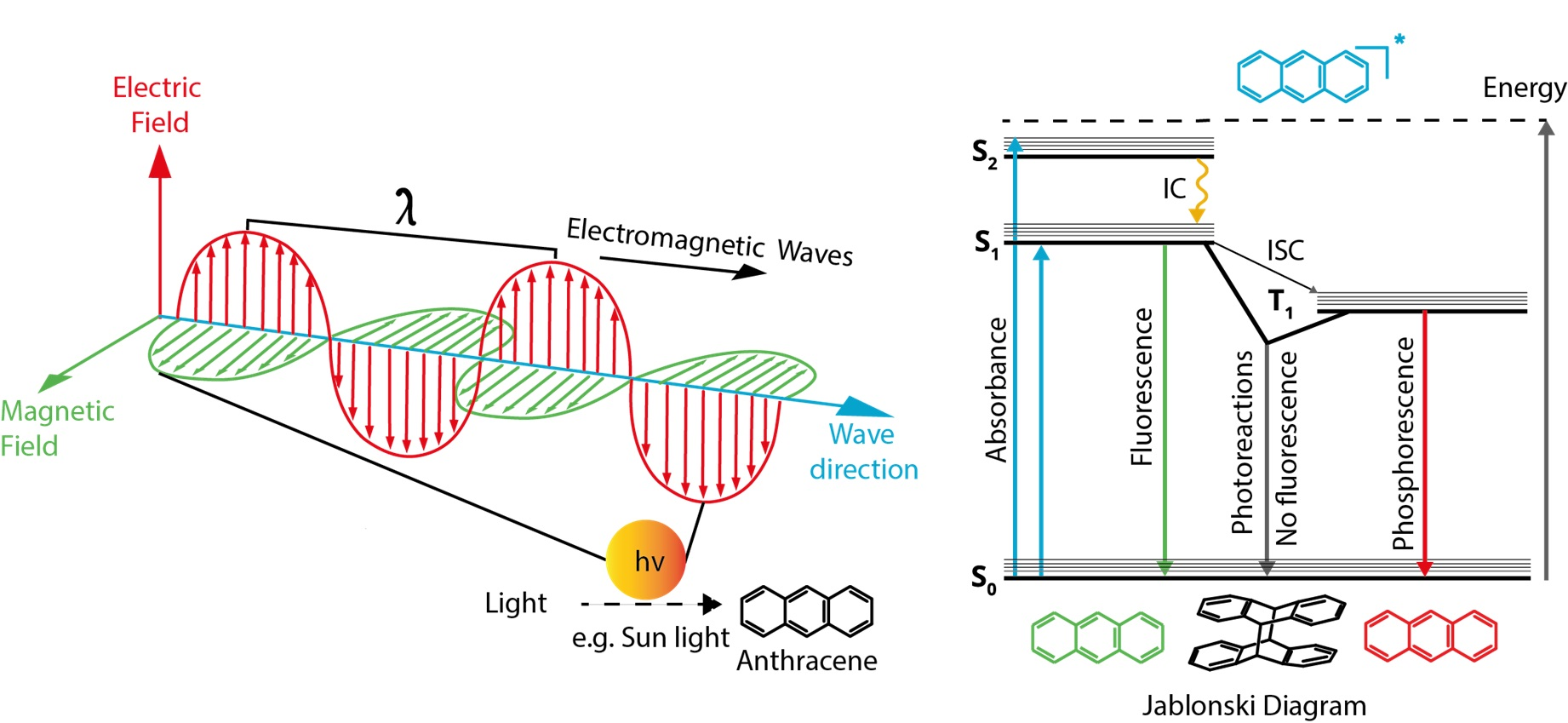
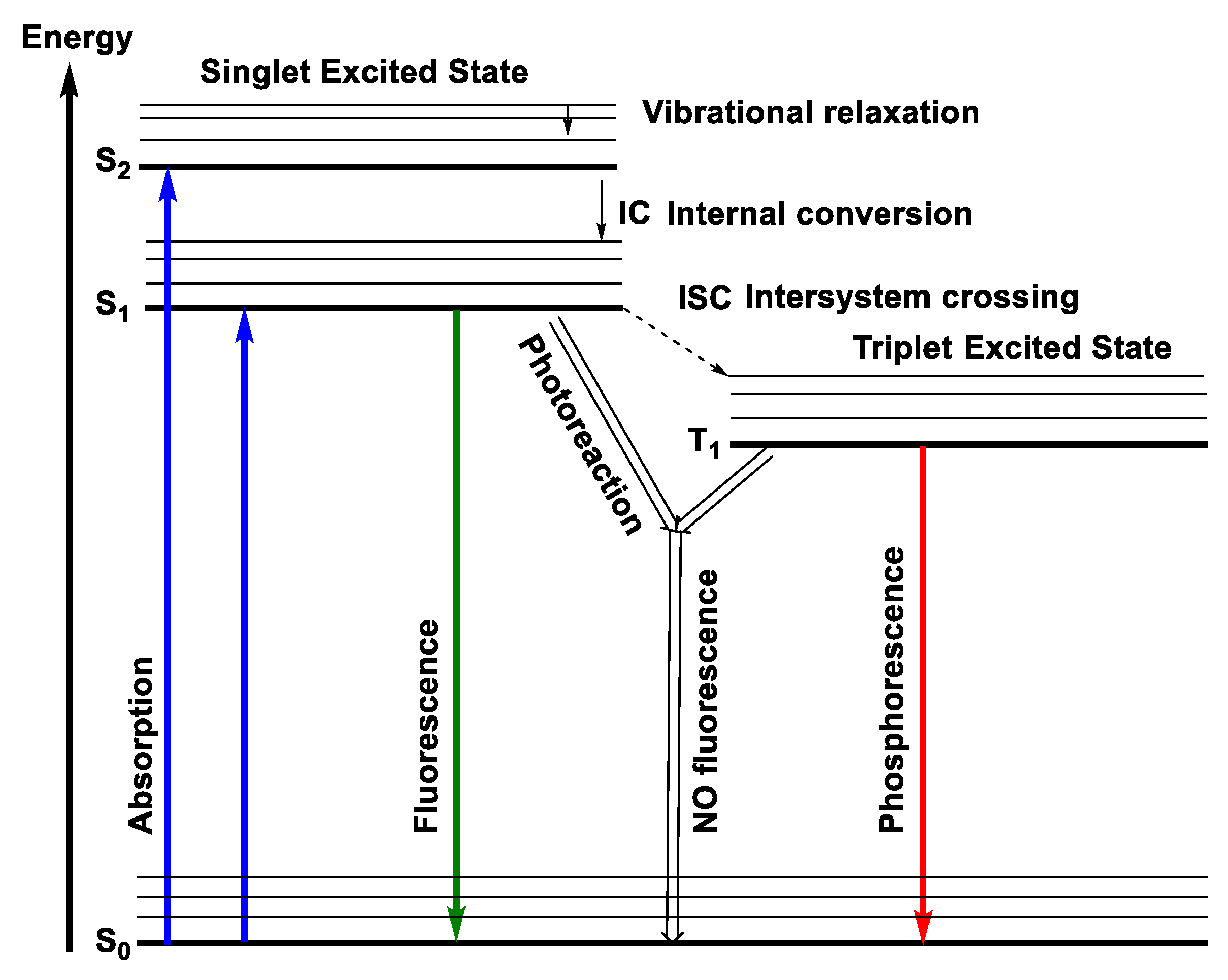
1.2.4. Emission
Jablonski Diagram
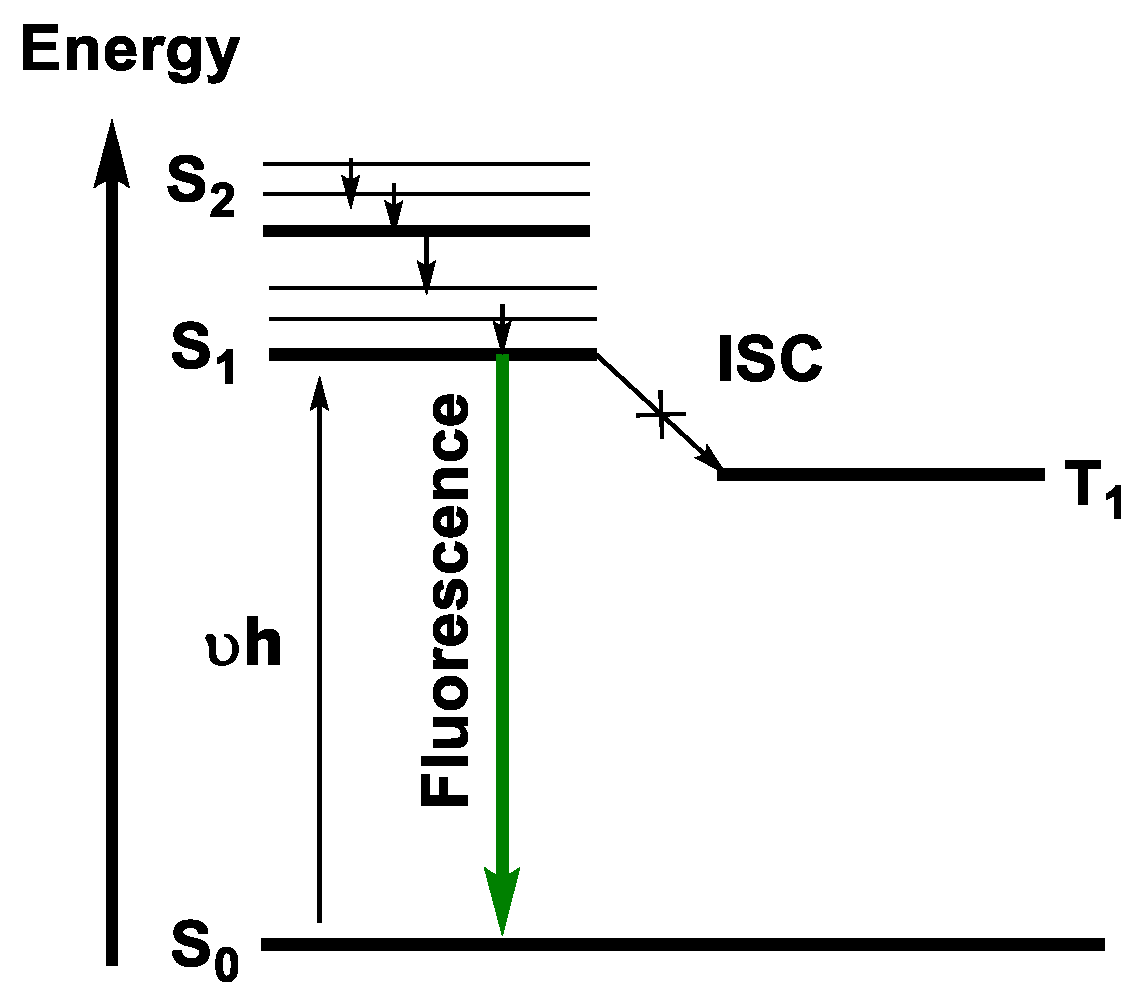
Fluorescence
Quenching Mechanism
1.2.5. Stern–Volmer Relationship
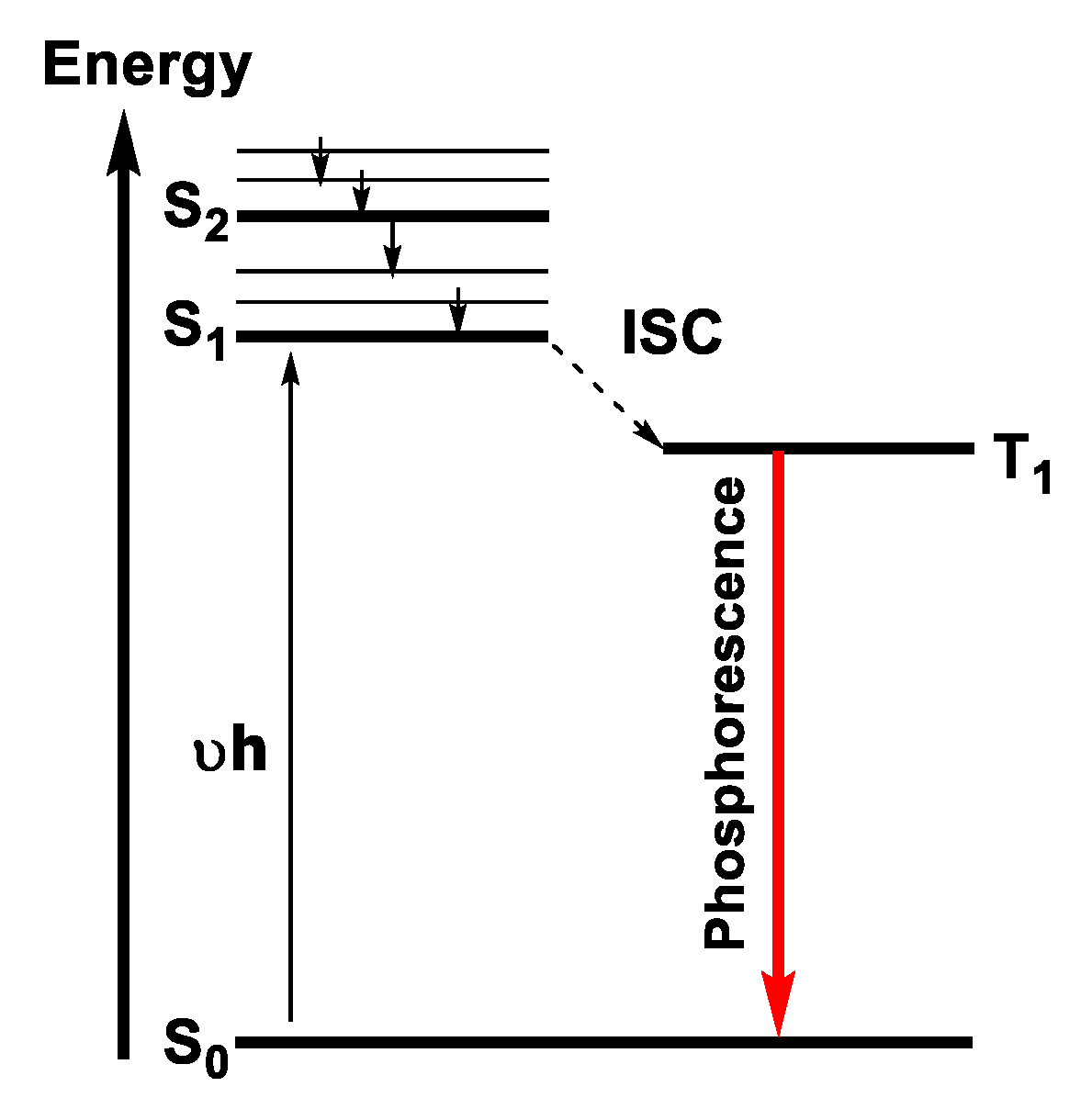
Phosphorescence
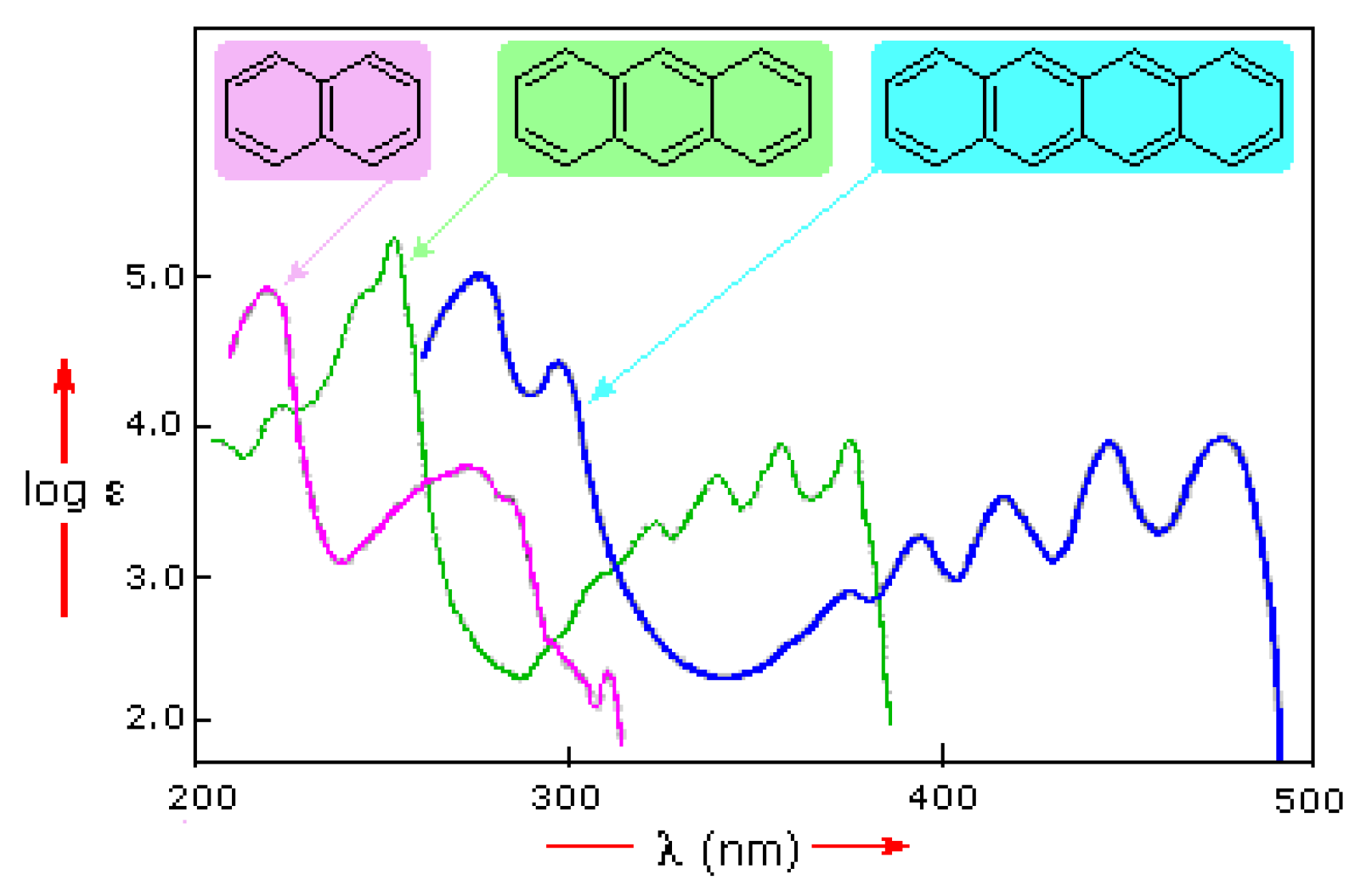
1.3. Anthracene and Its Derivatives
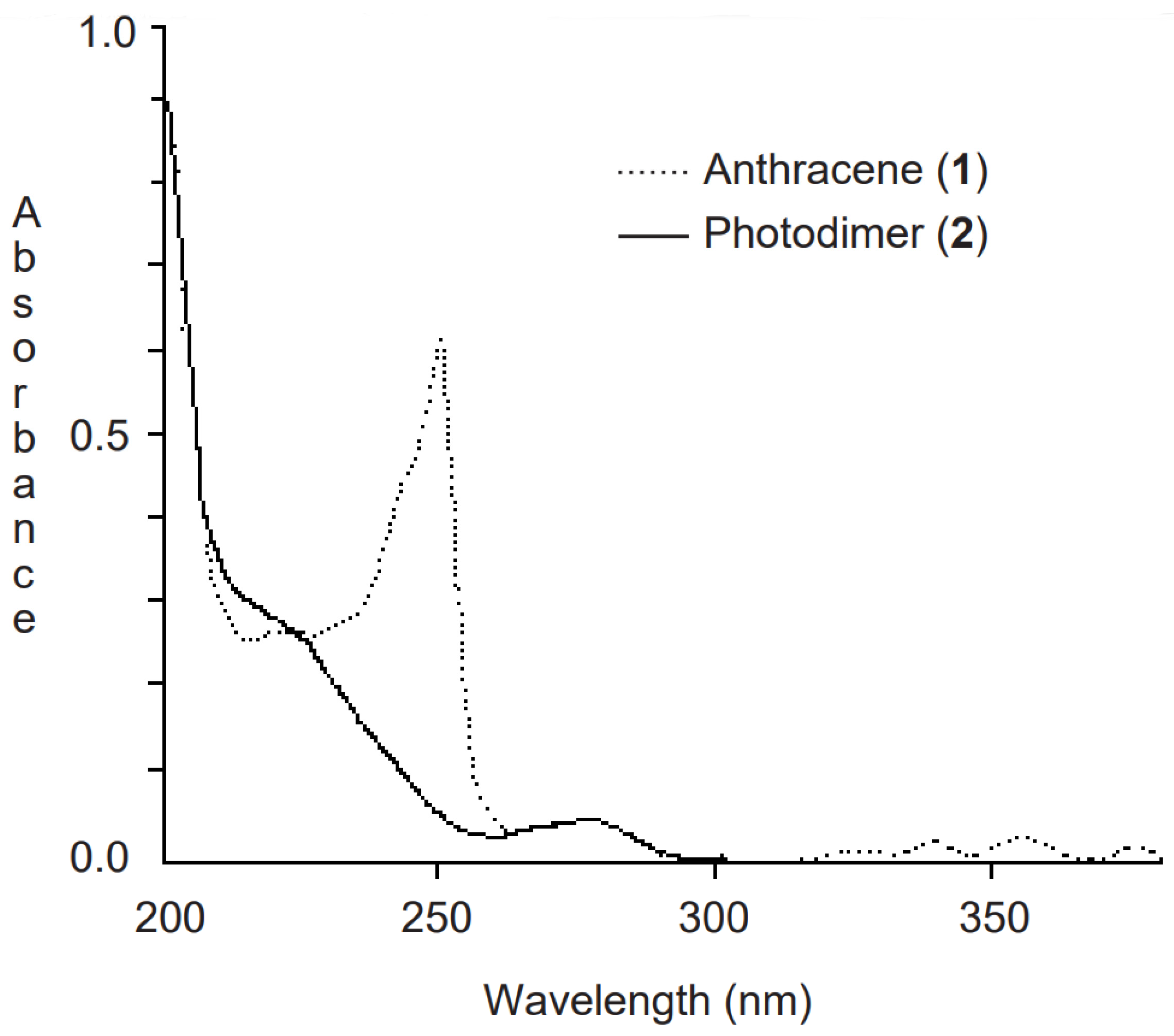
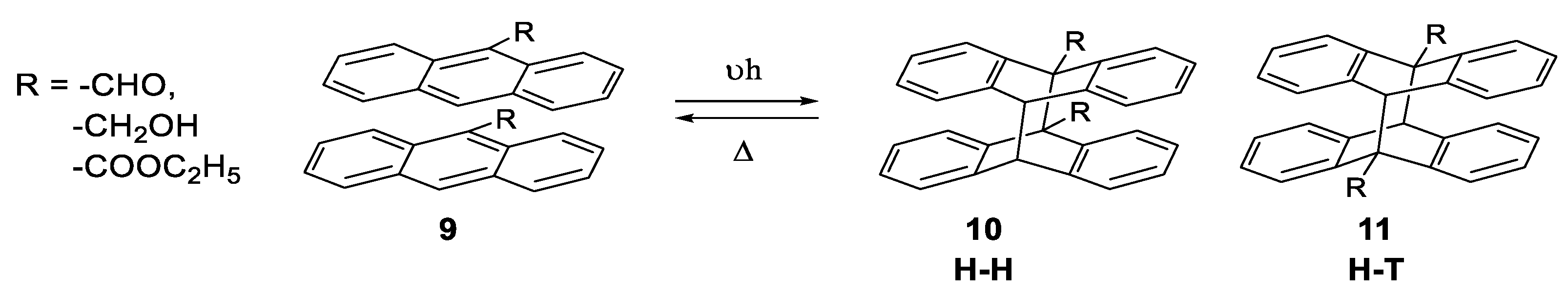
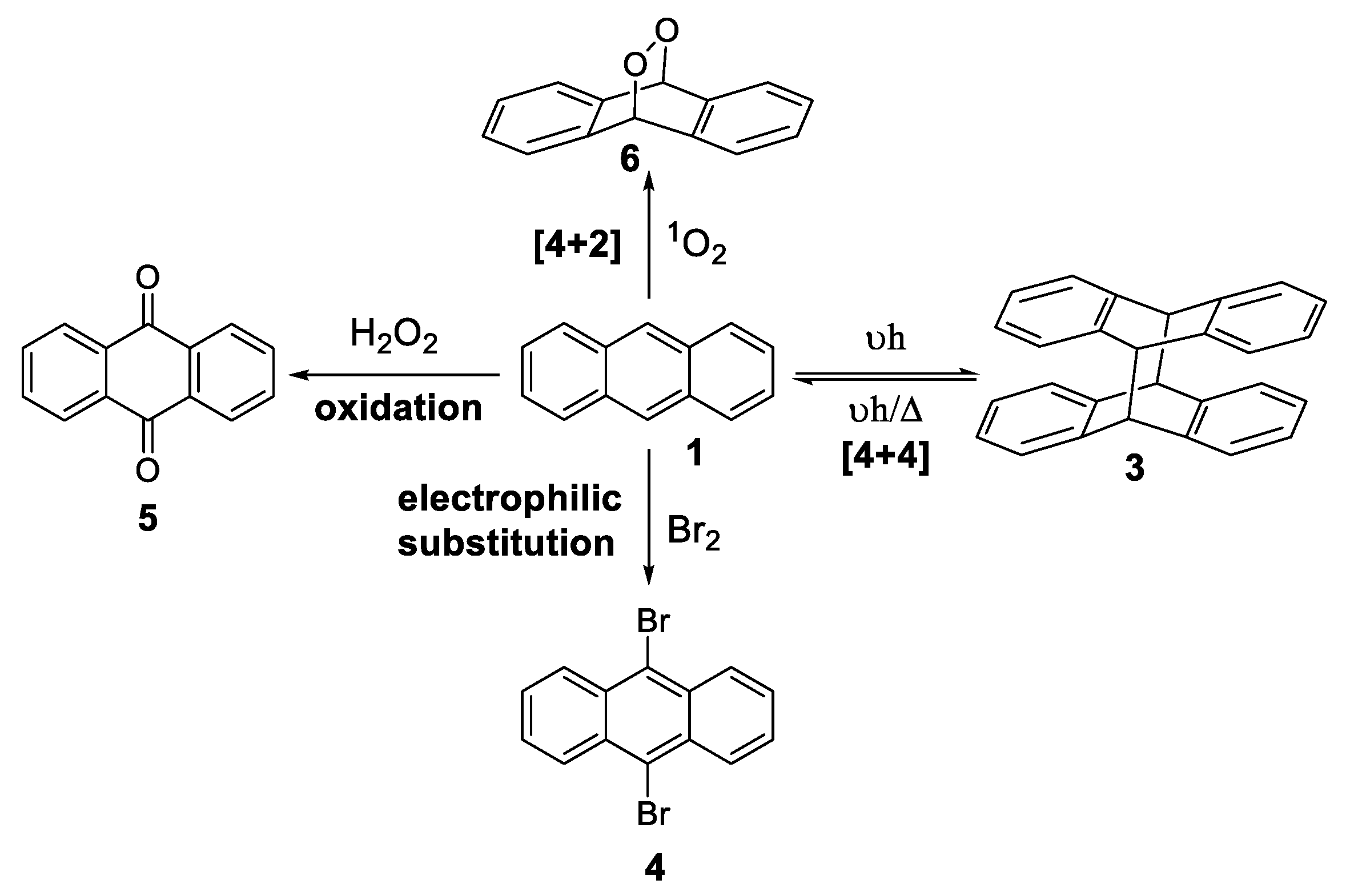
1.3.1. Photodimerization
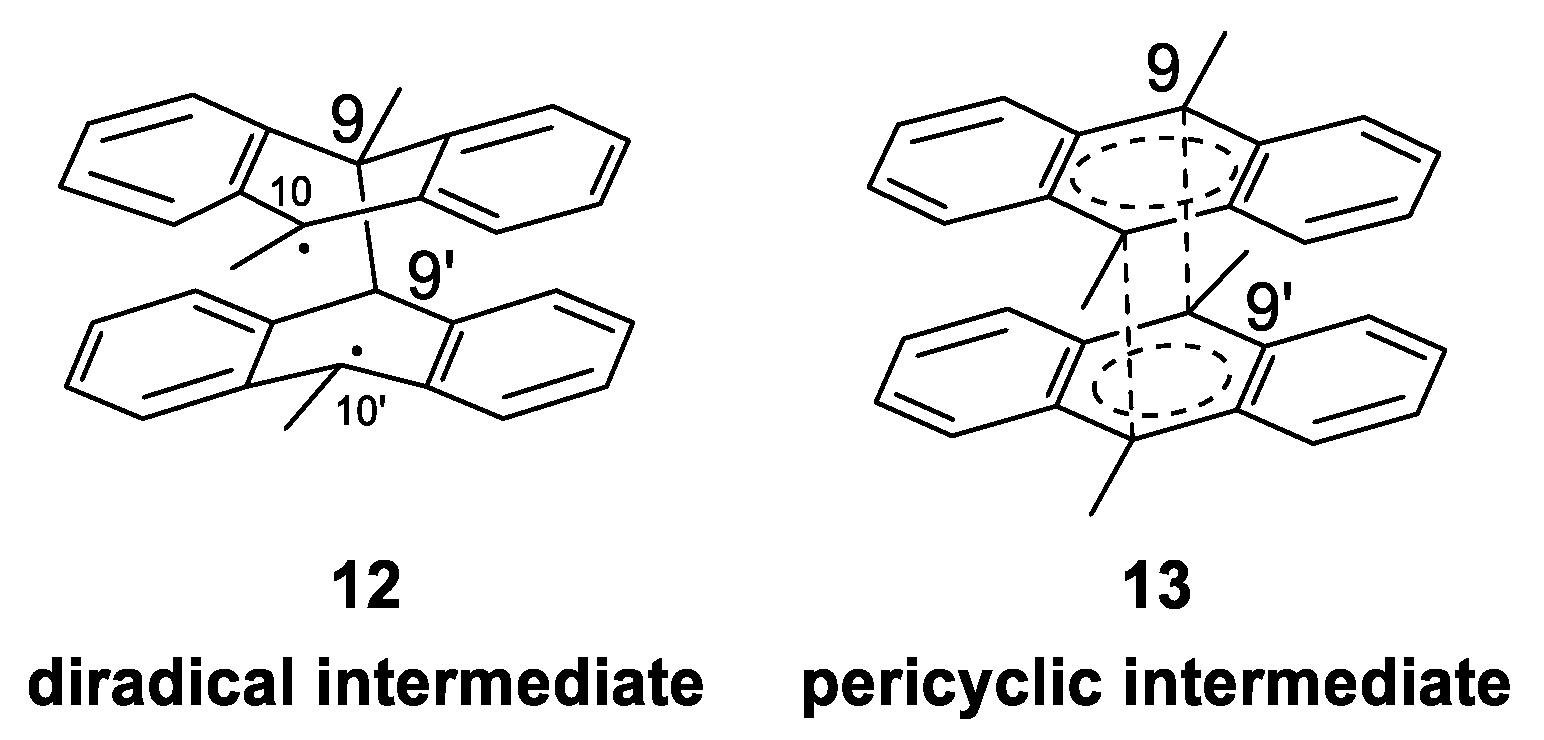
Mechanism of Photodimerization
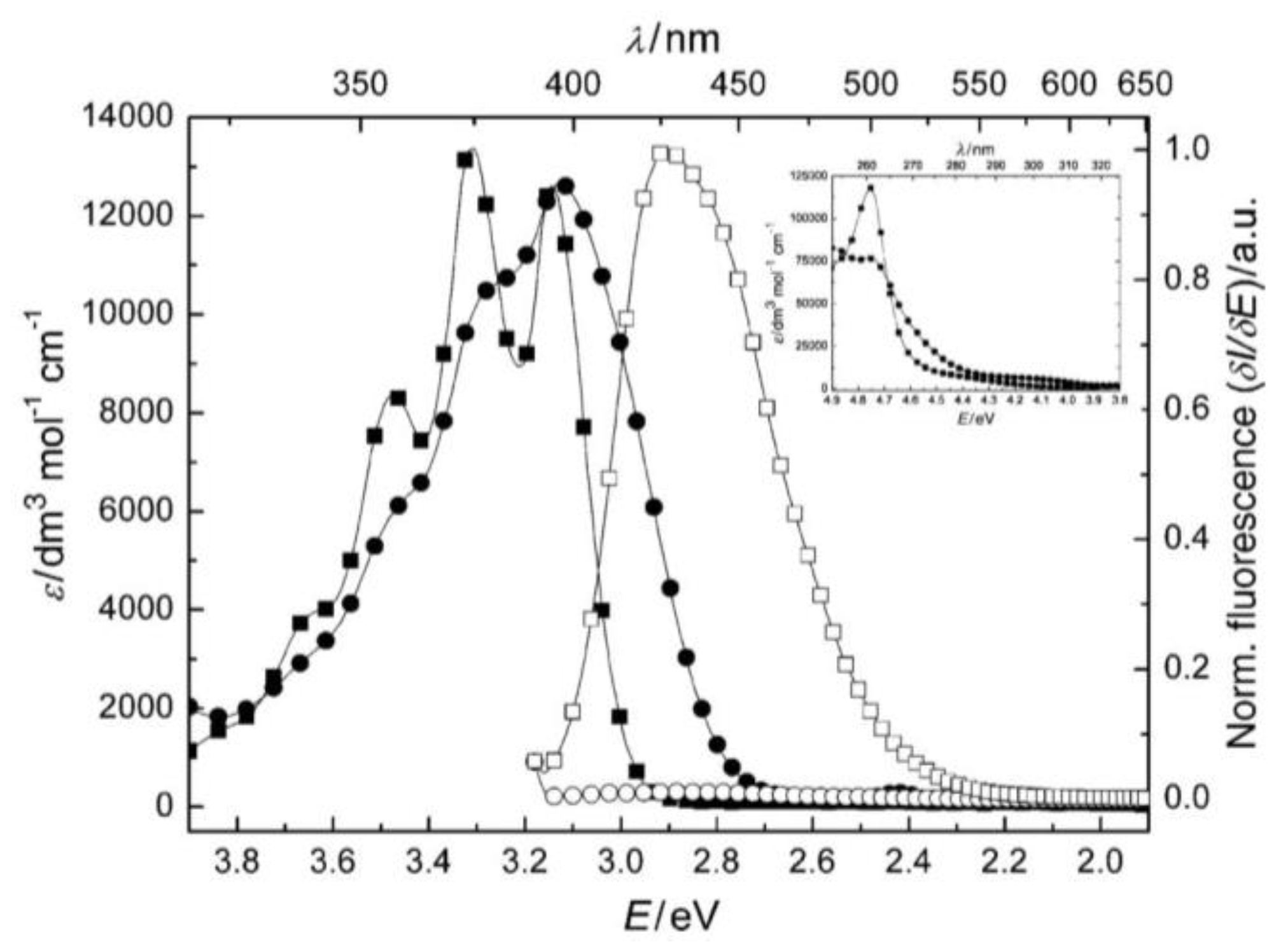
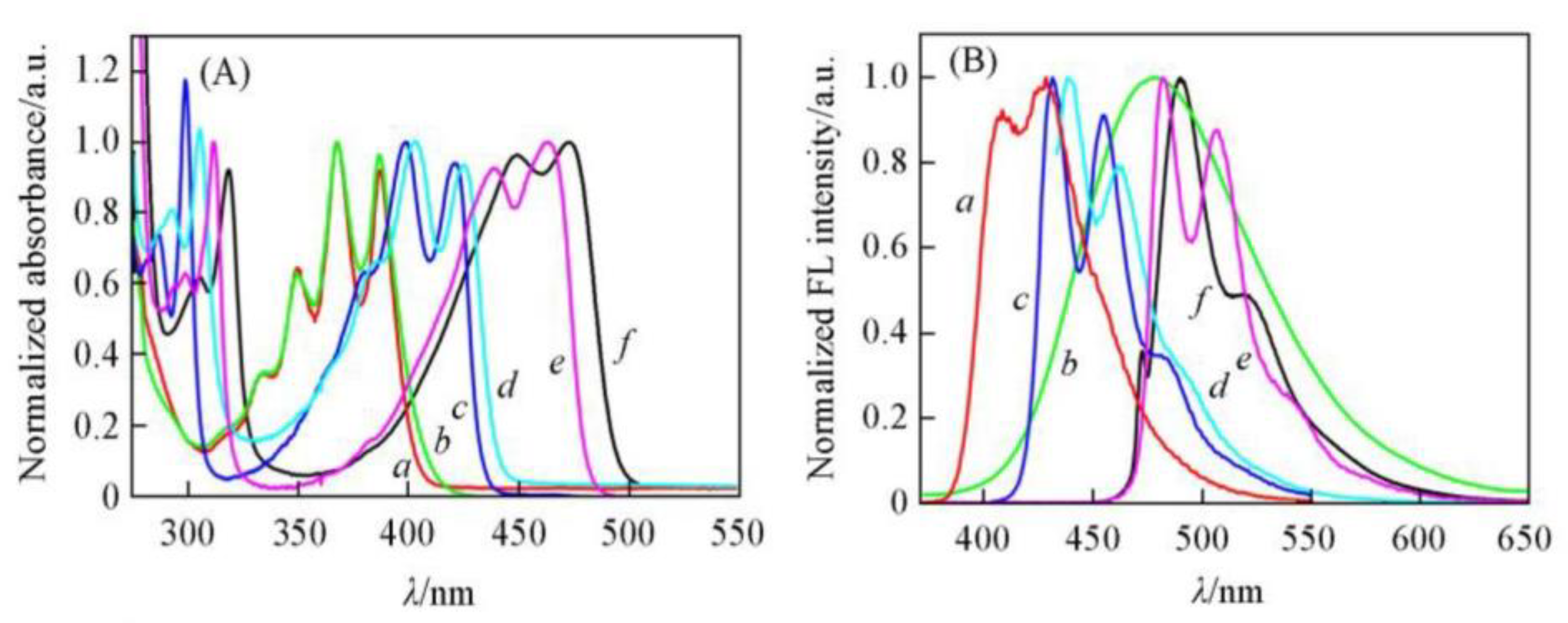
1.3.2. Photophysical Properties
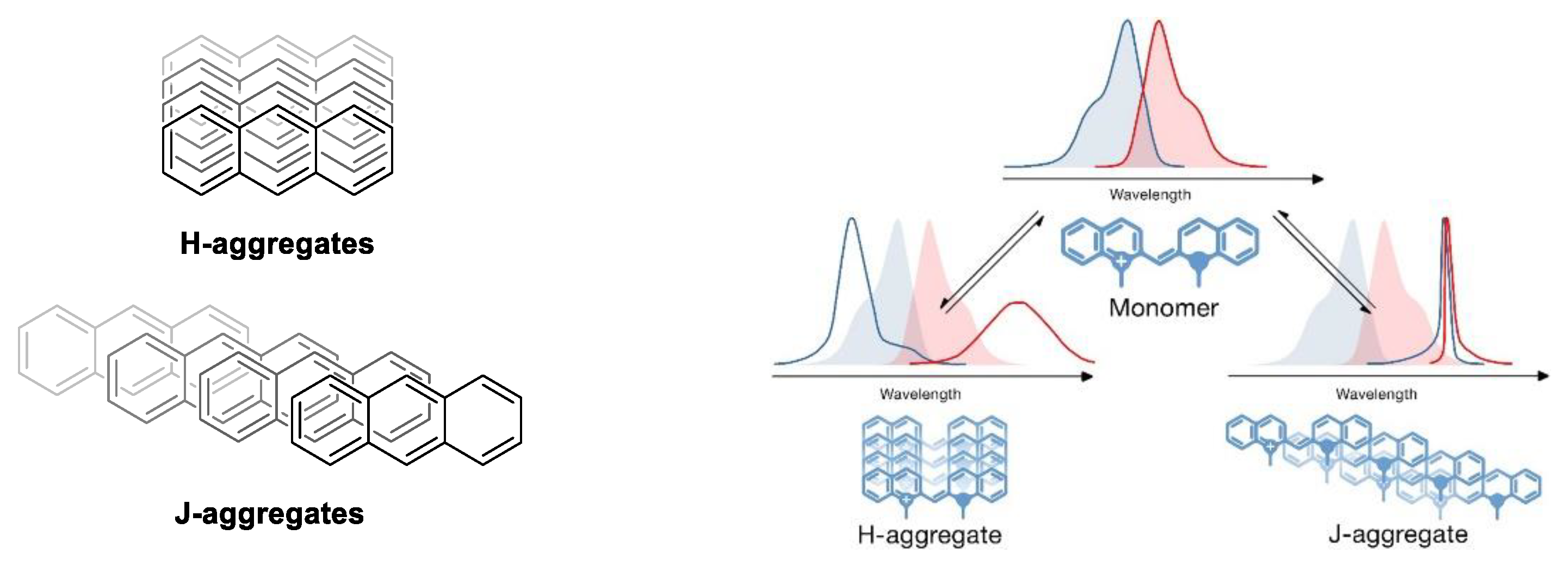
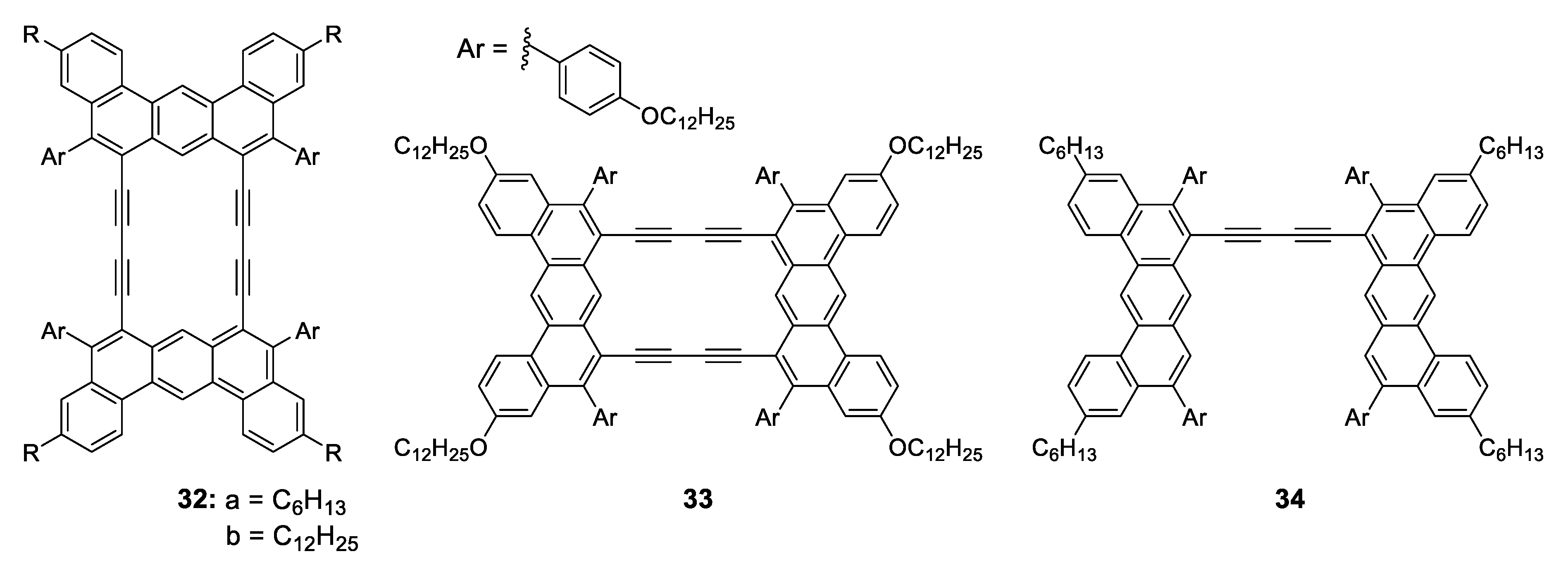
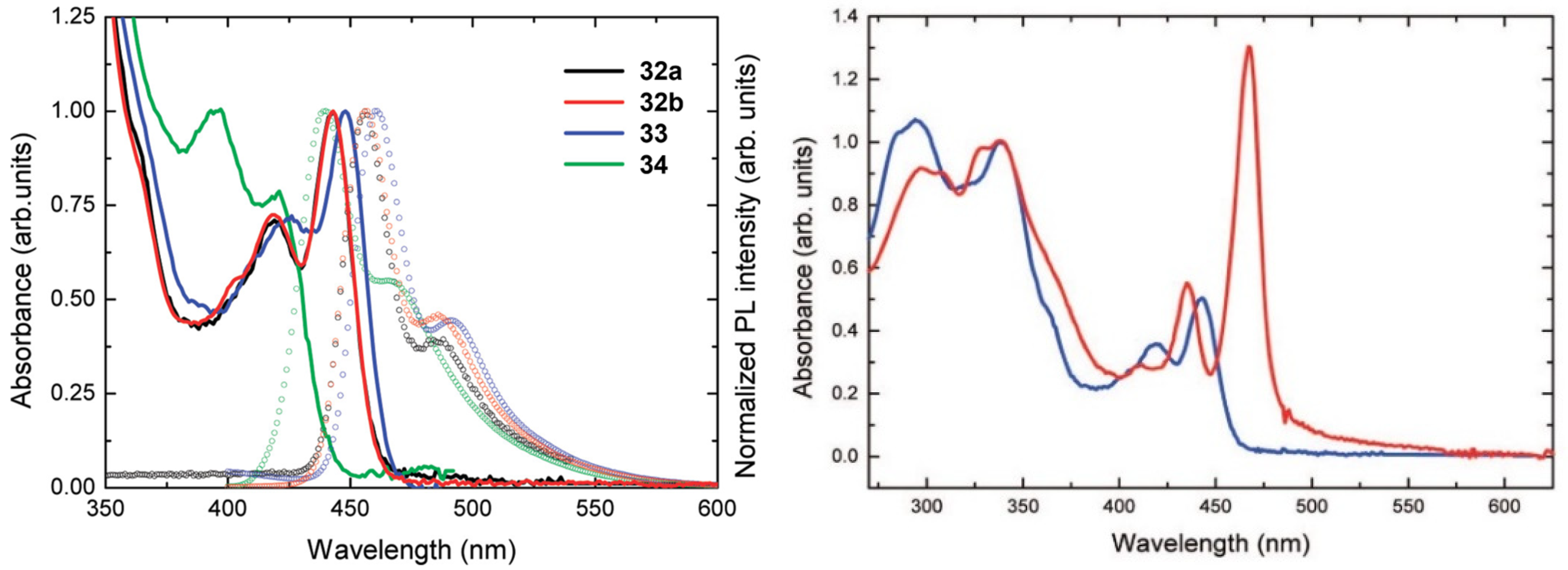
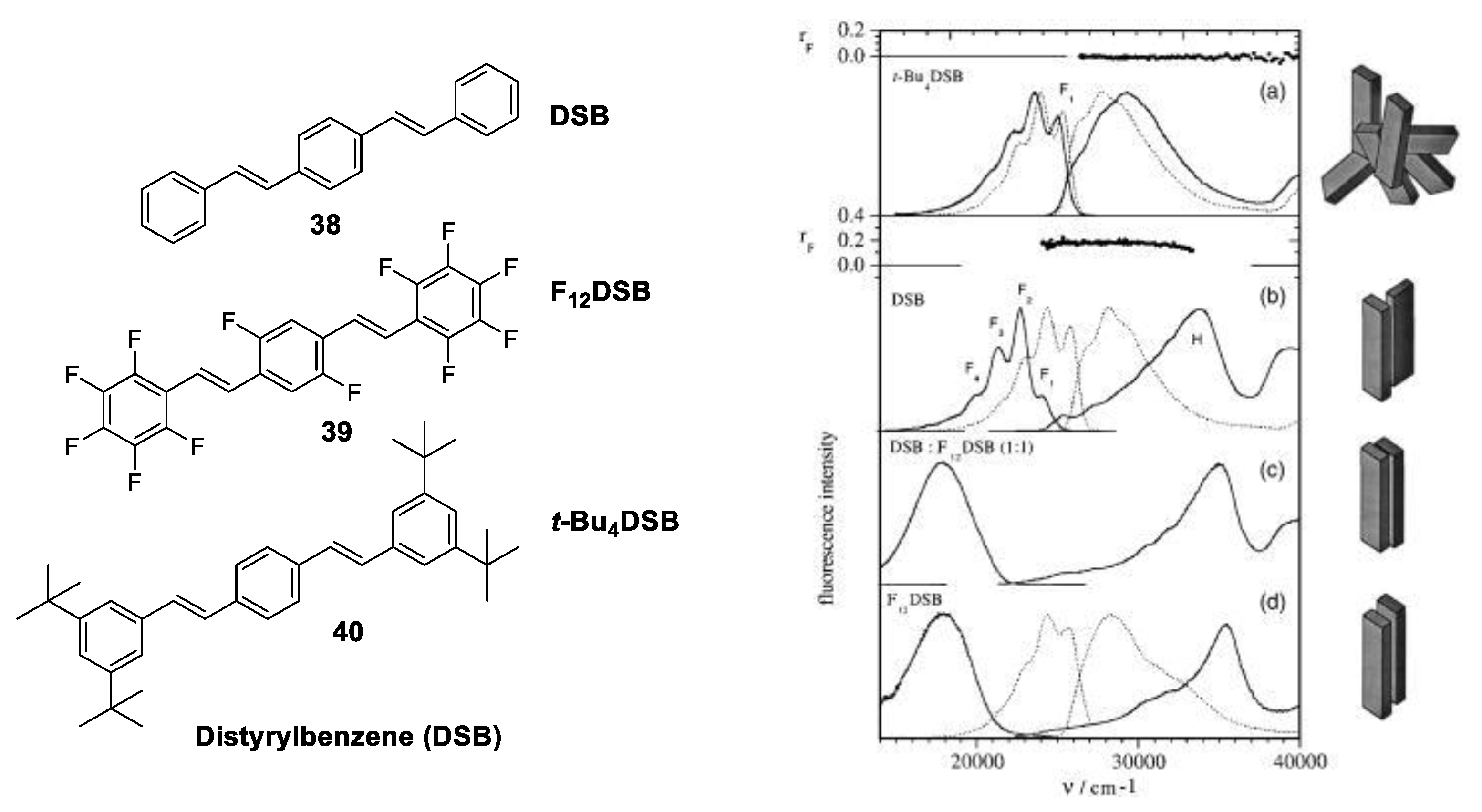
1.3.3. J-Aggregate
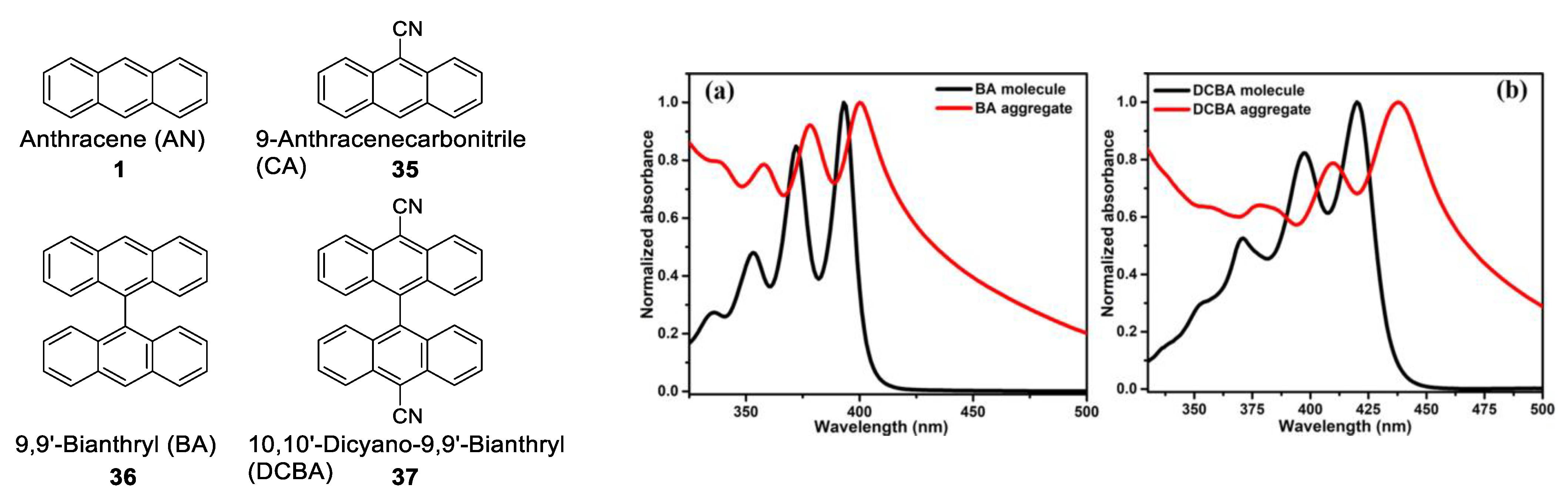
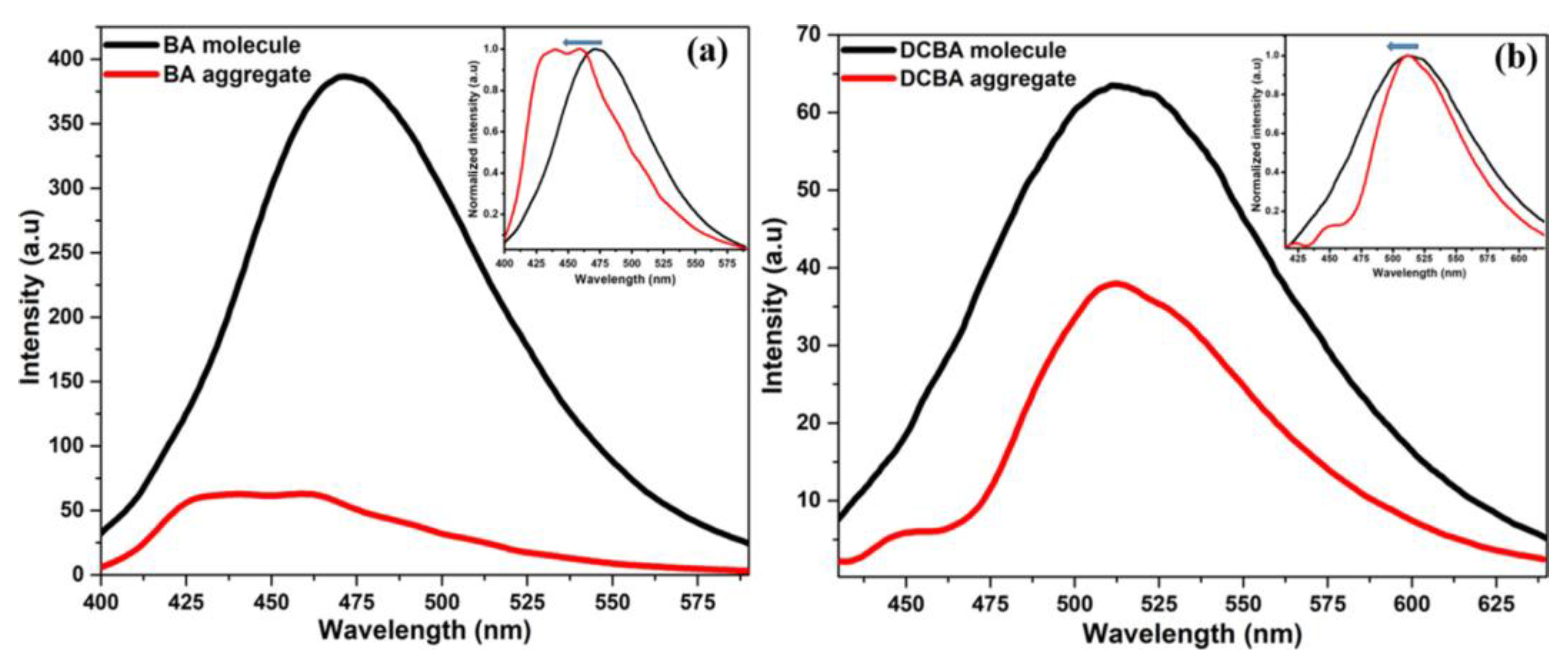
1.3.4. H-Aggregate
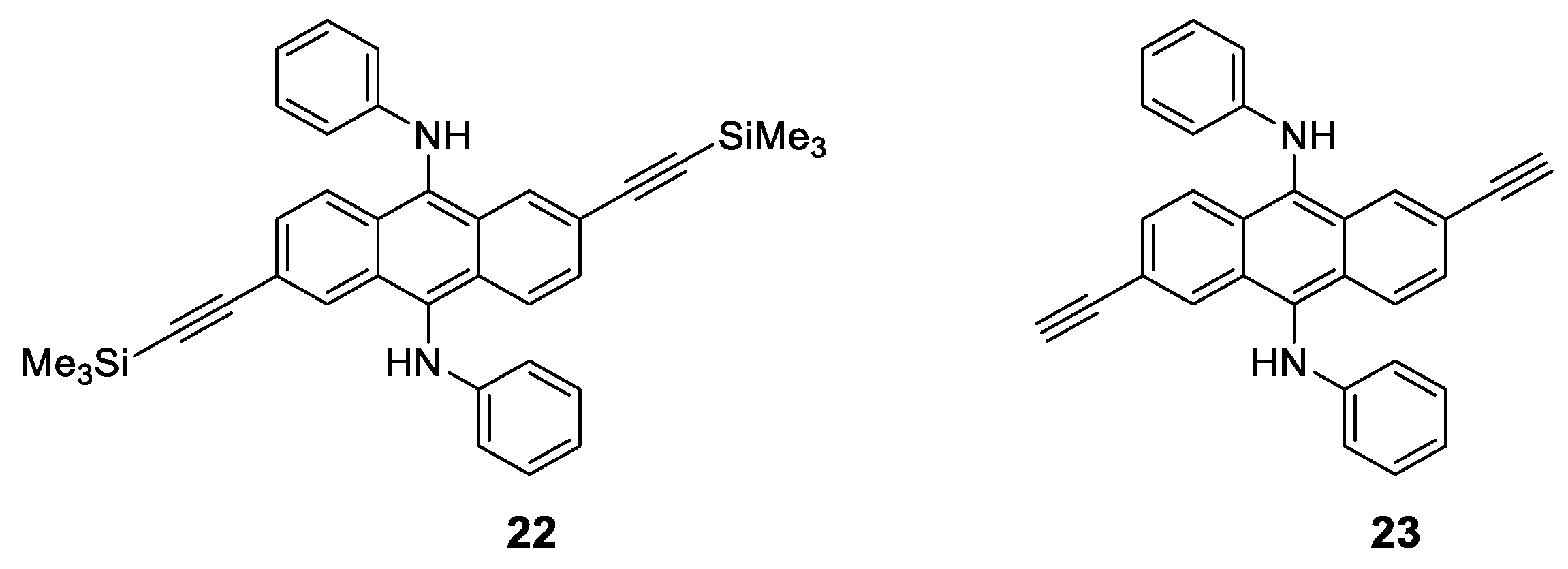
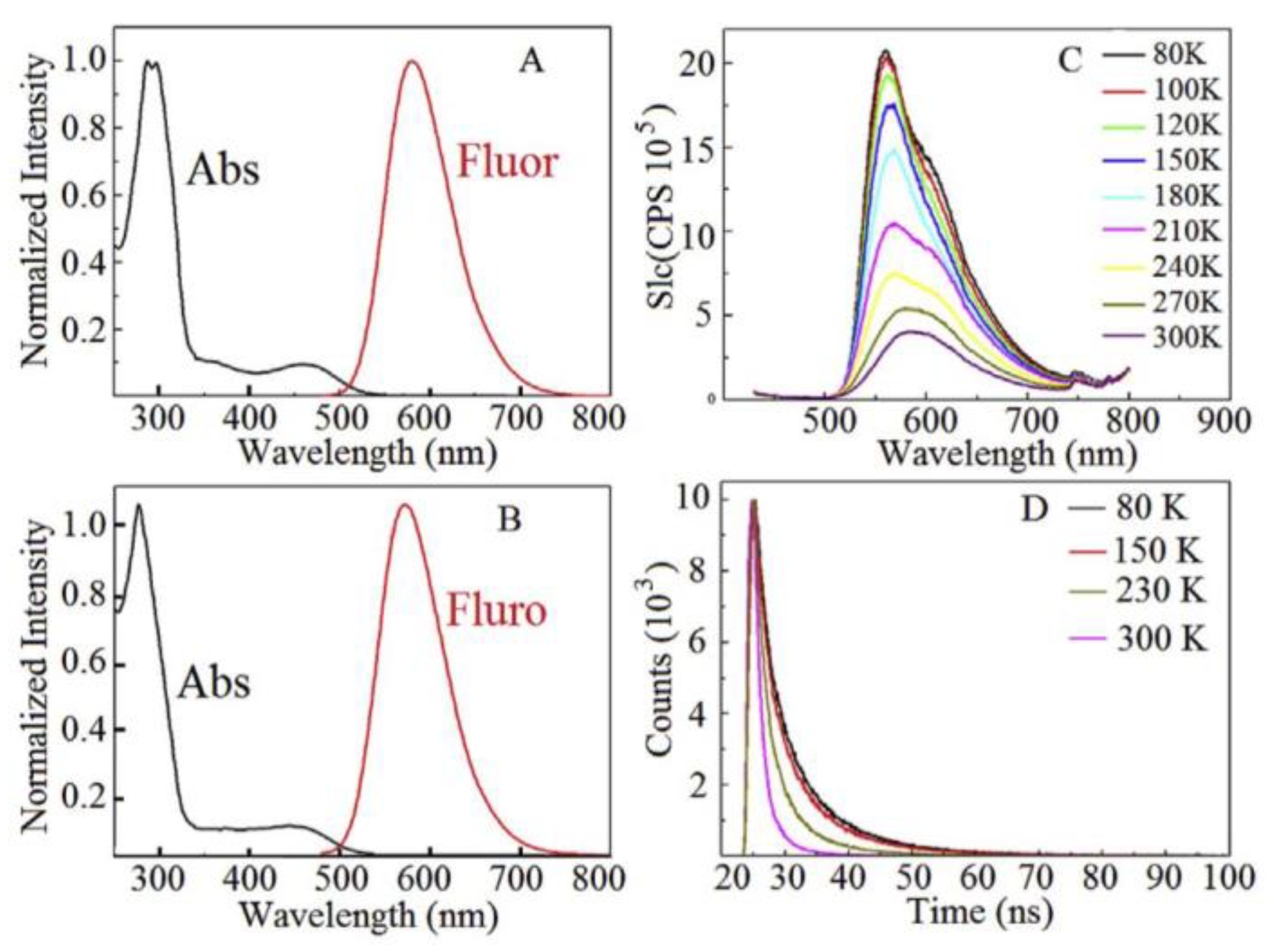
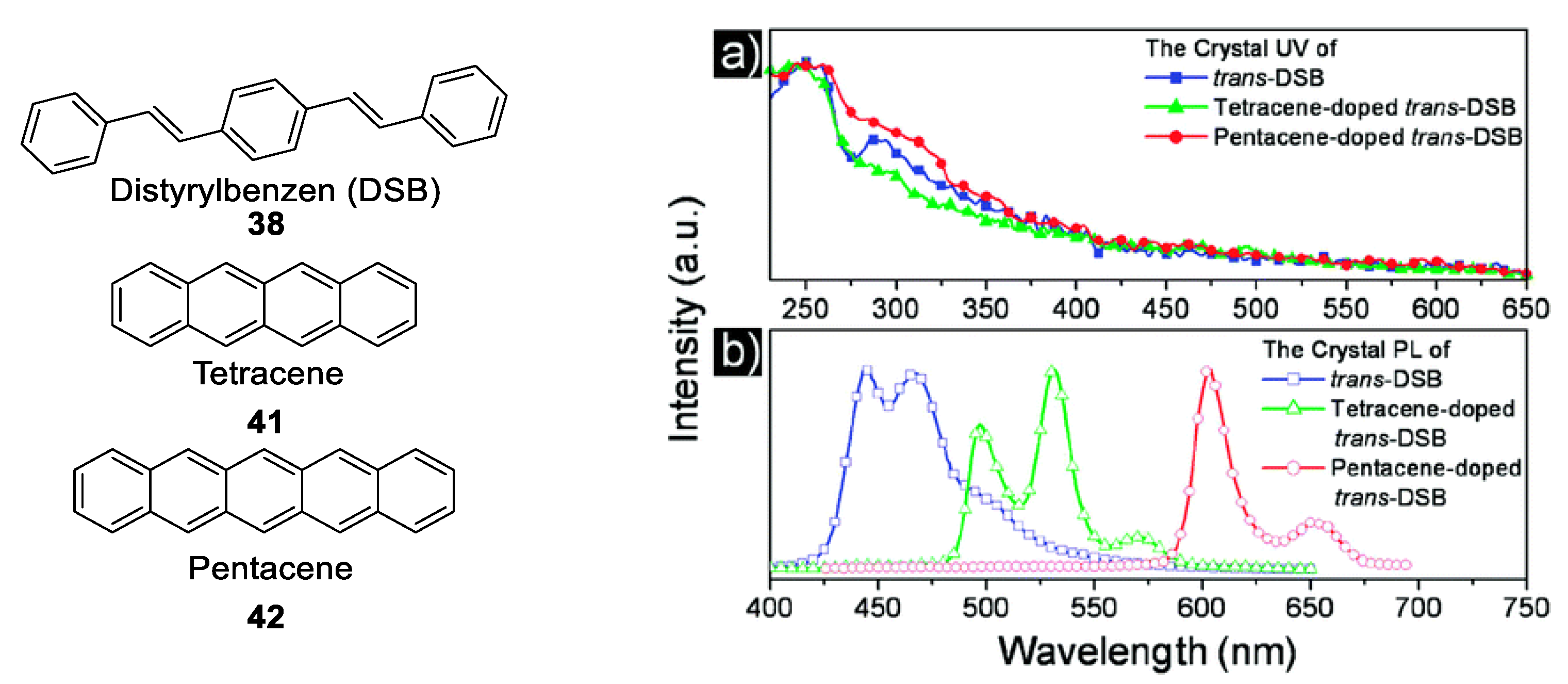
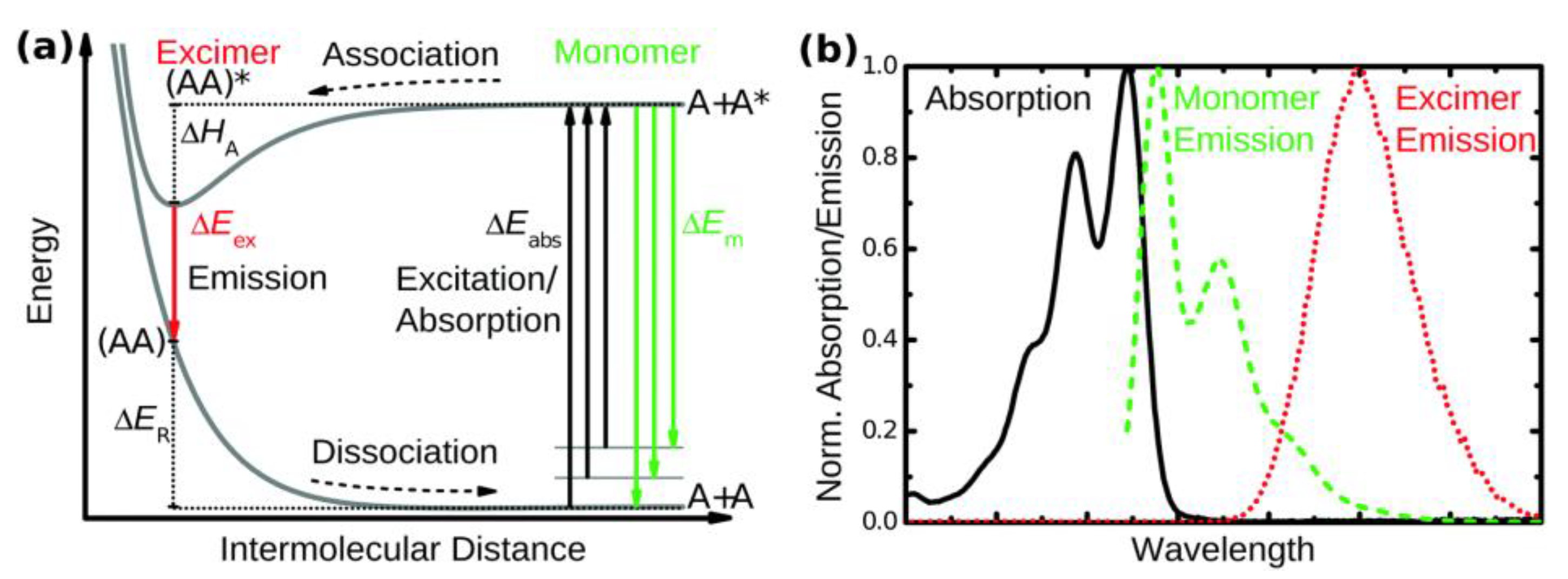
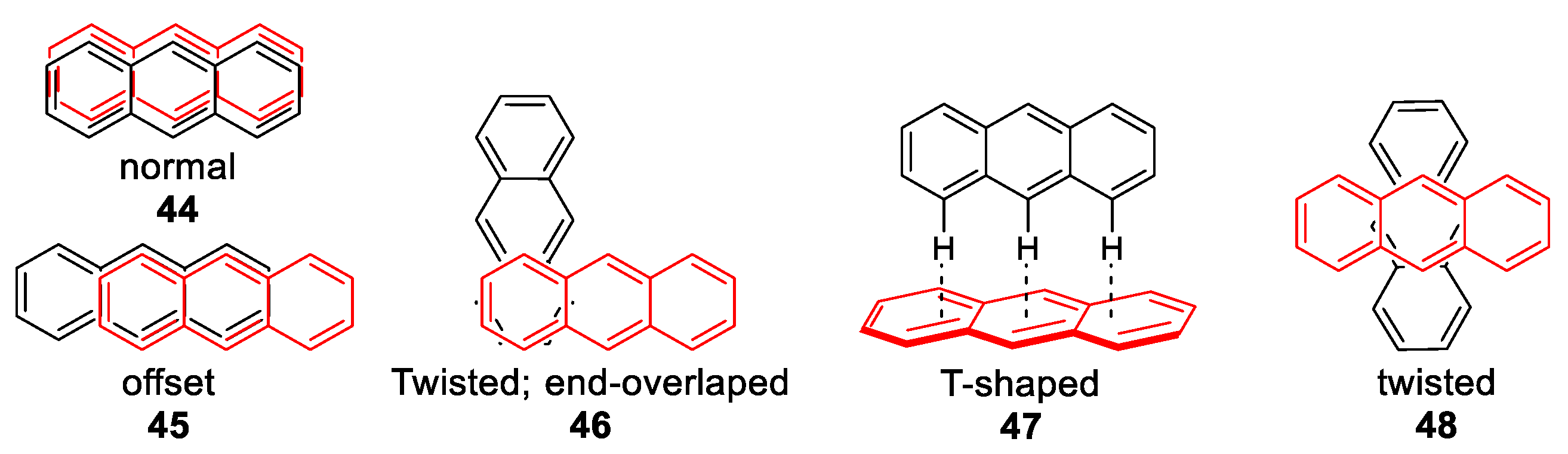
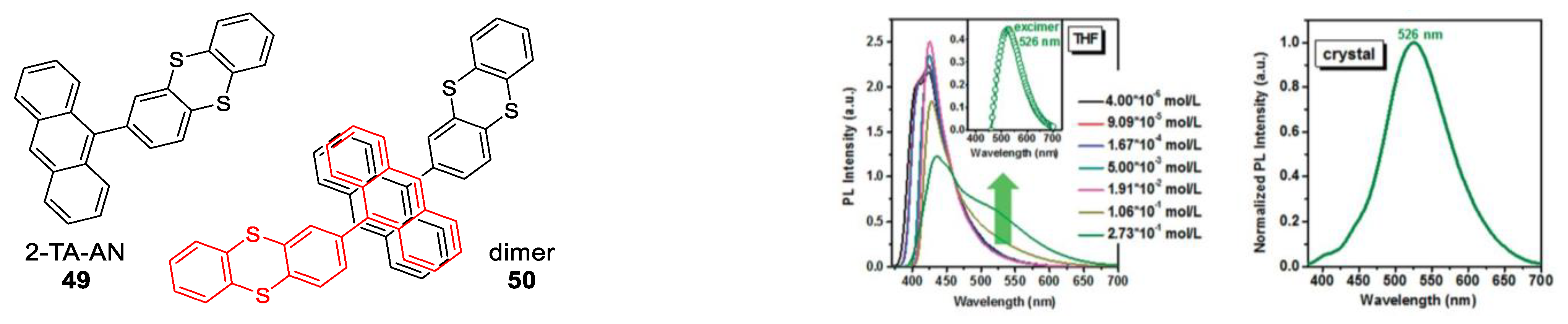
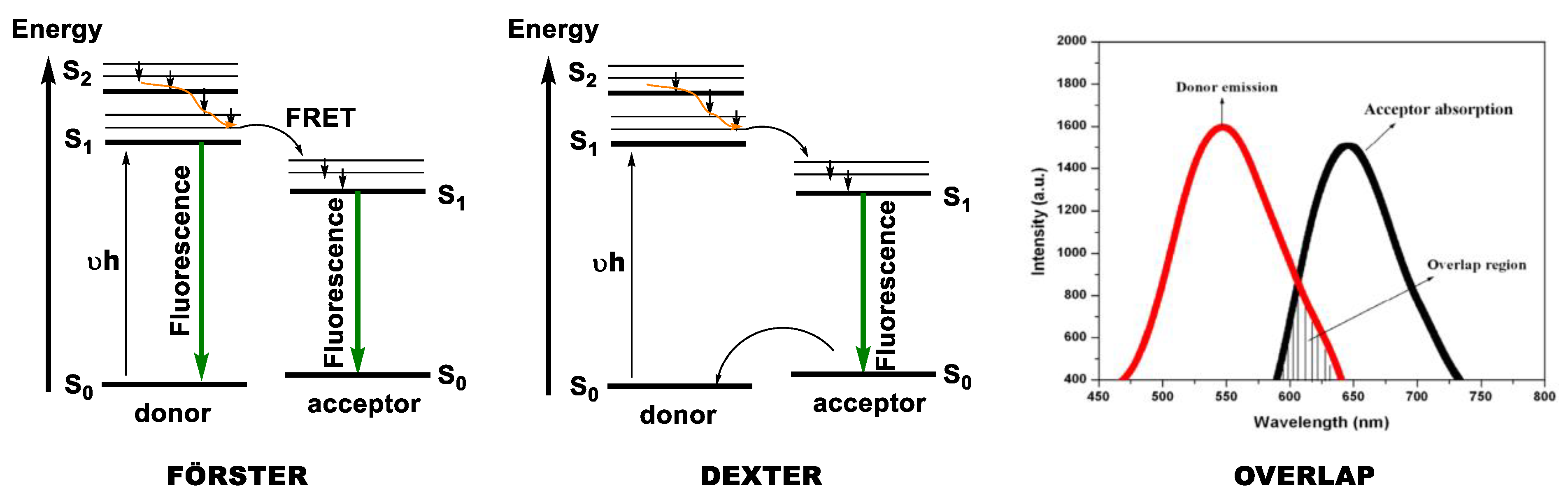
1.4. Excimers
1.5. Exciplexes
1.6. Dyad Emission
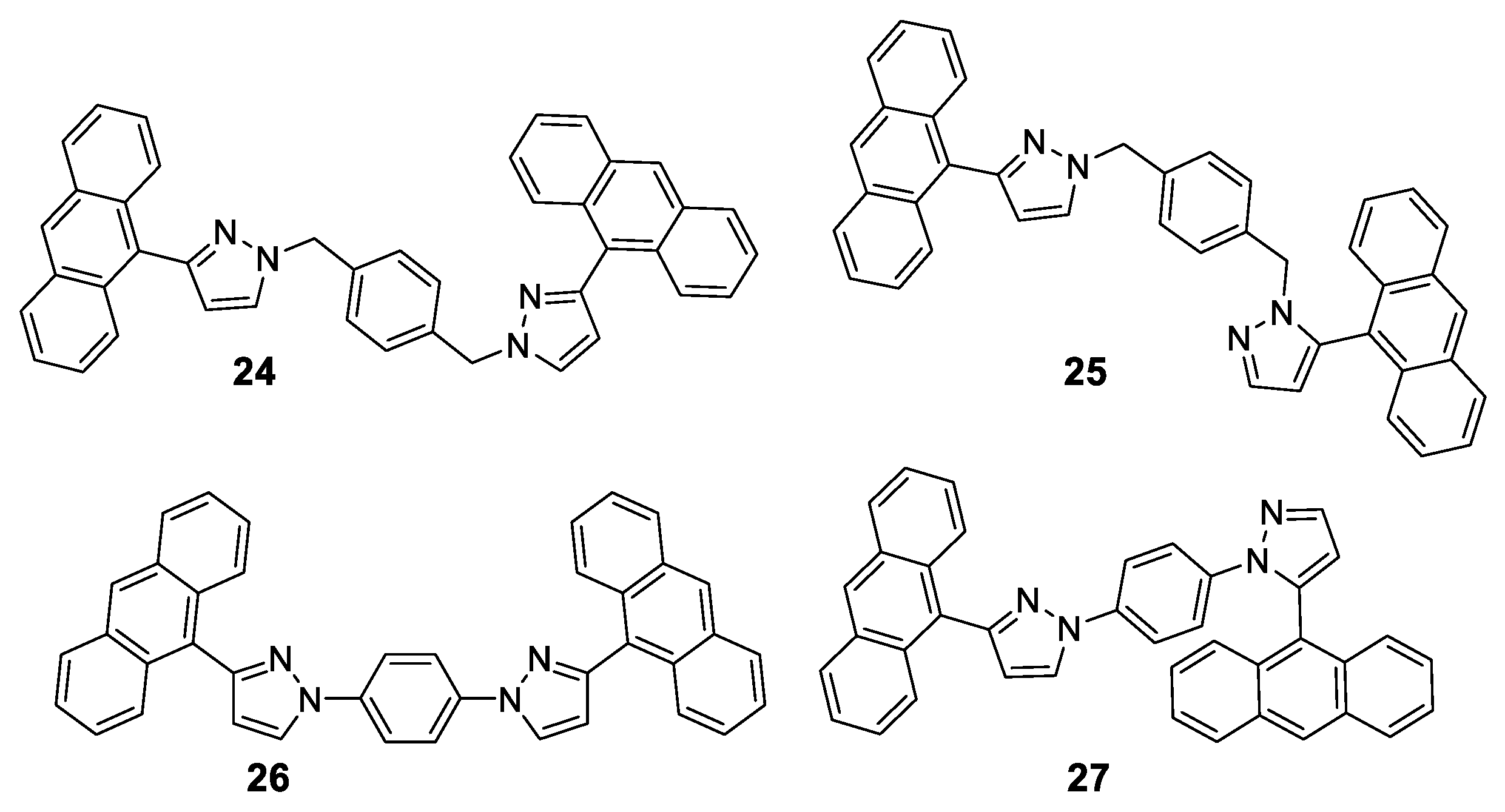
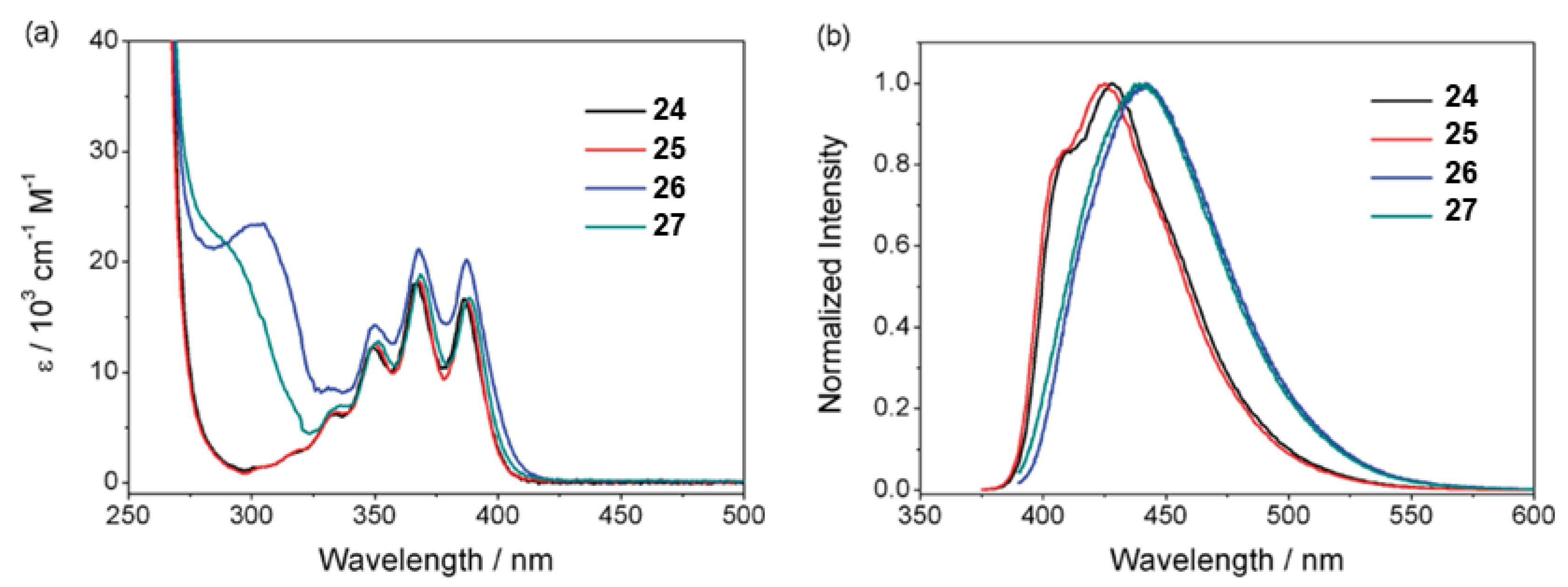
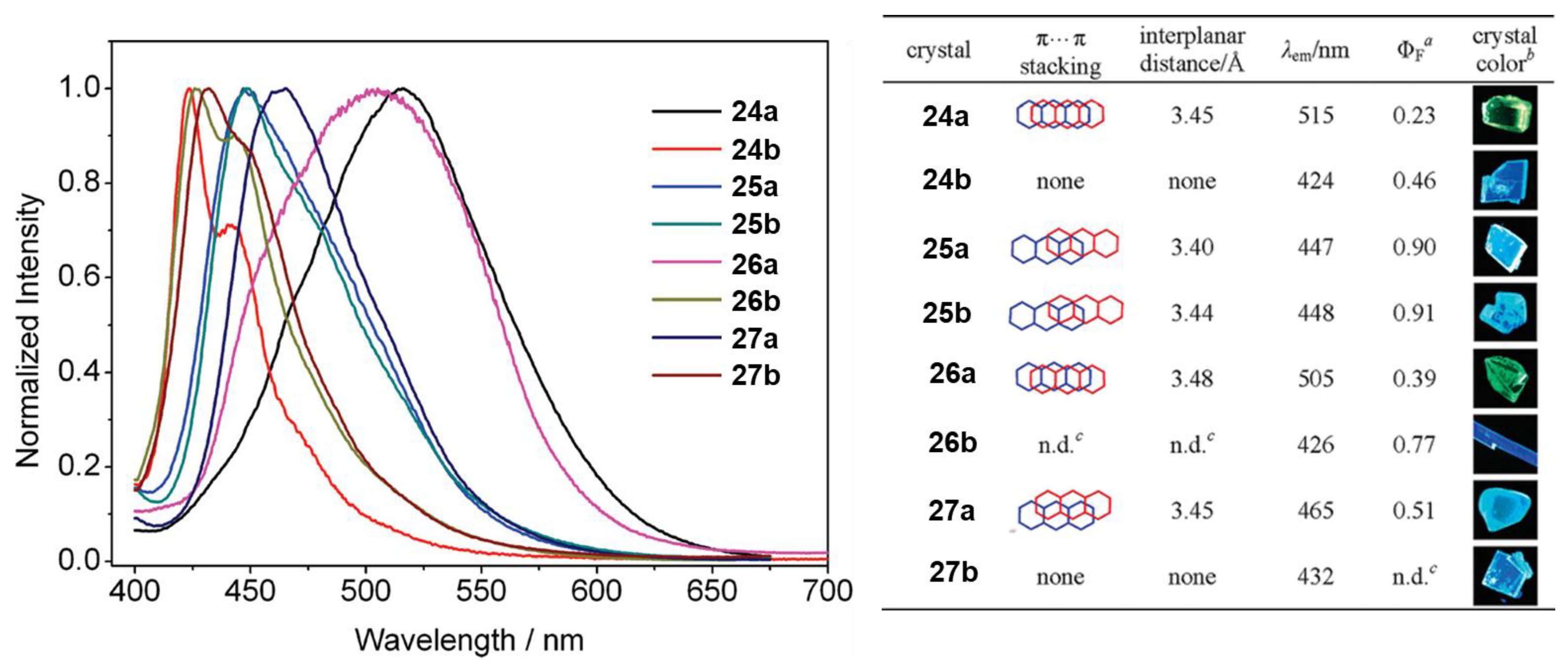
1.7. Triade Emission
1.8. Coordination Polymers (CPs)
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kumar, N. Comprehensive Physics for Class XII; Laxmi Publications: New Delhi, India, 2004. [Google Scholar]
- Tanaka, A.; Makino, A. Photosynthetic research in plant science. Plant Cell Physiol. 2009, 50, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Ciamician, G. The Photochemistry of the Future. Science 1912, 36, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Bayrakçeken, F. Triplet–triplet optical energy transfer from benzophenone to naphthalene in the vapor phase. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 71, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Contributors, P.L. UV-Visible Spectral Features of Benzine and Some PAHs. Available online: https://publiclab.org/notes/warren/8-5-2011/uv-visible-spectral-features-benzine-and-some-pahs (accessed on 9 September 2020).
- Jones, R.N. The Ultraviolet Absorption Spectra of Anthracene Derivatives. Chem. Rev. 1947, 41, 353–371. [Google Scholar] [CrossRef]
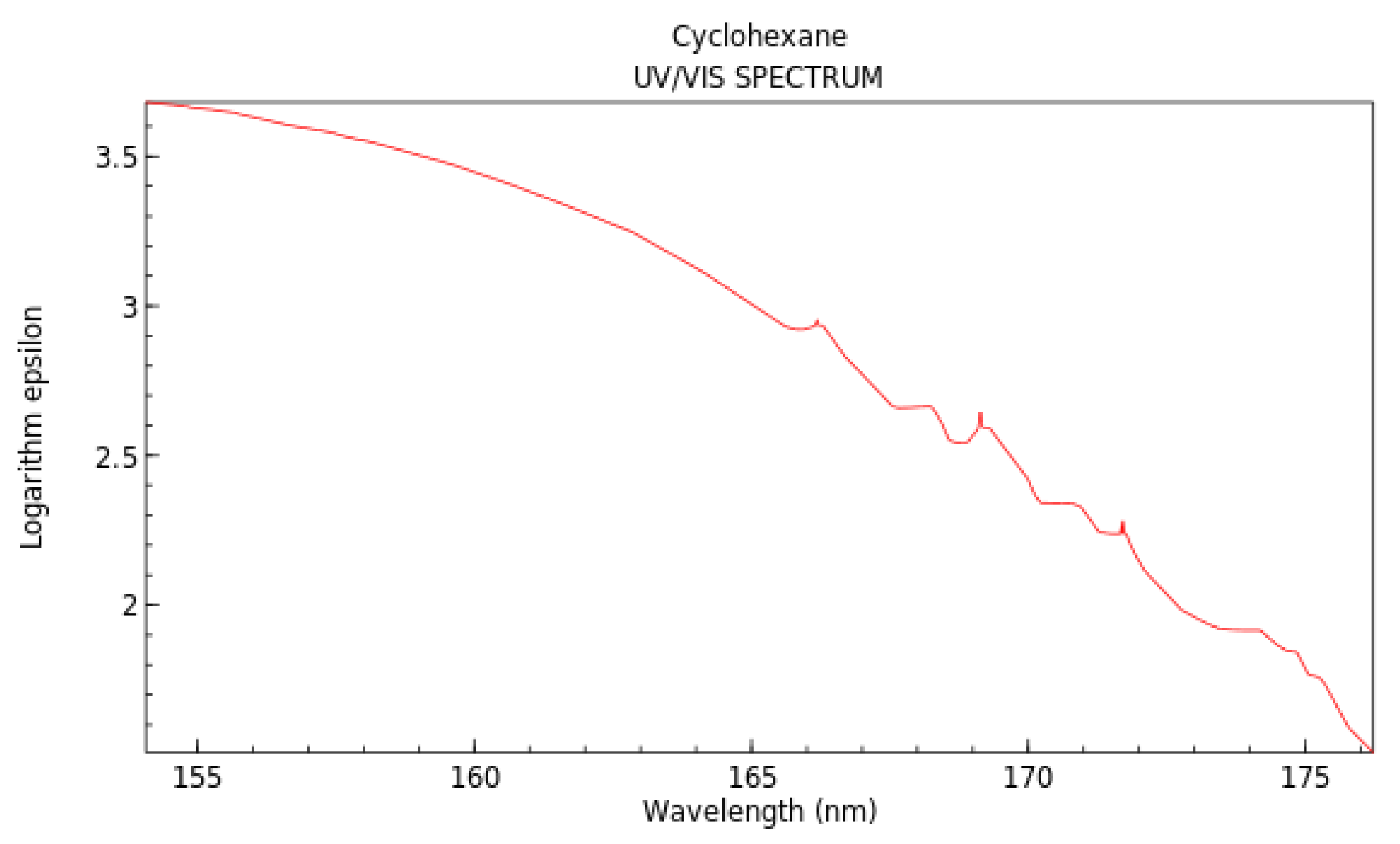
- Lucy, W.; Pickett, L.W.; Muntz, M.; McPherson, E.M. Vacuum Ultraviolet Absorption Spectra of Cyclic Compounds. I. Cyclohexane, Cyclohexene, Cyclopentane, Cyclopentene and Benzene1. J. Am. Chem. Soc. 1951, 73, 4862–4865. [Google Scholar]
- National Institute of Standards and Technology. Cyclohexane. Available online: https://webbook.nist.gov/cgi/cbook.cgi?ID=C110827&Mask=400#UV-Vis-Spec (accessed on 10 September 2020).
- Paris, J.P.; Brandt, W.W. Charge Transfer Luminescence of A Ruthenium(II) Chelate. J. Am. Chem. Soc. 1959, 81, 5001–5002. [Google Scholar] [CrossRef]
- Laporte, O.; Meggers, W.F. Some Rules of Spectral Structure*. J. Opt. Soc. Am. 1925, 11, 459–463. [Google Scholar] [CrossRef]
- Wardle, B. Principles and Applications of Photochemistry; John Wiley: Chichester, UK, 2009. [Google Scholar]
- Lavis, L.D.; Raines, R.T. Bright Ideas for Chemical Biology. ACS Chem. Biol. 2008, 3, 142–155. [Google Scholar] [CrossRef]
- Xu, H.; Chen, R.; Sun, Q.; Lai, W.; Su, Q.; Huang, W.; Liu, X. Recent progress in metal–organic complexes for optoelectronic applications. Chem. Soc. Rev. 2014, 43, 3259–3302. [Google Scholar] [CrossRef]
- Huang, J.; Su, J.-H.; Tian, H. The development of anthracene derivatives for organic light-emitting diodes. J. Mater. Chem. 2012, 22, 10977–10989. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Wang, X.-L.; Zhang, X.; Qin, C.; Li, P.; Su, Z.-M.; Zhu, D.-X.; Shan, G.-G.; Shao, K.-Z.; Wu, H.; et al. Efficient and tunable white-light emission of metal–organic frameworks by iridium-complex encapsulation. Nat. Commun. 2013, 4, 2717. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A. Aggregation-Induced Emission: A Tool for Sensitive Detection of Amines. ChemistrySelect 2019, 4, 12848–12860. [Google Scholar] [CrossRef]
- Jung, H.S.; Kwon, P.S.; Lee, J.W.; Kim, J.I.; Hong, C.S.; Kim, J.W.; Yan, S.; Lee, J.Y.; Lee, J.H.; Joo, T.; et al. Coumarin-Derived Cu2+-Selective Fluorescence Sensor: Synthesis, Mechanisms, and Applications in Living Cells. J. Am. Chem. Soc. 2009, 131, 2008–2012. [Google Scholar] [CrossRef]
- Rosenthal, J.; Lippard, S.J. Direct Detection of Nitroxyl in Aqueous Solution Using a Tripodal Copper(II) BODIPY Complex. J. Am. Chem. Soc. 2010, 132, 5536–5537. [Google Scholar] [CrossRef] [PubMed]
- Montalti, M.; Prodi, L.; Zaccheroni, N. Luminescent Chemosensors Based on Anthracene or Dioxyxanthone Derivatives. J. Fluoresc. 2000, 10, 71. [Google Scholar] [CrossRef]
- Räupke, A.; Palma-Cando, A.; Shkura, E.; Teckhausen, P.; Polywka, A.; Görrn, P.; Scherf, U.; Riedl, T. Highly sensitive gas-phase explosive detection by luminescent microporous polymer networks. Sci. Rep. 2016, 6, 29118. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.-Y.; Dai, M.; Young, D.J.; Ren, Z.-G.; Lang, J.-P. Luminescent Zn(II) Coordination Polymers for Highly Selective Sensing of Cr(III) and Cr(VI) in Water. Inorg. Chem. 2017, 56, 4668–4678. [Google Scholar] [CrossRef] [PubMed]
- Haldar, R.; Prasad, K.; Samanta, P.K.; Pati, S.; Maji, T.K. Luminescent Metal–Organic Complexes of Pyrene or Anthracene Chromophores: Energy Transfer Assisted Amplified Exciplex Emission and Al3+ Sensing. Cryst. Growth Des. 2016, 16, 82–91. [Google Scholar] [CrossRef]
- Vasylevskyi, S.I.; Bassani, D.M.; Fromm, K.M. Anion-Induced Structural Diversity of Zn and Cd Coordination Polymers Based on Bis-9,10-(pyridine-4-yl)-anthracene, Their Luminescent Properties, and Highly Efficient Sensing of Nitro Derivatives and Herbicides. Inorg. Chem. 2019, 58, 5646–5653. [Google Scholar] [CrossRef]
- Gong, W.-J.; Ren, Z.-G.; Li, H.-X.; Zhang, J.-G.; Lang, J.-P. Cadmium(II) Coordination Polymers of 4-Pyr-poly-2-ene and Carboxylates: Construction, Structure, and Photochemical Double [2 + 2] Cycloaddition and Luminescent Sensing of Nitroaromatics and Mercury(II) Ions. Cryst. Growth Des. 2017, 17, 870–881. [Google Scholar] [CrossRef]
- Shaligram, S.; Wadgaonkar, P.P.; Kharul, U.K. Fluorescent polymeric ionic liquids for the detection of nitroaromatic explosives. J. Mater. Chem. A 2014, 2, 13983–13989. [Google Scholar] [CrossRef]
- Vasylevskyi, S.I.; Regeta, K.; Ruggi, A.; Petoud, S.; Piguet, C.; Fromm, K.M. cis- and trans-9,10-di(1H-imidazol-1-yl)-anthracene based coordination polymers of ZnII and CdII: Synthesis, crystal structures and luminescence properties. Dalt Trans. 2018, 47, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Shanmugaraju, S.; Mukherjee, P.S. π-Electron rich small molecule sensors for the recognition of nitroaromatics. Chem. Commun. 2015, 51, 16014–16032. [Google Scholar] [CrossRef] [PubMed]
- Shanmugaraju, S.; Mukherjee, P.S. Self-Assembled Discrete Molecules for Sensing Nitroaromatics. Chem. Eur. J. 2015, 21, 6656–6666. [Google Scholar] [CrossRef] [PubMed]
- Nagarkar, S.S.; Joarder, B.; Chaudhari, A.K.; Mukherjee, S.; Ghosh, S.K. Highly Selective Detection of Nitro Explosives by a Luminescent Metal–Organic Framework. Angew. Chem. Int. Ed. 2013, 52, 2881–2885. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. Constructing and Exploiting the Fluorescent Protein Paintbox (Nobel Lecture). Angew. Chem. Int. Ed. 2009, 48, 5612–5626. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, O.V.; Stepanenko, O.V.; Shcherbakova, D.M.; Kuznetsova, I.M.; Turoverov, K.K.; Verkhusha, V.V. Modern fluorescent proteins: From chromophore formation to novel intracellular applications. BioTechniques 2011, 51, 313–327. [Google Scholar] [CrossRef]
- Drobizhev, M.; Makarov, N.S.; Tillo, S.E.; Hughes, T.E.; Rebane, A. Two-photon absorption properties of fluorescent proteins. Nat. Methods 2011, 8, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; van Weeren, L.; Hink, M.A.; Vischer, N.O.E.; Jalink, K.; Gadella, T.W.J. Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat. Methods 2010, 7, 137–139. [Google Scholar] [CrossRef]
- Zimmer, M. GFP: From jellyfish to the Nobel prize and beyond. Chem. Soc. Rev. 2009, 38, 2823–2832. [Google Scholar] [CrossRef]
- Rae, M.; Fedorov, A.; Berberan-Santos, M.N. Fluorescence quenching with exponential distance dependence: Application to the external heavy-atom effect. J. Chem. Phys. 2003, 119, 2223–2231. [Google Scholar] [CrossRef]
- Najbar, J.; Mac, M. Mechanisms of fluorescence quenching of aromatic molecules by potassium iodide and potassium bromide in methanol–ethanol solutions. J. Chem. Soc. Faraday Trans. 1991, 87, 1523–1529. [Google Scholar] [CrossRef]
- Probst, B.; Guttentag, M.; Rodenberg, A.; Hamm, P.; Alberto, R. Photocatalytic H2 Production from Water with Rhenium and Cobalt Complexes. Inorg. Chem. 2011, 50, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Förster, T. Fluorescence of Organic Compounds; Vandenhoeck und Ruprecht: Gottingen, Germany, 1951; p. 312. [Google Scholar]
- Chamorro-Garcia, A.; Merkoçi, A. Nanobiosensors in diagnostics. Nanobiomedicine 2016, 3, 1849543516663574. [Google Scholar] [CrossRef]
- Srivastava, R.; Kamalasanan, M.N.; Chauhan, G.; Kumar, A.; Tyagi, P.; Kumar, A. Organic Light Emitting Diodes for White Light Emission. In Organic Light Emitting Diode; Mazzeo, M., Ed.; InTech: London, UK, 2010; ISBN 978-953-307-140-4. [Google Scholar]
- Dexter, D.L. A Theory of Sensitized Luminescence in Solids. J. Chem. Phys. 1953, 21, 836–850. [Google Scholar] [CrossRef]
- Rehm, D.; Weller, A. Kinetik und Mechanismus der Elektronübertragung bei der Fluoreszenzlöschung in Acetonitril. Zeitschrift für Elektrochemie, Berichte der Bunsengesellschaft für physikalische Chemie (Zeitschrift für Elektrochemie, Berichte der Bunsengesellschaft für physikalische Chemie). Ber. Bunsenges. Phys. Chem. 1969, 73, 834–839. [Google Scholar]
- Watkins, A.R. Short-lived intermediates formed by the interaction between electronically excited molecules and inorganic ions. J. Phys. Chem. 1974, 78, 1885–1890. [Google Scholar] [CrossRef]
- Rhodes, A.A.; Swartz, B.L.; Hosler, E.R.; Snyder, D.L.; Benitez, K.M.; Chohan, B.S.; Basu, S. Static quenching of tryptophan fluorescence in proteins by a dioxomolybdenum(VI) thiolate complex. J. Photochem. Photobiol. A 2014, 293, 81–87. [Google Scholar] [CrossRef]
- Mehra, J.; Rechenberg, H. The Fundamental Equations of Quantum Mechanics, 1925–1926; 1st softcover print; Springer: New York, NY, USA, 2001; ISBN 0387951784. [Google Scholar]
- Franz, K.A.; Kehr, W.G.; Siggel, A.; Wieczoreck, J.; Adam, W. Luminescent materials, in Ullmann’s Encyclopedia of Industrial Chemistry; Ullmann, F., Ed.; Wiley: Weinheim, Germany, 2002; pp. 591–627. [Google Scholar]
- Clabau, F.; Rocquefelte, X.; Jobic, S.; Deniard, P.; Whangbo, M.-H.; Garcia, A.; Le Mercier, T. Mechanism of Phosphorescence Appropriate for the Long-Lasting Phosphors Eu2+-Doped SrAl2O4 with Codopants Dy3+ and B3+. Chem. Mater. 2005, 17, 3904–3912. [Google Scholar] [CrossRef]
- Zitoun, D.; Bernaud, L.; Manteghetti, A.; Filhol, J.-S. Microwave Synthesis of a Long-Lasting Phosphor. J. Chem. Educ. 2009, 86, 72. [Google Scholar] [CrossRef]
- Rocke, A.J. It Began with a Daydream: The 150th Anniversary of the Kekulé Benzene Structure. Angew. Chem. Int. Ed. 2015, 54, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Yeh-Yung Lin, R.; Lin, H.-W.; Yen, Y.-S.; Chang, C.-H.; Chou, H.-H.; Chen, P.-W.; Hsu, C.-Y.; Chen, Y.-C.; Lin, J.T.; Ho, K.-C. 2,6-Conjugated anthracene sensitizers for high-performance dye-sensitized solar cells. Energy Environ. Sci. 2013, 6, 2477–2486. [Google Scholar] [CrossRef]
- Teng, C.; Yang, X.; Yang, C.; Li, S.; Cheng, M.; Hagfeldt, A.; Sun, L. Molecular Design of Anthracene-Bridged Metal-Free Organic Dyes for Efficient Dye-Sensitized Solar Cells. J. Phys. Chem. C 2010, 114, 9101–9110. [Google Scholar] [CrossRef]
- Cho, I.; Kim, S.H.; Kim, J.H.; Park, S.; Park, S.Y. Highly efficient and stable deep-blue emitting anthracene-derived molecular glass for versatile types of non-doped OLED applications. J. Mater. Chem. 2012, 22, 123–129. [Google Scholar] [CrossRef]
- Uchimura, M.; Watanabe, Y.; Araoka, F.; Watanabe, J.; Takezoe, H.; Konishi, G. Development of Laser Dyes to Realize Low Threshold in Dye-Doped Cholesteric Liquid Crystal Lasers. Adv. Mater. 2010, 22, 4473–4478. [Google Scholar] [CrossRef]
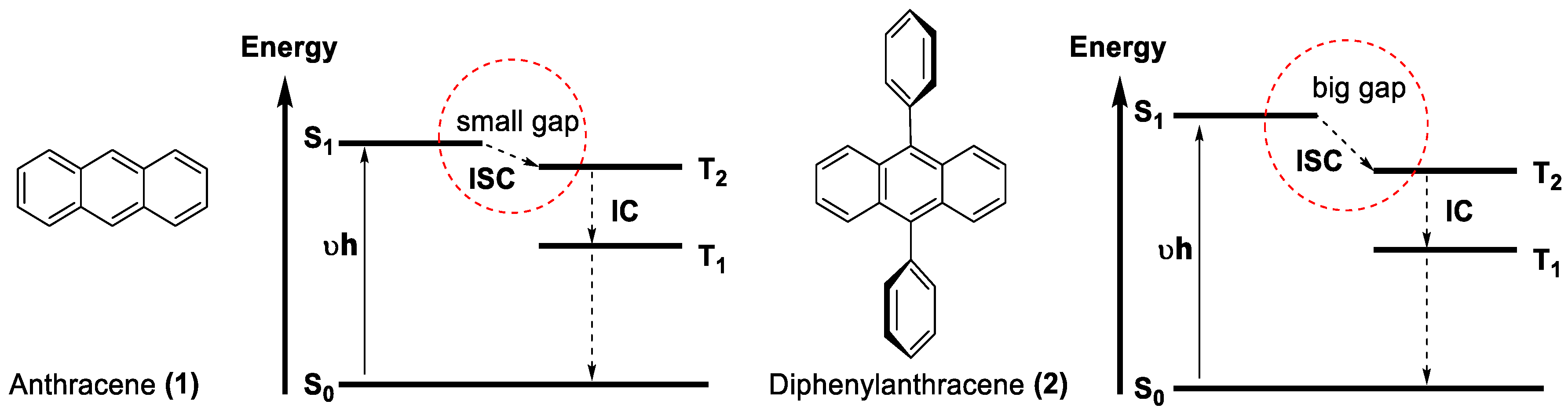
- Kanamaru, N. Radiationless Transition between Randomly Fluctuating Levels. S1-T2-T1 Intersystem Crossing in Condensed Phase. Bull. Chem. Soc. Jpn. 1982, 55, 3093–3096. [Google Scholar] [CrossRef]
- Yildiz, A.; Reilley, C.N. The Mechanism of Intersystem Crossing for Some Substituted Aromatic Hydrocarbons. Spectrosc. Lett 1968, 1, 335–343. [Google Scholar] [CrossRef]
- Coudret, C.; Mazenc, V. Heteroarylation of anthraquinone-triflate by suzuki cross-coupling. Tetrahedron Lett. 1997, 38, 5293–5296. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, K.; Hou, Y.; Che, Y.; Liu, L.; Jia, D. Recent progress in heavy atom-free organic compounds showing unexpected intersystem crossing (ISC) ability. Org. Biomol. Chem. 2018, 16, 3692–3701. [Google Scholar] [CrossRef]
- Musgrave, C.O. Oxidation of alkyl aryl ethers. Chem. Rev. 1968, 69, 499–531. [Google Scholar] [CrossRef]
- Heilbron, M.I.; Heaton, S.J. 9,10-Dibromoanthracene. Org. Synth. 1923, 3, 41. [Google Scholar]
- Jacob, J.; Espenson, J.H. Selective C-H bond activation of arenes catalyzed by methylrhenium trioxide. Inorg. Chim. Acta 1998, 270, 55–59. [Google Scholar] [CrossRef]
- Marshall, J.L.; Lehnherr, D.; Lindner, B.D.; Tykwinski, R.R. Reductive Aromatization/Dearomatization and Elimination Reactions to Access Conjugated Polycyclic Hydrocarbons, Heteroacenes, and Cumulenes. ChemPlusChem 2017, 82, 967–1001. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, W.E.; Chemerda, J.M. The Synthesis of 9,10-Dimethyl-1,2-benzanthracene, 9,10-Diethyl-1,2-benzanthracene and 5,9,10-Trimethyl-1,2-benzanthracene. J. Am. Chem. Soc. 1938, 60, 1023–1026. [Google Scholar] [CrossRef]
- Martinez, G.R.; Ravanat, J.-L.; Medeiros, M.H.G.; Cadet, J.; Di Mascio, P. Synthesis of a Naphthalene Endoperoxide as a Source of 18O-labeled Singlet Oxygen for Mechanistic Studies. J. Am. Chem. Soc. 2000, 122, 10212–10213. [Google Scholar] [CrossRef]
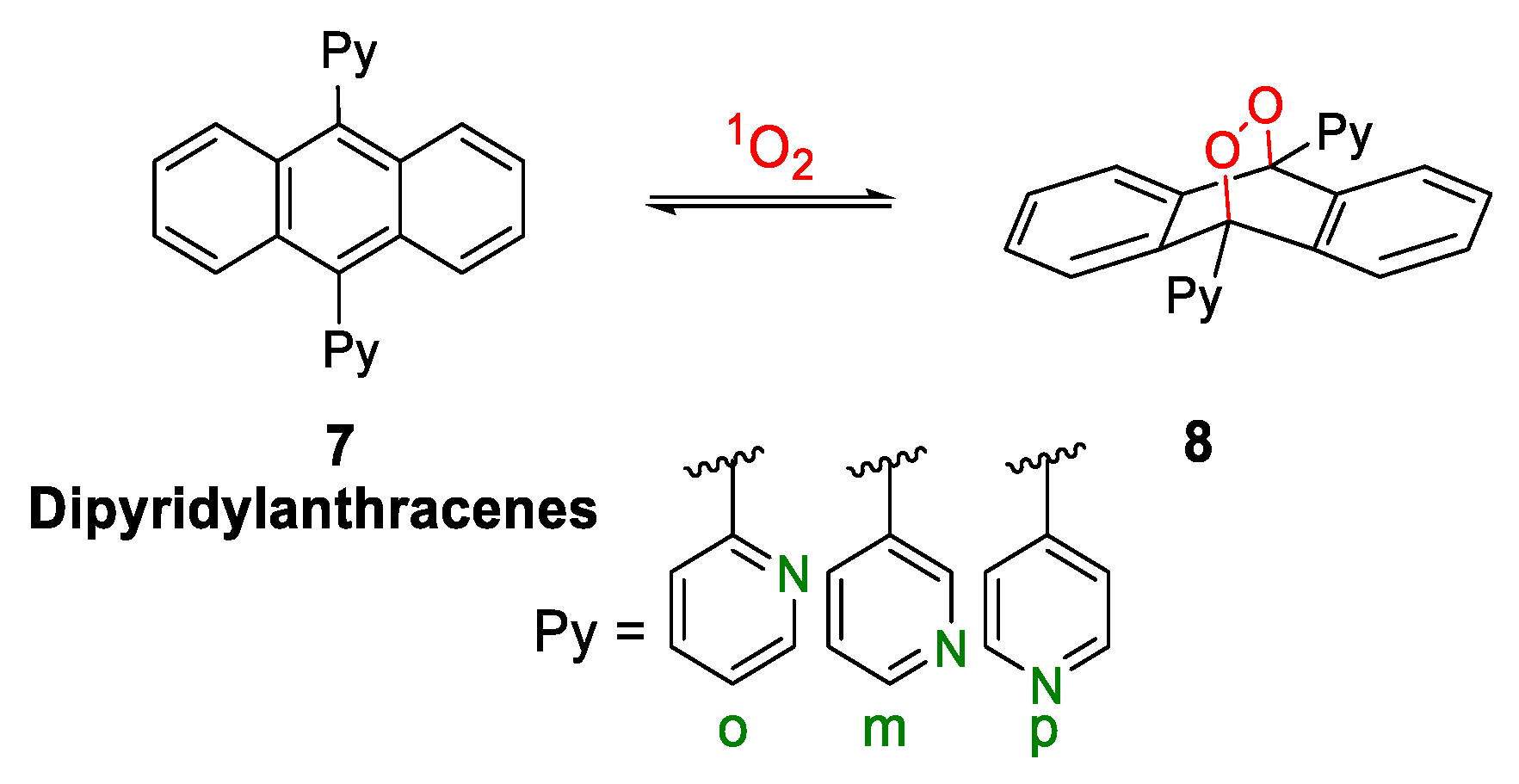
- Fudickar, W.; Linker, T. Synthesis of Pyridylanthracenes and Their Reversible Reaction with Singlet Oxygen to Endoperoxides. J. Org. Chem. 2017, 82, 9258–9262. [Google Scholar] [CrossRef]
- Fritzsche, J.J. Ueber die festen Kohlenwasserstoffe des Steinkohlentheers. Prakt. Chem. 1867, 101, 333. [Google Scholar] [CrossRef]
- Breton, G.W.; Vang, X. Photodimerization of Anthracene. J. Chem. Educ. 1998, 75, 81. [Google Scholar] [CrossRef]
- Greene, F.D.; Misrock, S.L.; Wolfe, J.R. The Structure of Anthracene Photodimers. J. Am. Chem. Soc. 1955, 77, 3852–3855. [Google Scholar] [CrossRef]
- Bhatnagar, S.S.; Kapur, P.L.; Kaur, G. Photopolymerisation of anthracene in benzene solution from the magnetic standpoint. Proc. Indian Acad. Sci. 1939, 10, 468. [Google Scholar] [CrossRef]
- Bouas-Laurent, H.; Castellan, A.; Desvergne, J.-P.; Lapouyade, R. Photodimerization of anthracenes in fluid solutions: (Part 2) Mechanistic aspects of the photocycloaddition and of the photochemical and thermal cleavage. Chem. Soc. Rev. 2001, 30, 248–263. [Google Scholar] [CrossRef]
- Becker, H.D. Unimolecular photochemistry of anthracenes. Chem. Rev. 1993, 93, 145–172. [Google Scholar] [CrossRef]
- Julian, M.M. Mechanism of photodimerization in single crystals of anthracene. Acta Cryst. 1973, 29, 116–120. [Google Scholar] [CrossRef]
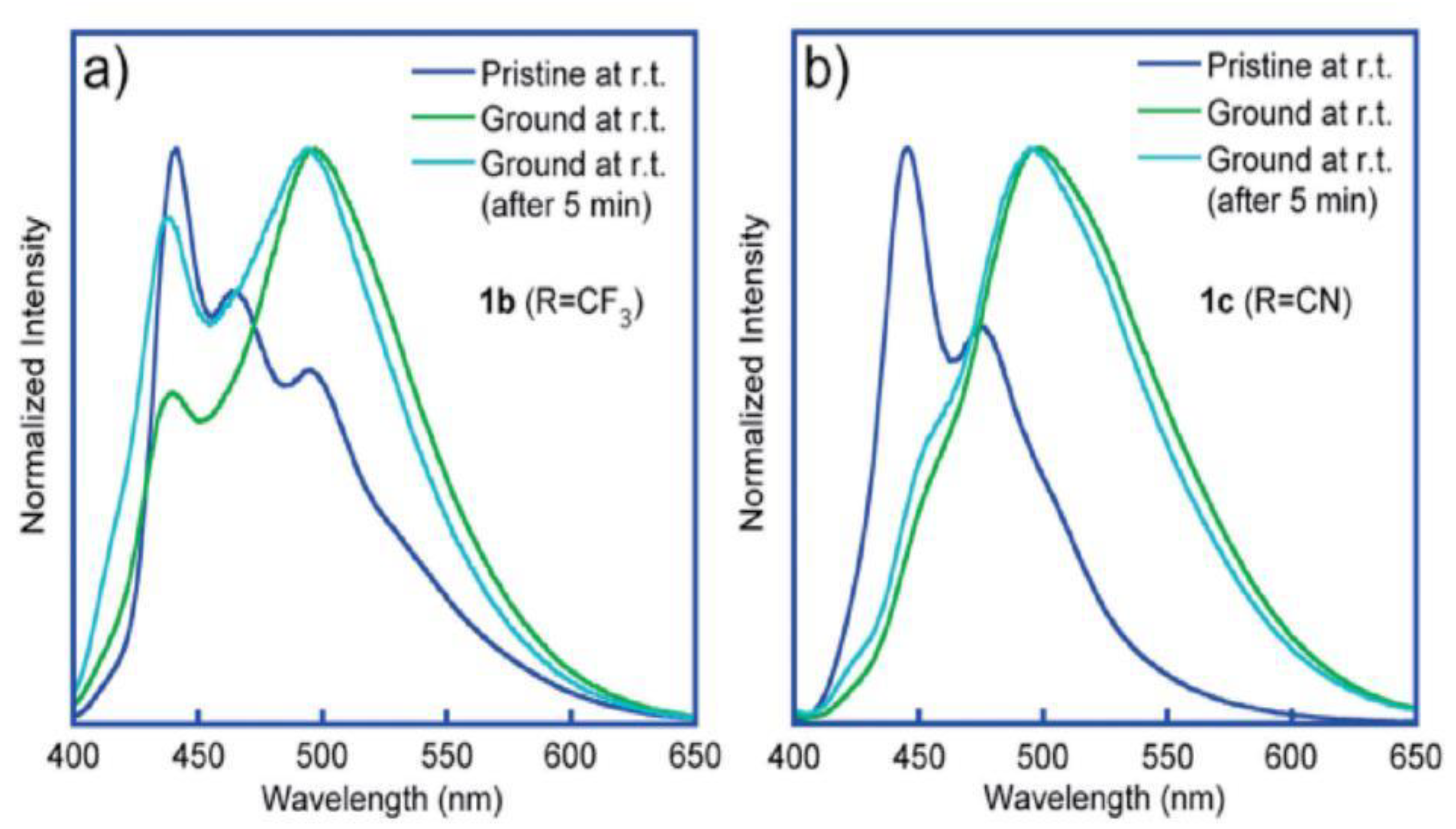
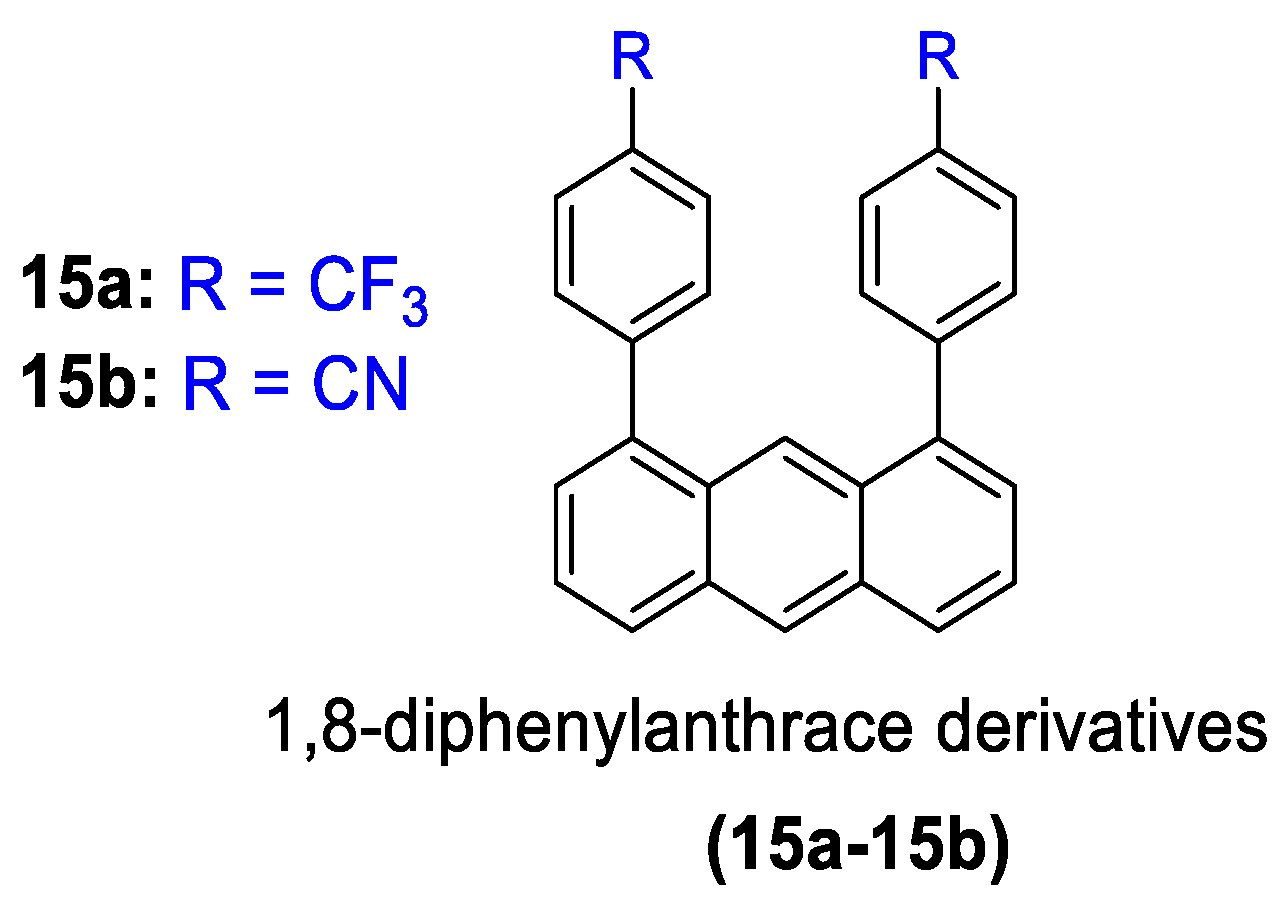
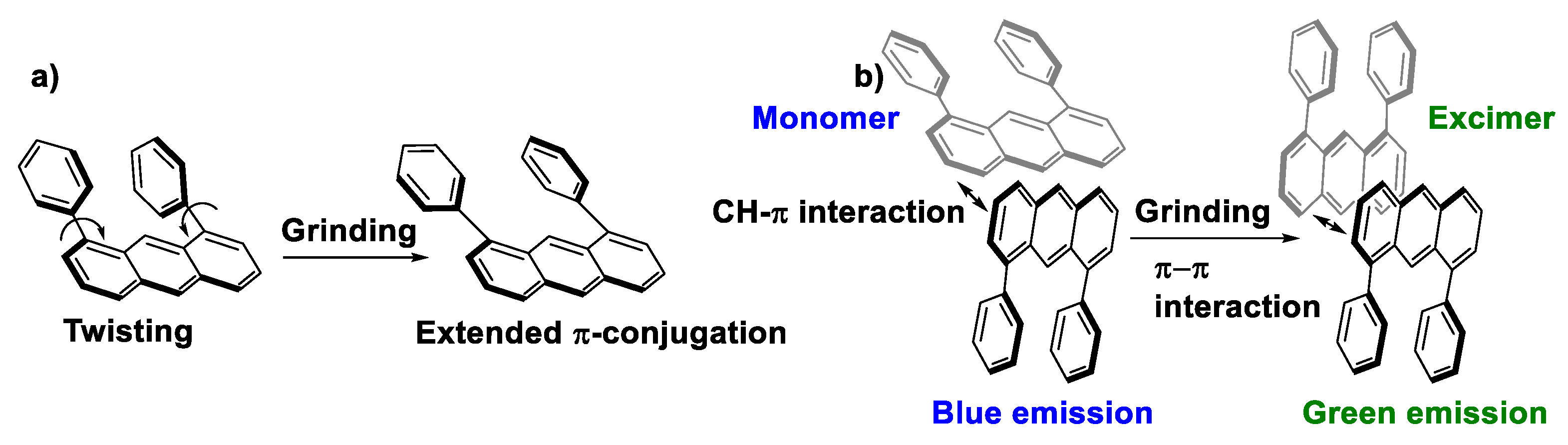
- Kusukawa, T.; Kojima, Y.; Kannen, F. Mechanofluorochromic Properties of 1,8-Diphenylanthracene Derivatives. Chem. Lett. 2019, 48, 1213–1216. [Google Scholar] [CrossRef]
- Luo, X.; Li, J.; Li, C.; Heng, L.; Dong, Y.Q.; Liu, Z.; Bo, Z.; Tang, B.Z. Reversible Switching of the Emission of Diphenyldibenzofulvenes by Thermal and Mechanical Stimuli. Adv. Mater. 2011, 23, 3261–3265. [Google Scholar] [CrossRef]
- Xue, P.; Yang, Z.; Chen, P. Hiding and revealing information using the mechanochromic system of a 2,5-dicarbazole-substituted terephthalate derivative. J. Mater. Chem. C 2018, 6, 4994–5000. [Google Scholar] [CrossRef]
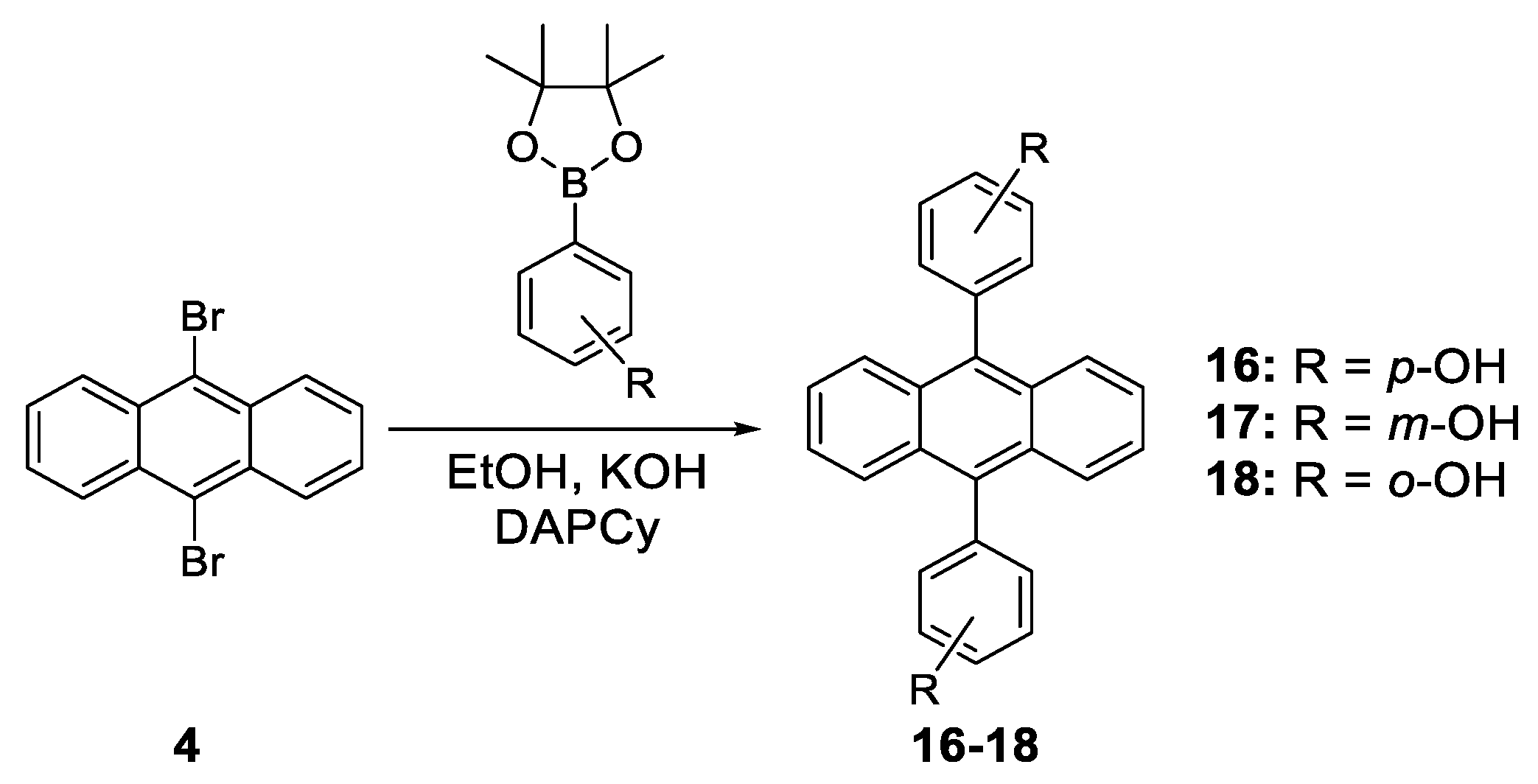
- Zelzer, M.; Kappaun, S.; Zojer, E.; Slugovc, C. Synthesis and Photo Physical Properties of 9,10-Bis(hydroxyphenyl)anthracene Derivatives. Monatsh. Chem. 2007, 138, 453–464. [Google Scholar] [CrossRef]
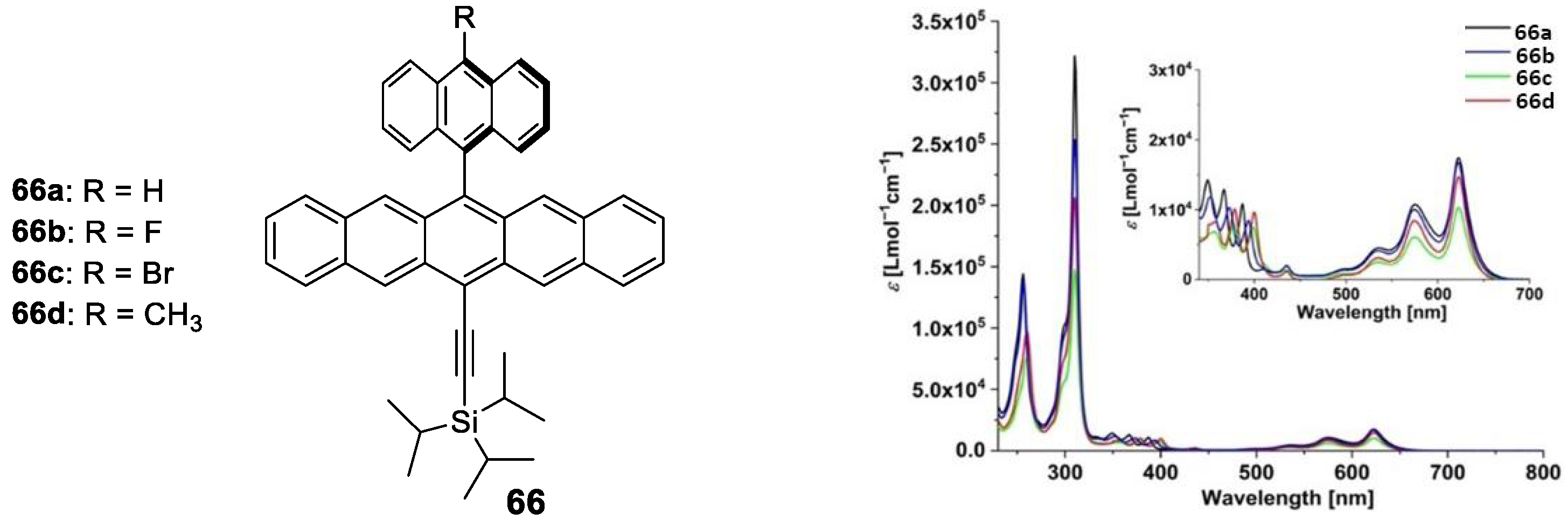
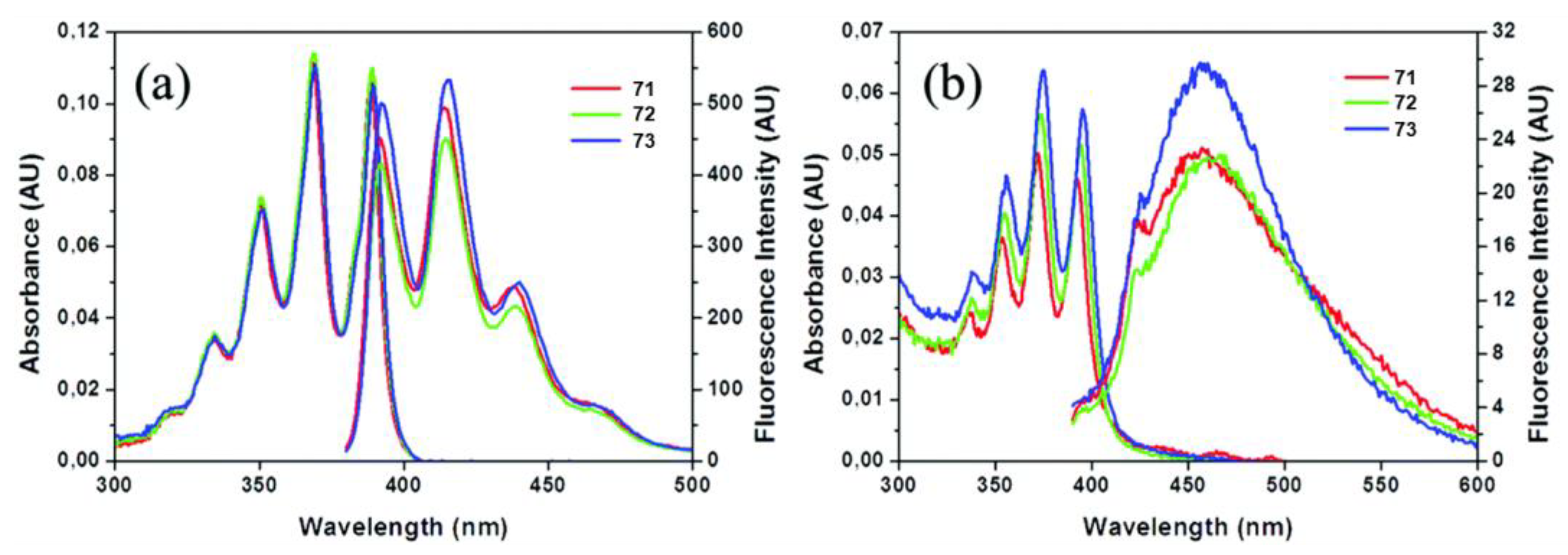
- Zhang, W.; Wang, Q.; Feng, X.; Yang, L.; Wu, Y.; Wei, X. Anthracene-based derivatives: Synthesis, photophysical properties and electrochemical properties. Chem. Res. Chin. Univ. 2017, 33, 603–610. [Google Scholar] [CrossRef]
- Hu, J.-Y.; Feng, X.; Seto, N.; Do, J.-H.; Zeng, X.; Tao, Z.; Yamato, T. Synthesis, structural and spectral properties of diarylamino-functionalized pyrene derivatives via Buchwald–Hartwig amination reaction. J. Mol. Struct. 2013, 1035, 19–26. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, S.; Chen, Y.; Wang, J. Synthesis and photophysical properties of multilayer emitting π-p-π fluorophores. Spectrochim. Acta A 2020, 227, 117680. [Google Scholar] [CrossRef]
- Fan, Y.; Zhao, Y.; Ye, L.; Li, B.; Yang, G.; Wang, Y. Polymorphs and Pseudopolymorphs of N, N -Di(n -butyl)Quinacridone: Structures and Solid-State Luminescence Properties. Cryst. Growth Des. 2009, 9, 1421–1430. [Google Scholar] [CrossRef]
- Kohmoto, S.; Tsuyuki, R.; Masu, H.; Azumaya, I.; Kishikawa, K. Polymorphism-dependent fluorescence of 9,10-bis(pentafluorobenzoyloxy)anthracene. Tetrahedron Lett. 2008, 49, 39–43. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Yao, D.; Bi, H.; Javed, I.; Fan, Y.; Zhang, H.; Wang, Y. Anthracene-Arrangement-Dependent Emissions of Crystals of 9-Anthrylpyrazole Derivatives. Cryst. Growth Des. 2009, 9, 5069–5076. [Google Scholar] [CrossRef]
- Venkataramana, G.; Sankararaman, S. Synthesis and Spectroscopic Investigation of Aggregation through Cooperative π−π and C−H···O Interactions in a Novel Pyrene Octaaldehyde Derivative. Org. Lett. 2006, 8, 2739–2742. [Google Scholar] [CrossRef]
- Curtis, M.D.; Cao, J.; Kampf, J.W. Solid-State Packing of Conjugated Oligomers: From π-Stacks to the Herringbone Structure. J. Am. Chem. Soc. 2004, 126, 4318–4328. [Google Scholar] [CrossRef]
- Dong, J.; Solntsev, K.M.; Tolbert, L.M. Activation and Tuning of Green Fluorescent Protein Chromophore Emission by Alkyl Substituent-Mediated Crystal Packing. J. Am. Chem. Soc. 2009, 131, 662–670. [Google Scholar] [CrossRef]
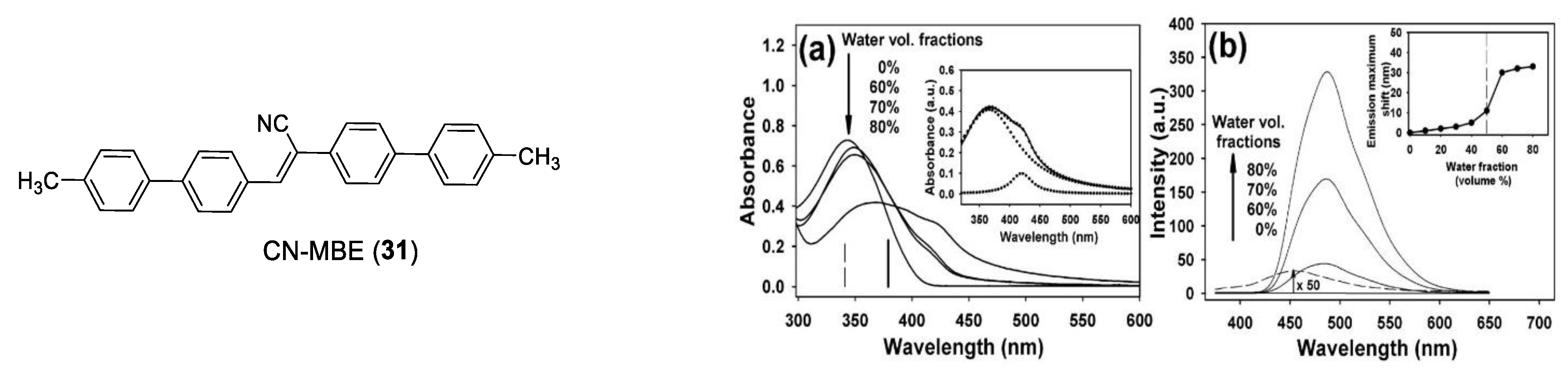
- An, B.-K.; Kwon, S.-K.; Jung, S.-D.; Park, S.Y. Enhanced Emission and Its Switching in Fluorescent Organic Nanoparticles. J. Am. Chem. Soc. 2002, 124, 14410–14415. [Google Scholar] [CrossRef]
- Bricks, J.L.; Slominskii, Y.L.; Panas, I.D.; Demchenko, A.P. Fluorescent J-aggregates of cyanine dyes: Basic research and applications review. Methods Appl. Fluoresc. 2017, 6, 12001. [Google Scholar] [CrossRef]
- Xiao, J.; Yin, Z.; Yang, B.; Liu, Y.; Ji, L.; Guo, J.; Huang, L.; Liu, X.; Yan, Q.; Zhang, H.; et al. Preparation, characterization, physical properties, and photoconducting behaviour of anthracene derivative nanowires. Nanoscale 2011, 3, 4720–4723. [Google Scholar] [CrossRef]
- Hoeben, F.J.; Jonkheijm, P.; Meijer, E.W.; Schenning, A.P. About Supramolecular Assemblies of π-Conjugated Systems. Chem. Rev. 2005, 105, 1491–1546. [Google Scholar] [CrossRef]
- Ajayaghosh, A.; Praveen, V.K. π-Organogels of Self-Assembled p-Phenylenevinylenes: Soft Materials with Distinct Size, Shape, and Functions. Acc. Chem. Res. 2007, 40, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Grimsdale, A.C.; Müllen, K. The Chemistry of Organic Nanomaterials. Angew. Chem. Int. Ed. 2005, 44, 5592–5629. [Google Scholar] [CrossRef]
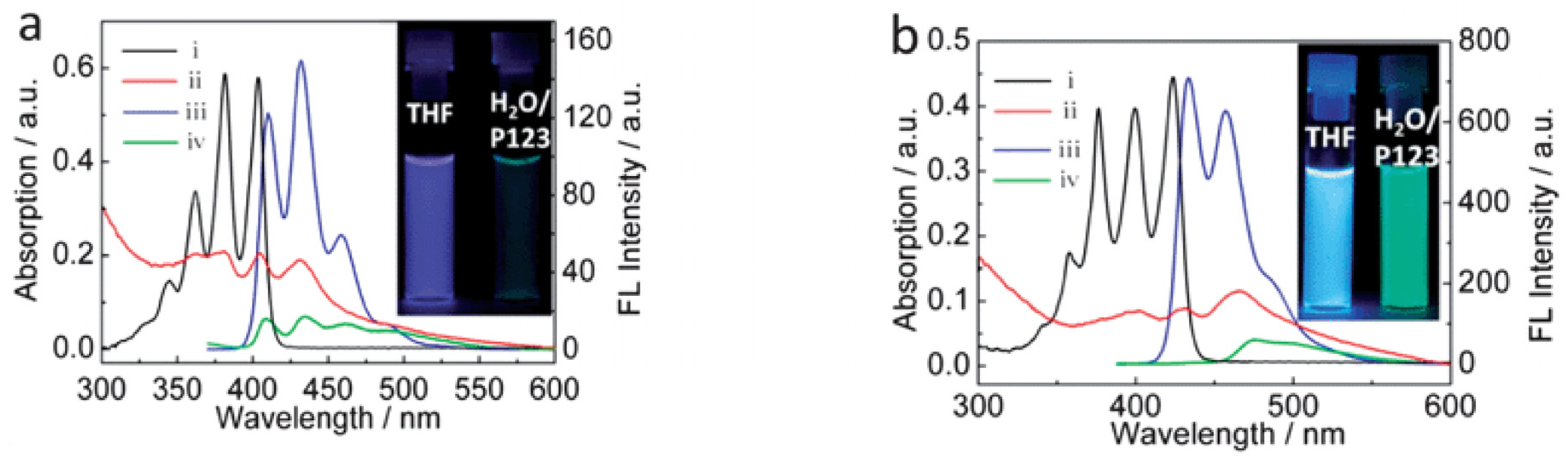
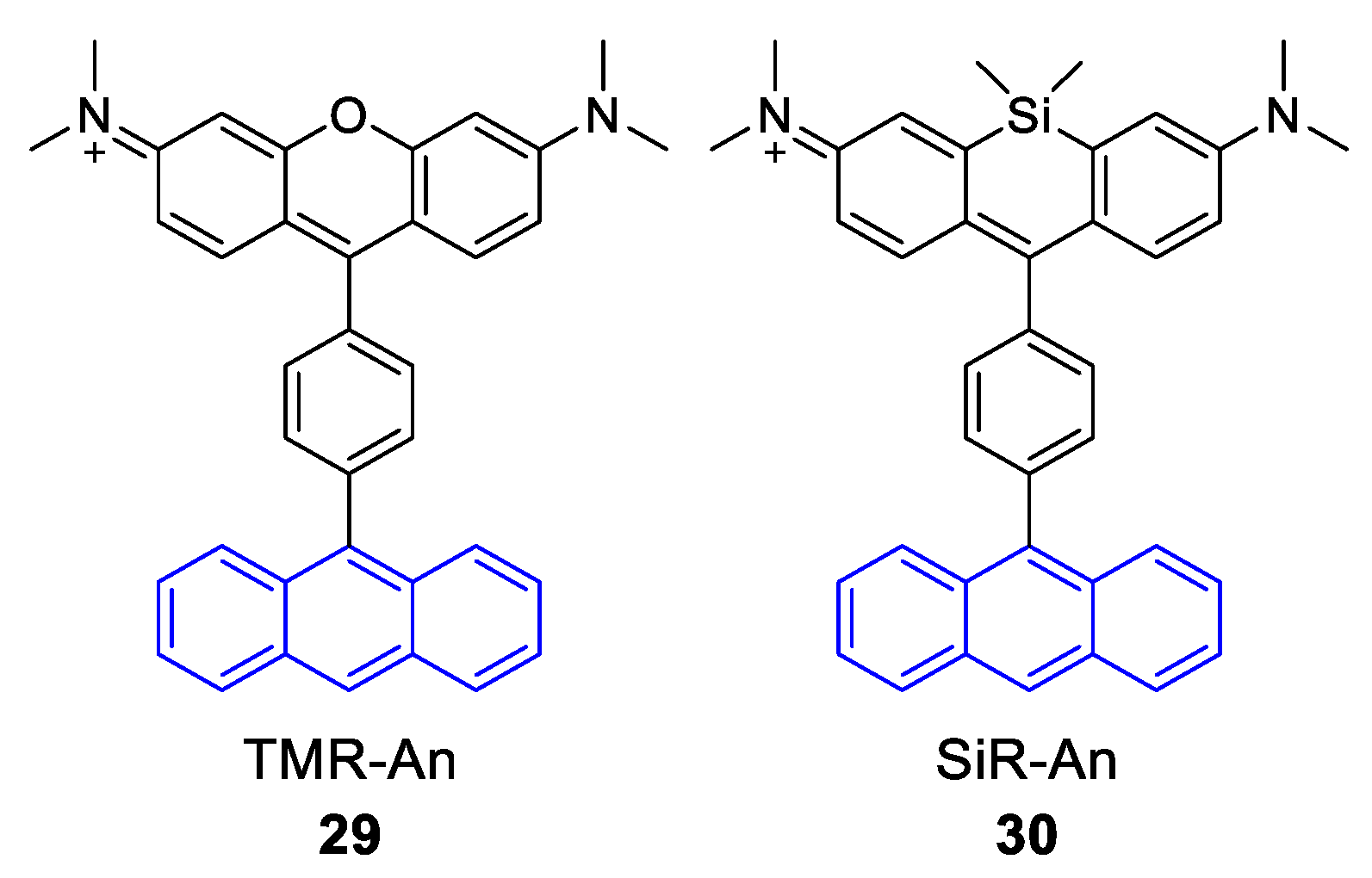
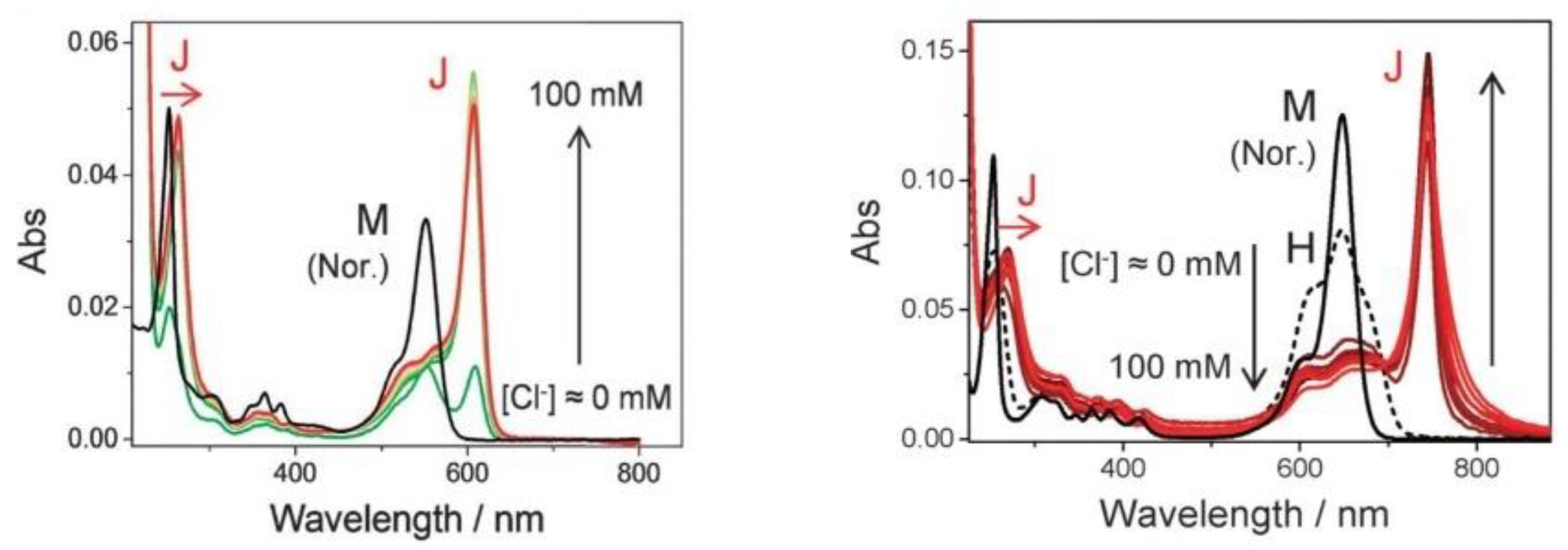
- Kim, S.; Fujitsuka, M.; Tohnai, N.; Tachikawa, T.; Hisaki, I.; Miyata, M.; Majima, T. The unprecedented J-aggregate formation of rhodamine moieties induced by 9-phenylanthracenyl substitution. Chem. Commun. 2015, 51, 11580–11583. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, K.; Yamaguchi, S.; Tahara, T. Formation and Dissociation of Rhodamine 800 Dimers in Water: Steady-State and Ultrafast Spectroscopic Study. J. Phys. Chem. A 2006, 110, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
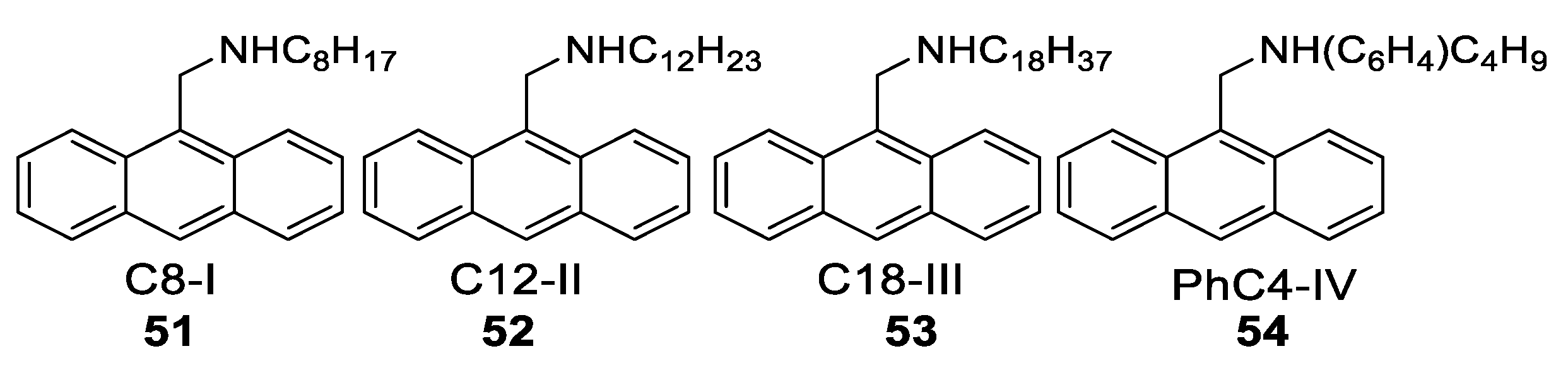
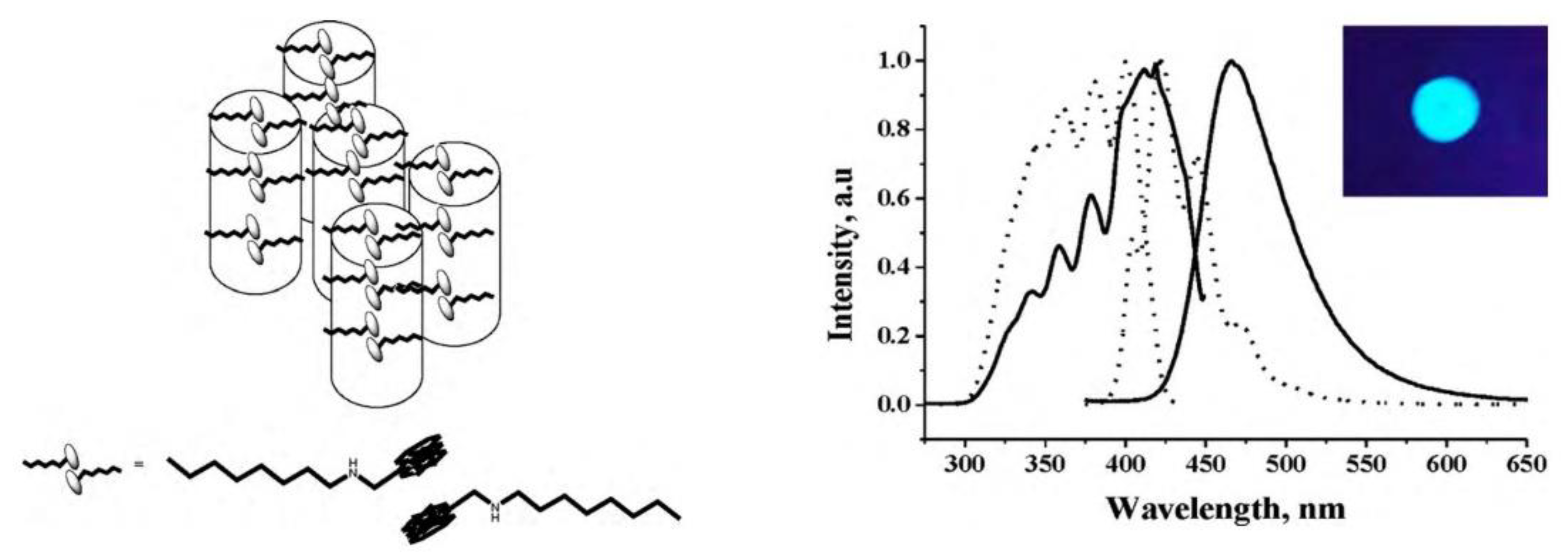
- Xue, S.; Qiu, X.; Sun, Q.; Yang, W. Alkyl length effects on solid-state fluorescence and mechanochromic behavior of small organic luminophores. J. Mater. Chem. C 2016, 4, 1568–1578. [Google Scholar] [CrossRef]
- Chan, J.M.W.; Tischler, J.R.; Kooi, S.E.; Bulović, V.; Swager, T.M. Synthesis of J-Aggregating Dibenz[a,j]anthracene-Based Macrocycles. J. Am. Chem. Soc. 2009, 131, 5659–5666. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Both, A.K.; Sarkar, M. Probing the Aggregation and Signaling Behavior of Some Twisted 9,9′-Bianthryl Derivatives: Observation of Aggregation-Induced Blue-Shifted Emission. ACS Omega 2018, 3, 15709–15724. [Google Scholar] [CrossRef]
- Gruszecki, W.I. Structural characterization of the aggregated forms of violaxanthin. J. Biol. Phys. 1991, 18, 99–109. [Google Scholar] [CrossRef]
- Gierschner, J.; Ehni, M.; Egelhaaf, H.-J.; Milián Medina, B.; Beljonne, D.; Benmansour, H.; Bazan, G.C. Solid-state optical properties of linear polyconjugated molecules: π-stack contra herringbone. J. Chem. Phys. 2005, 123, 144914. [Google Scholar] [CrossRef]
- Gierschner, J.; Lüer, L.; Milián-Medina, B.; Oelkrug, D.; Egelhaaf, H.-J. Highly Emissive H-Aggregates or Aggregation-Induced Emission Quenching? The Photophysics of All-Trans para-Distyrylbenzene. J. Phys. Chem. Lett. 2013, 4, 2686–2697. [Google Scholar] [CrossRef]
- Wang, H.; Li, F.; Gao, B.; Xie, Z.; Liu, S.; Wang, C.; Hu, D.; Shen, F.; Xu, Y.; Shang, H.; et al. Doped Organic Crystals with High Efficiency, Color-Tunable Emission toward Laser Application. Cryst. Growth Des. 2009, 9, 4945–4950. [Google Scholar] [CrossRef]
- Wu, D.-E.; Wang, M.-N.; Luo, Y.-H.; Zhang, Y.-W.; Ma, Y.-H.; Sun, B.-W. Tuning the structures and photophysical properties of 9,10-distyrylanthrance (DSA) via fluorine substitution. New J. Chem. 2017, 41, 4220–4233. [Google Scholar] [CrossRef]
- Ferguson, J. Absorption spectroscopy of sandwich dimers and cyclophanes. Chem. Rev. 1986, 86, 957–982. [Google Scholar] [CrossRef]
- Vollbrecht, J. Excimers in organic electronics. New J. Chem. 2018, 42, 11249–11254. [Google Scholar] [CrossRef]
- Dey, S.; Mondal, P.; Rath, S.P. Aggregation-controlled excimer emission in an axial anthracene–Sn(iv)porphyrin–anthracene triad in the solid and solution phases. New J. Chem. 2015, 39, 4100–4108. [Google Scholar] [CrossRef]
- Wannasiri, C.; Chanmungkalakul, S.; Bunchuay, T.; Chuenchom, L.; Uraisin, K.; Ervithayasuporn, V.; Kiatkamjornwong, S. Cross-Linking Silsesquioxane Cages with Polyaromatics as Fluorescent Porous Polymers for Fluoride Sensing and Removal. ACS Appl. Polym. Mater. 2020, 2, 1244–1255. [Google Scholar] [CrossRef]
- Birks, J.B. Excimers. Rep. Prog. Phys. 1975, 38, 903–974. [Google Scholar] [CrossRef]
- Chen, J.; Neels, A.; Fromm, K.M. Excimer formation in crystalline and nanostructured coordination polymers. Chem. Commun. 2010, 46, 8282–8284. [Google Scholar] [CrossRef]
- Yamane, S.; Sagara, Y.; Kato, T. Steric effects on excimer formation for photoluminescent smectic liquid-crystalline materials. Chem. Commun. 2013, 49, 3839–3841. [Google Scholar] [CrossRef]
- Hinoue, T.; Shigenoi, Y.; Sugino, M.; Mizobe, Y.; Hisaki, I.; Miyata, M.; Tohnai, N. Regulation of π-Stacked Anthracene Arrangement for Fluorescence Modulation of Organic Solid from Monomer to Excited Oligomer Emission. Chem. Eur. J. 2012, 18, 4634–4643. [Google Scholar] [CrossRef]
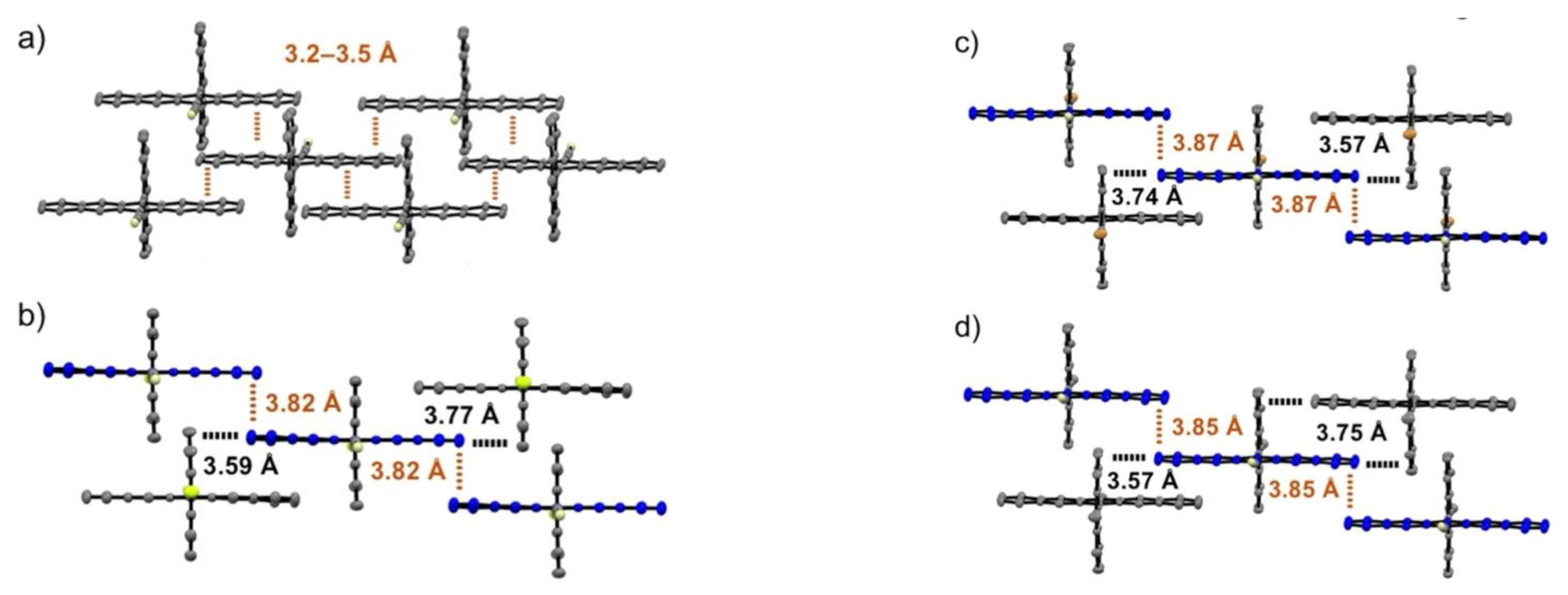
- Gao, Y.; Liu, H.; Zhang, S.; Gu, Q.; Shen, Y.; Ge, Y.; Yang, B. Excimer formation and evolution of excited state properties in discrete dimeric stacking of an anthracene derivative: A computational investigation. Phys. Chem. Chem. Phys. 2018, 20, 12129–12137. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yao, L.; Li, B.; Chen, X.; Gao, Y.; Zhang, S.; Li, W.; Lu, P.; Yang, B.; Ma, Y. Excimer-induced high-efficiency fluorescence due to pairwise anthracene stacking in a crystal with long lifetime. Chem. Commun. 2016, 52, 7356–7359. [Google Scholar] [CrossRef] [PubMed]
- Jaseer, M.; Prasad, E. Room temperature anthracene excimer emission from self-assembled (aminomethyl)anthracene derivatives in plastic crystalline phase. J. Photochem. Photobiol. A 2010, 214, 248–256. [Google Scholar] [CrossRef]
- Luo, D.; Liao, C.-W.; Chang, C.-H.; Tsai, C.-C.; Lu, C.-W.; Chuang, T.C.; Chang, H.-H. Approach to Fast Screen the Formation of an Exciplex. J. Phys. Chem. C 2020, 124, 10175–10184. [Google Scholar] [CrossRef]
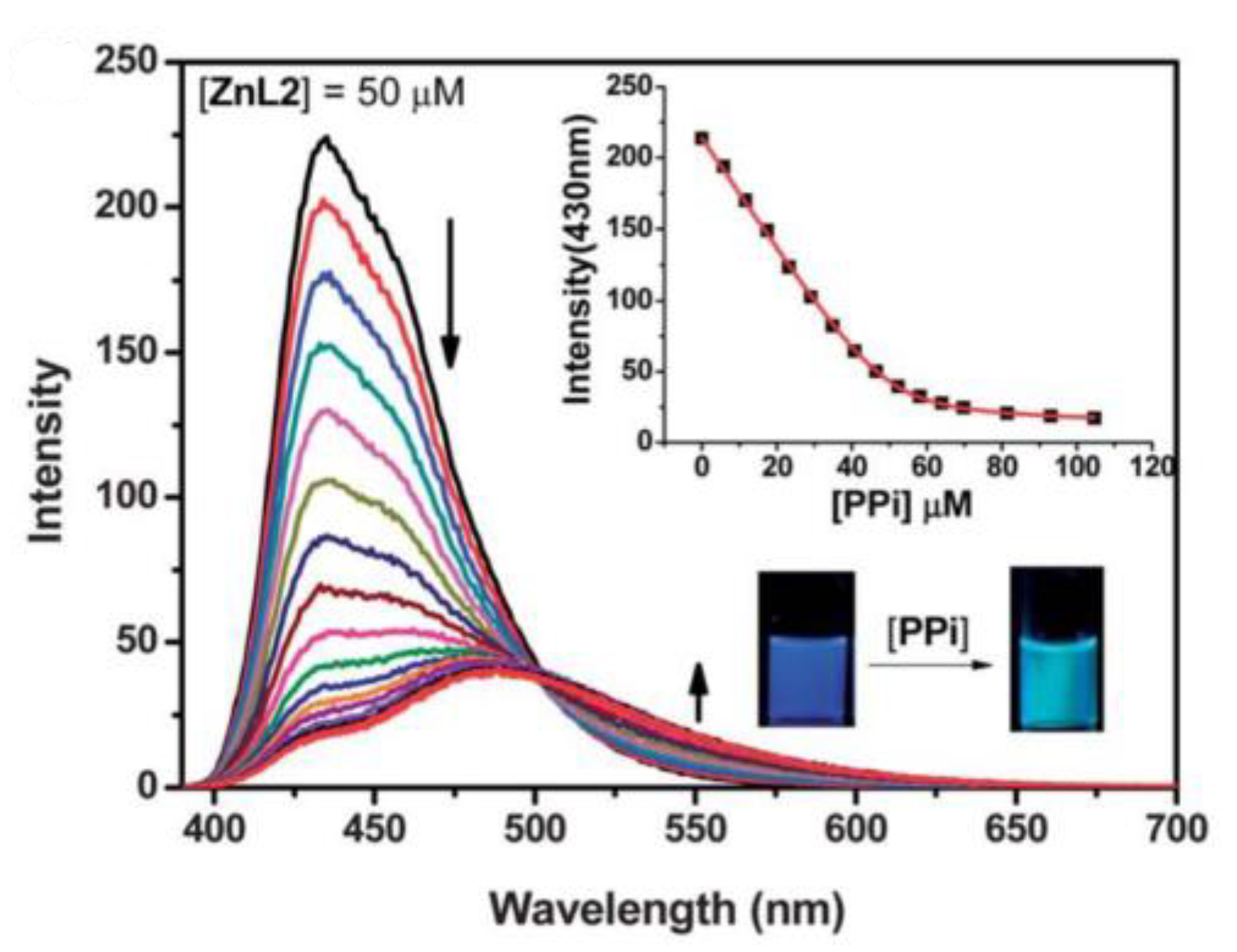
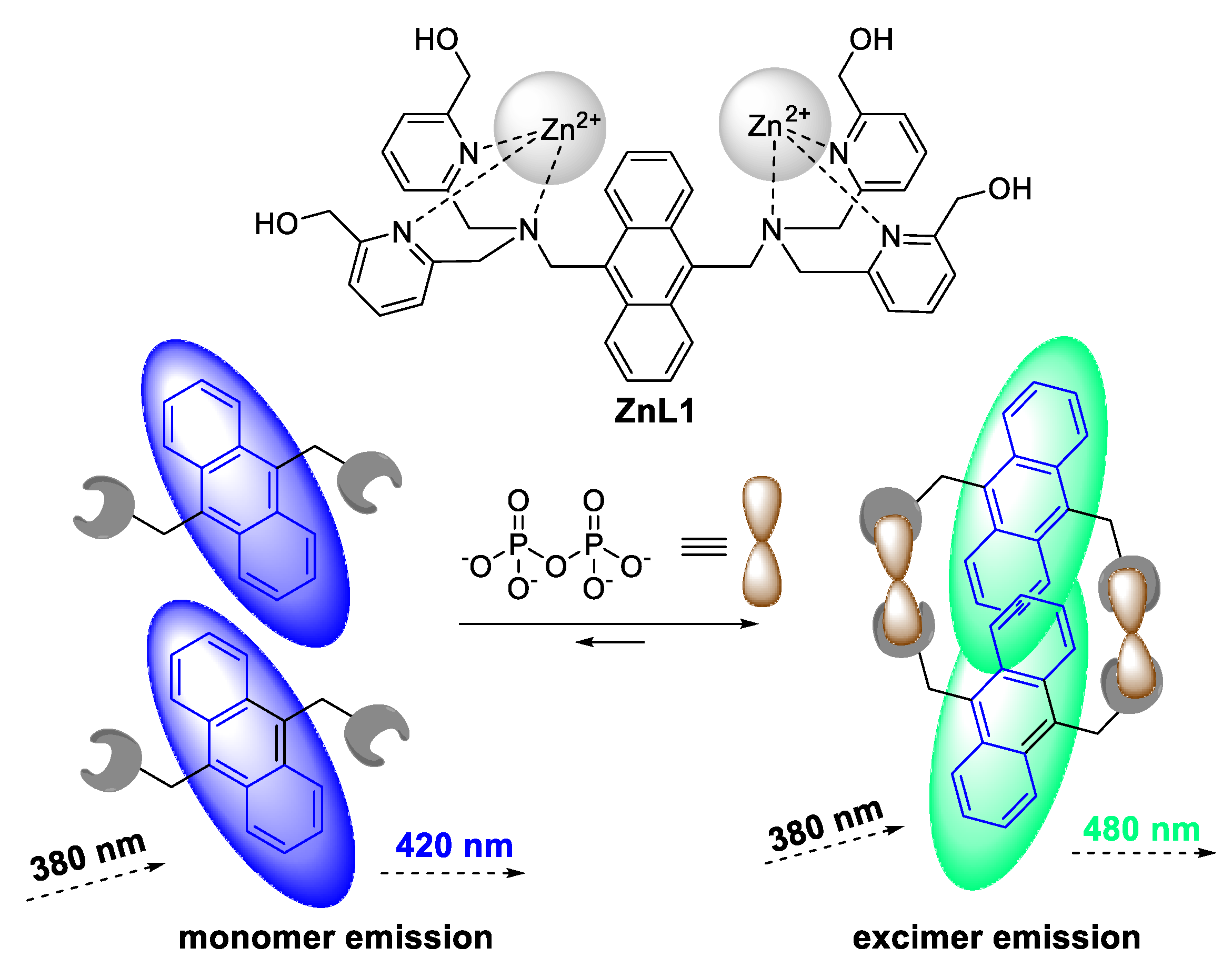
- Huang, F.; Feng, G. Highly selective and controllable pyrophosphate induced anthracene-excimer formation in water. RSC Adv. 2014, 4, 484–487. [Google Scholar] [CrossRef]
- Ban, X.; Sun, K.; Sun, Y.; Huang, B.; Jiang, W. Enhanced Electron Affinity and Exciton Confinement in Exciplex-Type Host: Power Efficient Solution-Processed Blue Phosphorescent OLEDs with Low Turn-on Voltage. ACS Appl. Mater. Interfaces 2016, 8, 2010–2016. [Google Scholar] [CrossRef]
- Kim, J.-M.; Lee, C.-H.; Kim, J.-J. Mobility balance in the light-emitting layer governs the polaron accumulation and operational stability of organic light-emitting diodes. Appl. Phys. Lett. 2017, 111, 203301. [Google Scholar] [CrossRef]
- Zhang, Y.; Mali, B.L.; Geddes, C.D. Metal-enhanced fluorescence exciplex emission. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 85, 134–138. [Google Scholar] [CrossRef]
- Chen, L.-Y.; Shiu, Y.-J.; Wu, Y.-J.; Huang, W.-Y. Simple structured color tunable white organic light-emitting diodes utilizing an ambipolar anthracene derivative with low-lying LUMO. Org. Electron. 2020, 76, 105454. [Google Scholar] [CrossRef]
- Bowen, E.J. Fluorescence quenching in solution and in the vapour state. Trans. Faraday Soc. 1954, 50, 97–102. [Google Scholar] [CrossRef]
- Labianca, D.A.; Taylor, G.N.; Hammond, G.S. Structure-reactivity factors in the quenching of fluorescence from naphthalenes by conjugated dienes. J. Am. Chem. Soc. 1972, 94, 3679–3683. [Google Scholar] [CrossRef]
- Weller, A. Electron-transfer and complex formation in the excited state. Pure Appl. Chem. 1968, 16, 115–124. [Google Scholar] [CrossRef]
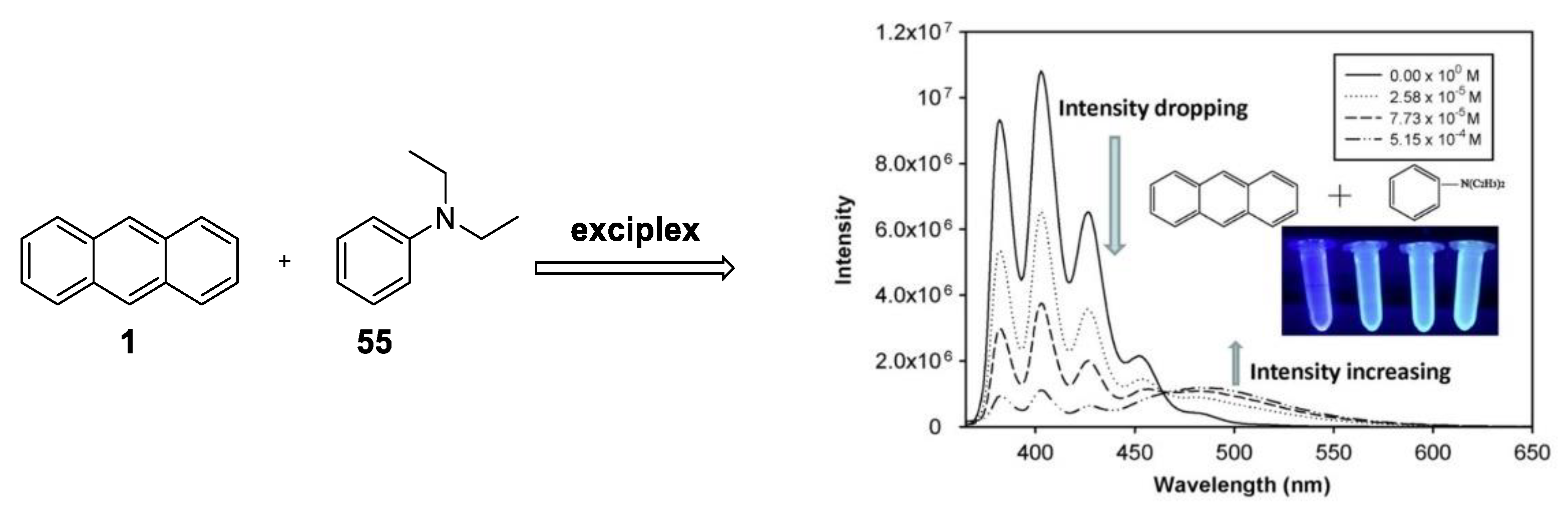
- Ohshiro, I.; Ikegami, M.; Nishimura, Y.; Arai, T. Exciplex Formation of Intermolecularly Hydrogen-Bonded System between Anthracene and N,N-Dimethylaniline Derivatives. Bull. Chem. Soc. Jpn. 2006, 79, 1950–1954. [Google Scholar] [CrossRef]
- Yang, N.C.; Shold, D.M.; Kim, B. Chemistry of exciplexes. 5. Photochemistry of anthracene in the presence and absence of dimethylaniline. J. Am. Chem. Soc. 1976, 98, 6587–6596. [Google Scholar] [CrossRef]
- Yang, N.C.; Shold, D.M.; McVey, J.K. Chemistry of exciplexes. III. Exciplex fluorescence from anthracene and substituted anthracenes in the presence of 2,5-dimethyl-2,4-hexadiene. J. Am. Chem. Soc. 1975, 97, 5004–5005. [Google Scholar] [CrossRef]
- Mizuno, K.; Pac, C.; Sakurai, H. Photochemical reactions of aromatic compounds. XIX. Photocycloaddition of olefins to 9-cyanophenanthrene. Singlet exciplex or triplet mechanism depending on olefins. J. Am. Chem. Soc. 1974, 96, 2993–2994. [Google Scholar] [CrossRef]
- Cann, J.R.; Cabanetos, C.; Welch, G.C. Synthesis of Molecular Dyads and Triads Based Upon N-Annulated Perylene Diimide Monomers and Dimers. Eur. J. Org. Chem. 2018, 2018, 6933–6943. [Google Scholar] [CrossRef]
- Bureš, F. Fundamental aspects of property tuning in push–pull molecules. RSC Adv. 2014, 4, 58826–58851. [Google Scholar] [CrossRef]
- Brunner, K.; Van Dijken, A.; Börner, H.; Bastiaansen, J.J.; Kiggen, N.M.; Langeveld, B.M. Carbazole Compounds as Host Materials for Triplet Emitters in Organic Light-Emitting Diodes: Tuning the HOMO Level without Influencing the Triplet Energy in Small Molecules. J. Am. Chem. Soc. 2004, 126, 6035–6042. [Google Scholar] [CrossRef]
- Matteucci, E.; Baschieri, A.; Sambri, L.; Monti, F.; Pavoni, E.; Bandini, E.; Armaroli, N. Carbazole-Terpyridine Donor-Acceptor Dyads with Rigid π-Conjugated Bridges. ChemPlusChem 2019, 84, 1353–1365. [Google Scholar] [CrossRef]
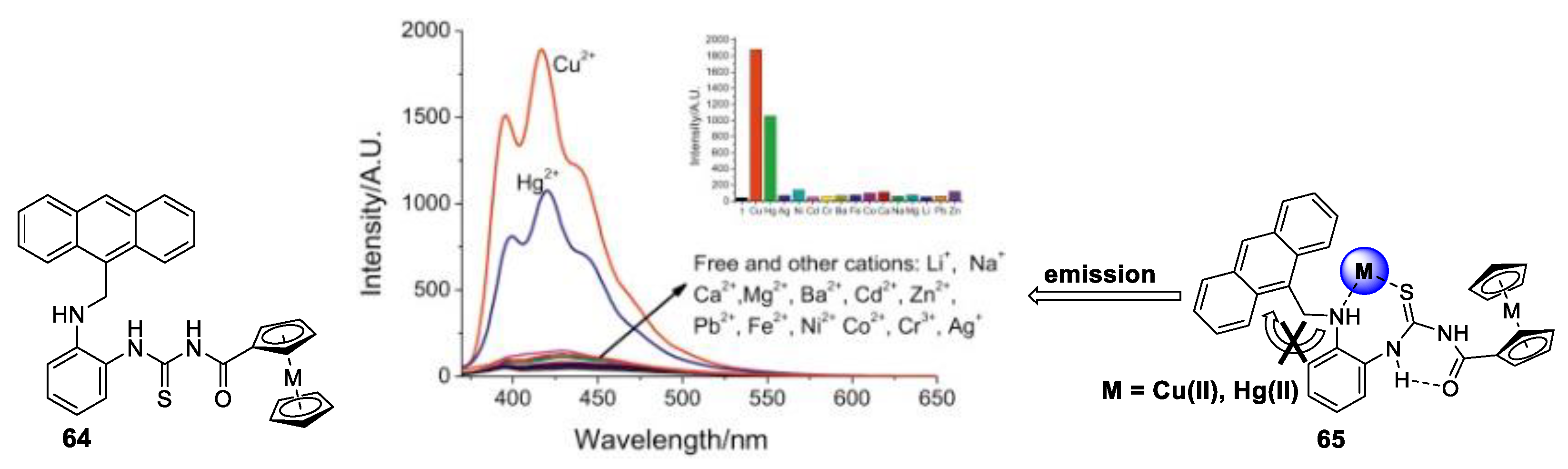
- Ge, J.-Z.; Zou, Y.; Yan, Y.-H.; Lin, S.; Zhao, X.-F.; Cao, Q.-Y. A new ferrocene–anthracene dyad for dual-signaling sensing of Cu(II) and Hg(II). J. Photochem. Photobiol. A Chem. 2016, 315, 67–75. [Google Scholar] [CrossRef]
- Hauschild, M.; Chen, L.; Etschel, S.H.; Ferguson, M.J.; Hampel, F.; Halik, M.; Tykwinski, R.R. Anthracene-Pentacene Dyads: Synthesis and OFET Characterization. ChemPlusChem 2020, 85, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Etschel, S.H.; Waterloo, A.R.; Margraf, J.T.; Amin, A.Y.; Hampel, F.; Jäger, C.M.; Clark, T.; Halik, M.; Tykwinski, R.R. An unsymmetrical pentacene derivative with ambipolar behavior in organic thin-film transistors. Chem. Commun. 2013, 49, 6725–6727. [Google Scholar] [CrossRef] [PubMed]
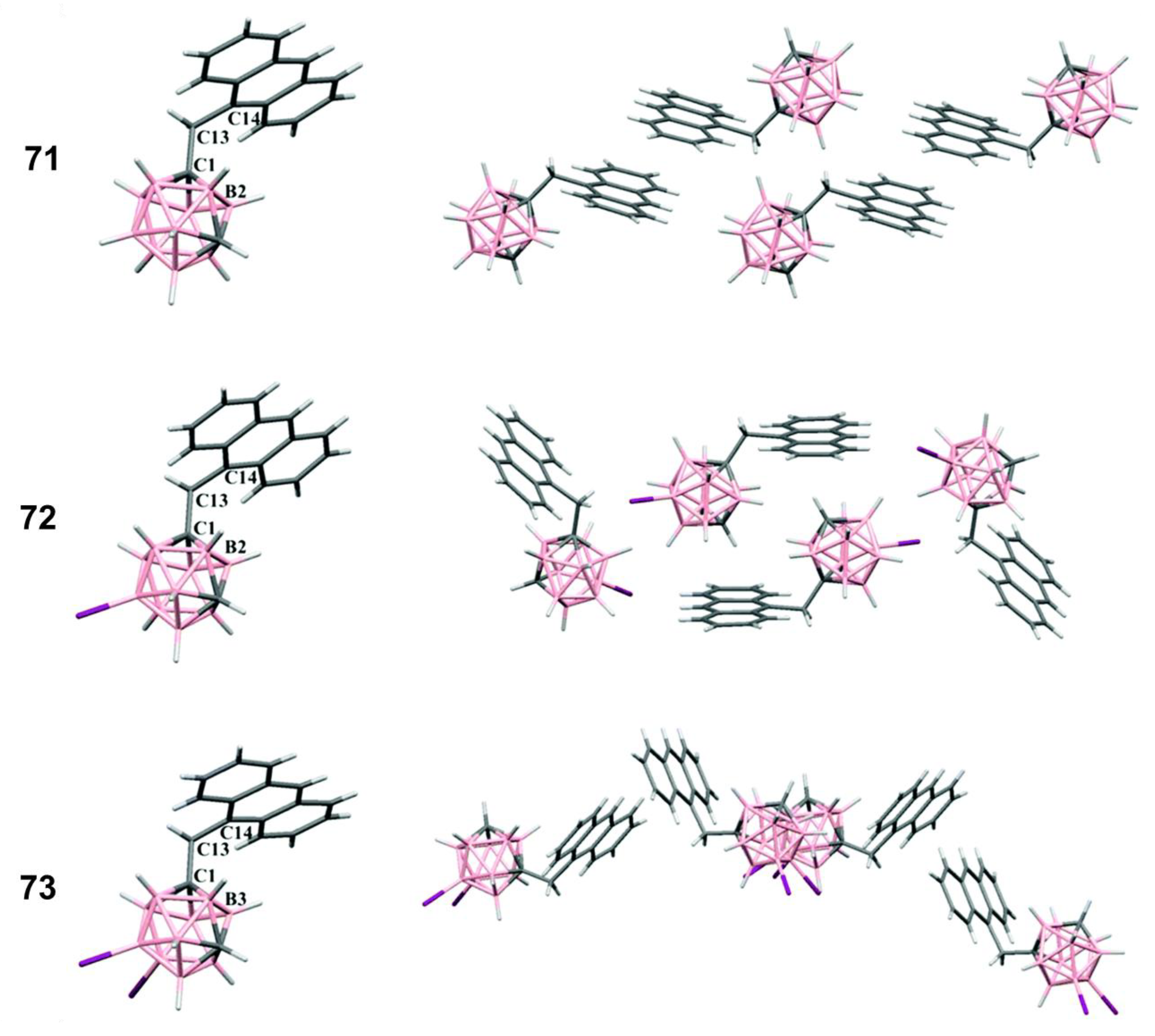
- Chaari, M.; Kelemen, Z.; Choquesillo-Lazarte, D.; Gaztelumendi, N.; Teixidor, F.; Viñas, C.; Nogués, C.; Núñez, R. Efficient blue light emitting materials based on m-carborane–anthracene dyads. Structure, photophysics and bioimaging studies. Biomater. Sci. 2019, 7, 5324–5337. [Google Scholar] [CrossRef] [PubMed]
- Chaari, M.; Kelemen, Z.; Planas, J.G.; Teixidor, F.; Choquesillo-Lazarte, D.; Ben Salah, A.; Viñas, C.; Núñez, R. Photoluminescence in m -carborane–anthracene triads: A combined experimental and computational study. J. Mater. Chem. C 2018, 6, 11336–11347. [Google Scholar] [CrossRef]
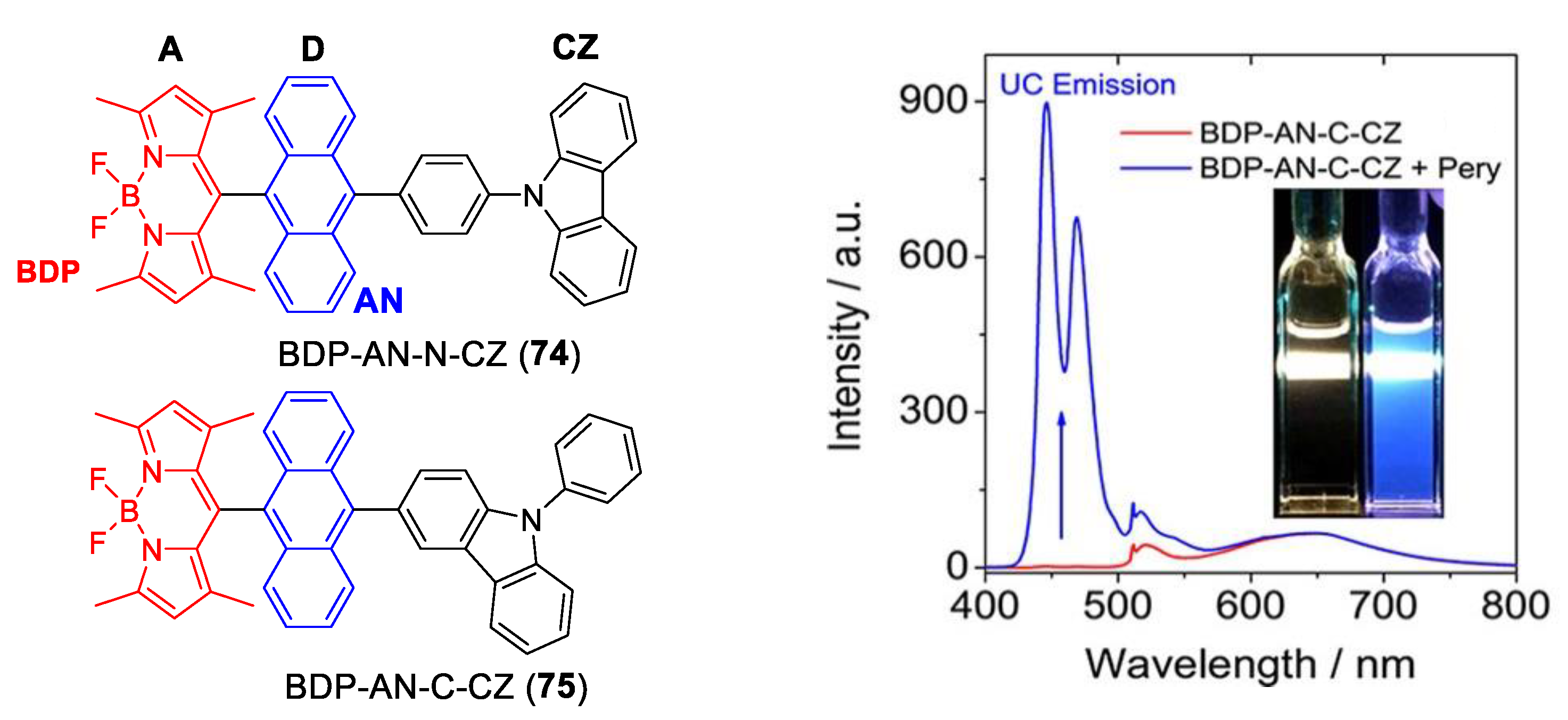
- Mahmood, Z.; Taddei, M.; Rehmat, N.; Bussotti, L.; Doria, S.; Guan, Q.; Ji, S.; Zhao, J.; Di Donato, M.; Huo, Y.; et al. Color-Tunable Delayed Fluorescence and Efficient Spin–Orbit Charge Transfer Intersystem Crossing in Compact Carbazole-Anthracene-Bodipy Triads Employing the Sequential Electron Transfer Approach. J. Phys. Chem. C 2020, 124, 5944–5957. [Google Scholar] [CrossRef]
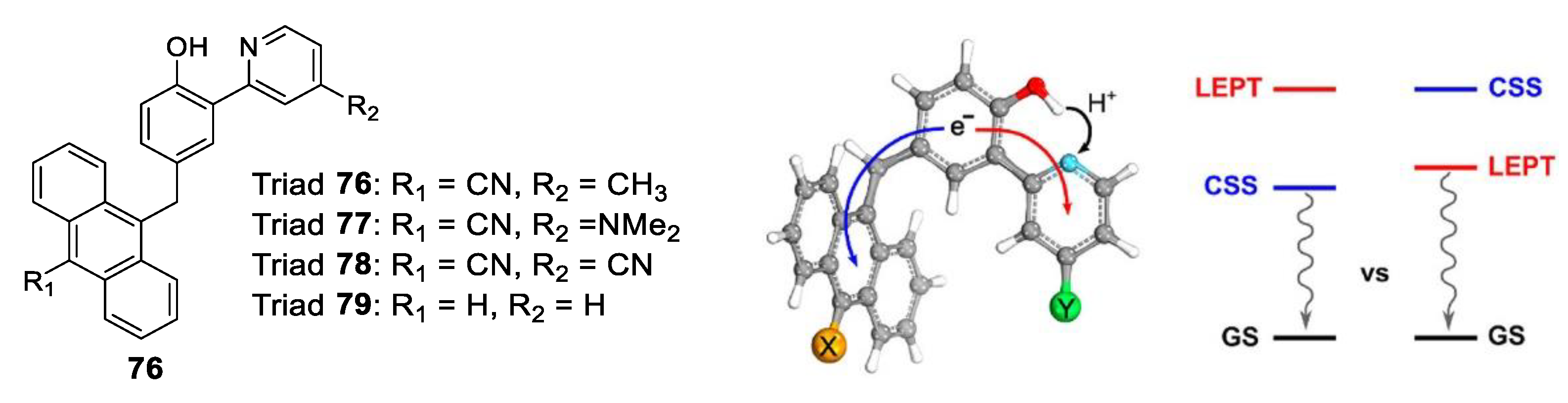
- Sayfutyarova, E.R.; Hammes-Schiffer, S. Substituent Effects on Photochemistry of Anthracene–Phenol–Pyridine Triads Revealed bCd((y Multireference Calculations. J. Am. Chem. Soc. 2020, 142, 487–494. [Google Scholar] [CrossRef]
- Turro, N.J.; Ramamurthy, V.; Scaiano, J.C. Modern Molecular Photochemistry of Organic Molecules; University Science Books: Sausalito, CA, USA, 2010. [Google Scholar]
- Parada, G.A.; Goldsmith, Z.K.; Kolmar, S.; Pettersson Rimgard, B.; Mercado, B.Q.; Hammarström, L.; Hammes-Schiffer, S.; Mayer, J.M. Concerted proton-electron transfer reactions in the Marcus inverted region. Science 2019, 364, 471–475. [Google Scholar] [CrossRef]
- Ariga, K.; Hill, J.P.; Lee, M.V.; Vinu, A.; Charvet, R.; Acharya, S. Challenges and breakthroughs in recent research on self-assembly. Sci. Technol. Adv. Mater. 2008, 9, 14109. [Google Scholar] [CrossRef]
- Caulder, D.L.; Raymond, K.N. Supermolecules by Design. Acc. Chem. Res. 1999, 32, 975–982. [Google Scholar] [CrossRef]
- Batten, S.R.; Champness, N.R.; Chen, X.-M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’Keeffe, M.; Suh, M.P.; Reedijk, J. Coordination polymers, metal–organic frameworks and the need for terminology guidelines. CrystEngComm 2012, 14, 3001–3004. [Google Scholar] [CrossRef]
- Cui, X.; Khlobystov, A.N.; Chen, X.; Marsh, D.H.; Blake, A.J.; Lewis, W.; Champness, N.R.; Roberts, C.J.; Schröder, M. Dynamic Equilibria in Solvent-Mediated Anion, Cation and Ligand Exchange in Transition-Metal Coordination Polymers: Solid-State Transfer or Recrystallisation? Chem. Euro J. 2009, 15, 8861–8873. [Google Scholar] [CrossRef] [PubMed]
- Yaghi, O.M.; Li, G.; Li, H. Selective binding and removal of guests in a microporous metal–organic framework. Nature 1995, 378, 703–706. [Google Scholar] [CrossRef]
- Dalgarno, S.J.; Tucker, S.A.; Bassil, D.B.; Atwood, J.L. Fluorescent Guest Molecules Report Ordered Inner Phase of Host Capsules in Solution. Science 2005, 309, 2037–2039. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-H.; Li, G.-Y.; Liu, X.-J.; Feng, R.; Zhang, H.-J.; Zhang, S.-Y.; Zhang, Y.-H. A fluorescent anthracene-based metal–organic framework for highly selective detection of nitroanilines. Inorg. Chim. Acta 2018, 473, 70–74. [Google Scholar] [CrossRef]
- Wu, Z.-F.; Velasco, E.; Shan, C.; Tan, K.; Zhang, Z.-Z.; Hu, Q.; Xing, K.; Huang, X.; Li, J. Robust fluorescent calcium coordination polymers as Cu2+ sensors with high sensitivity and fast response. J. Mater. Chem. C 2020, 8, 6820–6825. [Google Scholar] [CrossRef]
- Aleksovska, A.; Lönnecke, P.; Hey-Hawkins, E. Zn- and Cd-based coordination polymers with a novel anthracene dicarboxylate ligand for highly selective detection of hydrogen peroxide. Dalt Trans. 2020, 49, 4817–4823. [Google Scholar] [CrossRef] [PubMed]
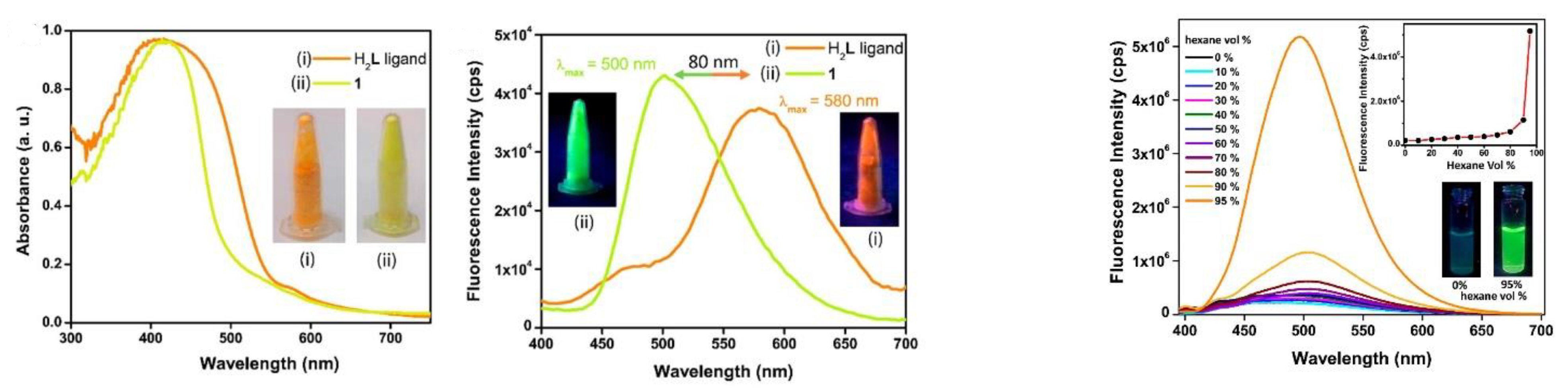
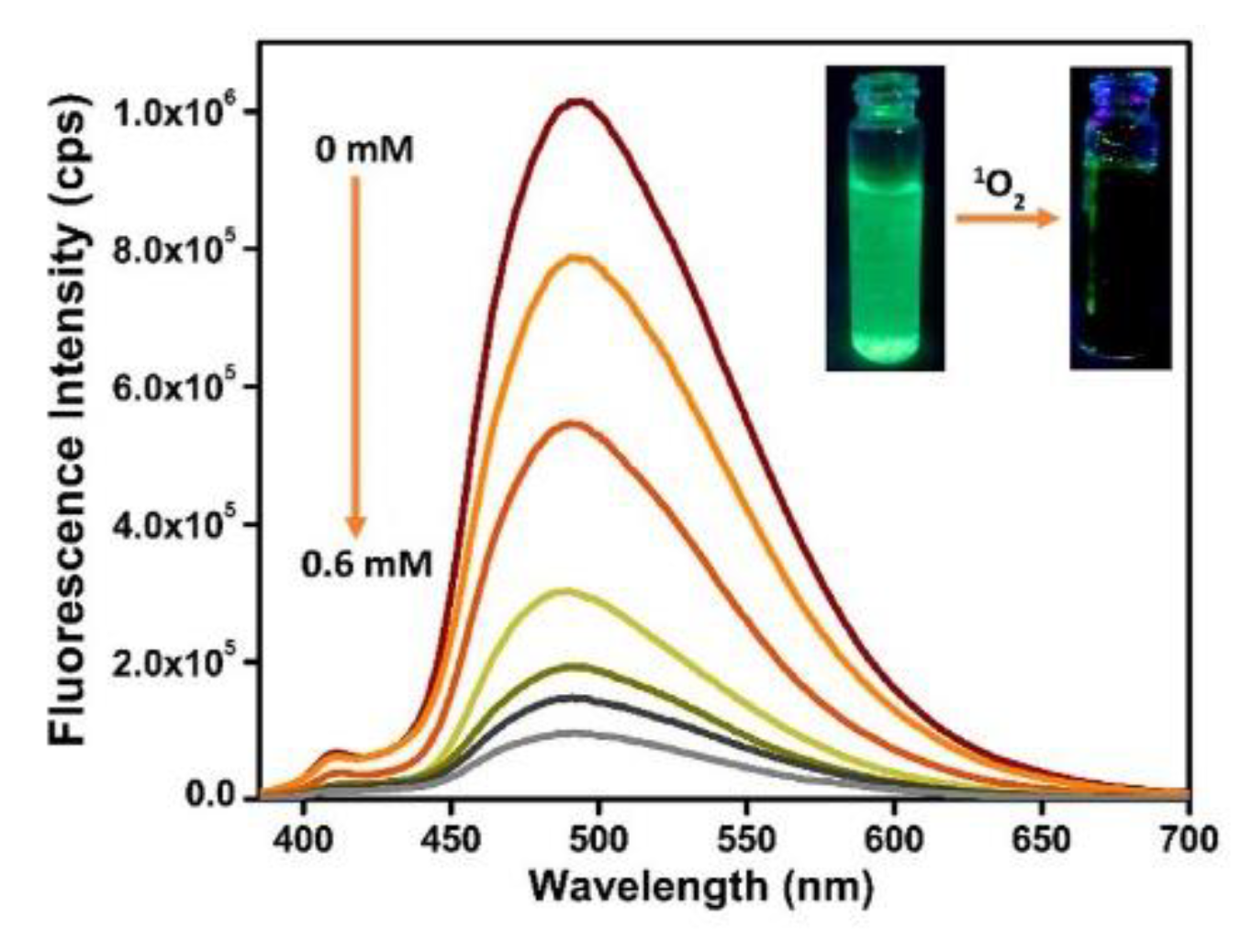
- Dalapati, R.; Nandi, S.; van Hecke, K.; Biswas, S. Fluorescence Modulation of an Aggregation-Induced Emission Active Ligand via Rigidification in a Coordination Polymer and Its Application in Singlet Oxygen Sensing. Cryst. Growth Des. 2019, 19, 6388–6397. [Google Scholar] [CrossRef]
- Pedersen, S.K.; Holmehave, J.; Blaikie, F.H.; Gollmer, A.; Breitenbach, T.; Jensen, H.H.; Ogilby, P.R. Aarhus sensor green: A fluorescent probe for singlet oxygen. J. Org. Chem. 2014, 79, 3079–3087. [Google Scholar] [CrossRef]
- Frausto, F.; Thomas, S.W. Ratiometric Singlet Oxygen Detection in Water Using Acene-Doped Conjugated Polymer Nanoparticles. ACS Appl. Mater. Interfaces 2017, 9, 15768–15775. [Google Scholar] [CrossRef]
- Kubát, P.; Henke, P.; Berzediová, V.; Štěpánek, M.; Lang, K.; Mosinger, J. Nanoparticles with Embedded Porphyrin Photosensitizers for Photooxidation Reactions and Continuous Oxygen Sensing. ACS Appl. Mater. Interfaces 2017, 9, 36229–36238. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Martinez, G.R.; Medeiros, M.H.G.; Di Mascio, P. Singlet molecular oxygen generated from lipid hydroperoxides by the russell mechanism: Studies using 18(O)-labeled linoleic acid hydroperoxide and monomol light emission measurements. J. Am. Chem. Soc. 2003, 125, 6172–6179. [Google Scholar] [CrossRef]
- Li, X.; Zhang, G.; Ma, H.; Zhang, D.; Li, J.; Zhu, D. 4,5-dimethylthio-4′-2-(9-anthryloxy)ethylthiotetrathiafulvalene, a highly selective and sensitive chemiluminescence probe for singlet oxygen. J. Am. Chem. Soc. 2004, 126, 11543–11548. [Google Scholar] [CrossRef] [PubMed]
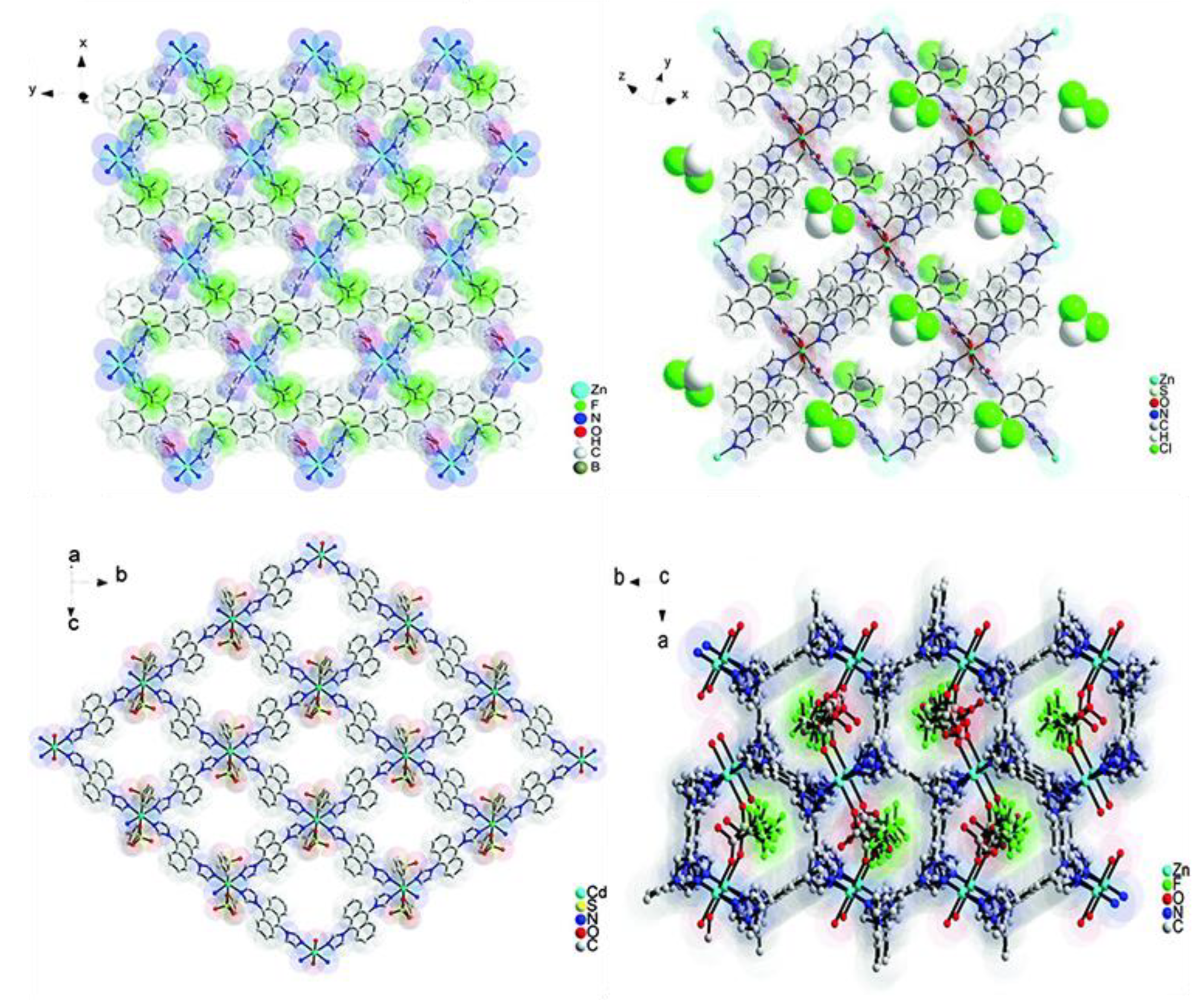
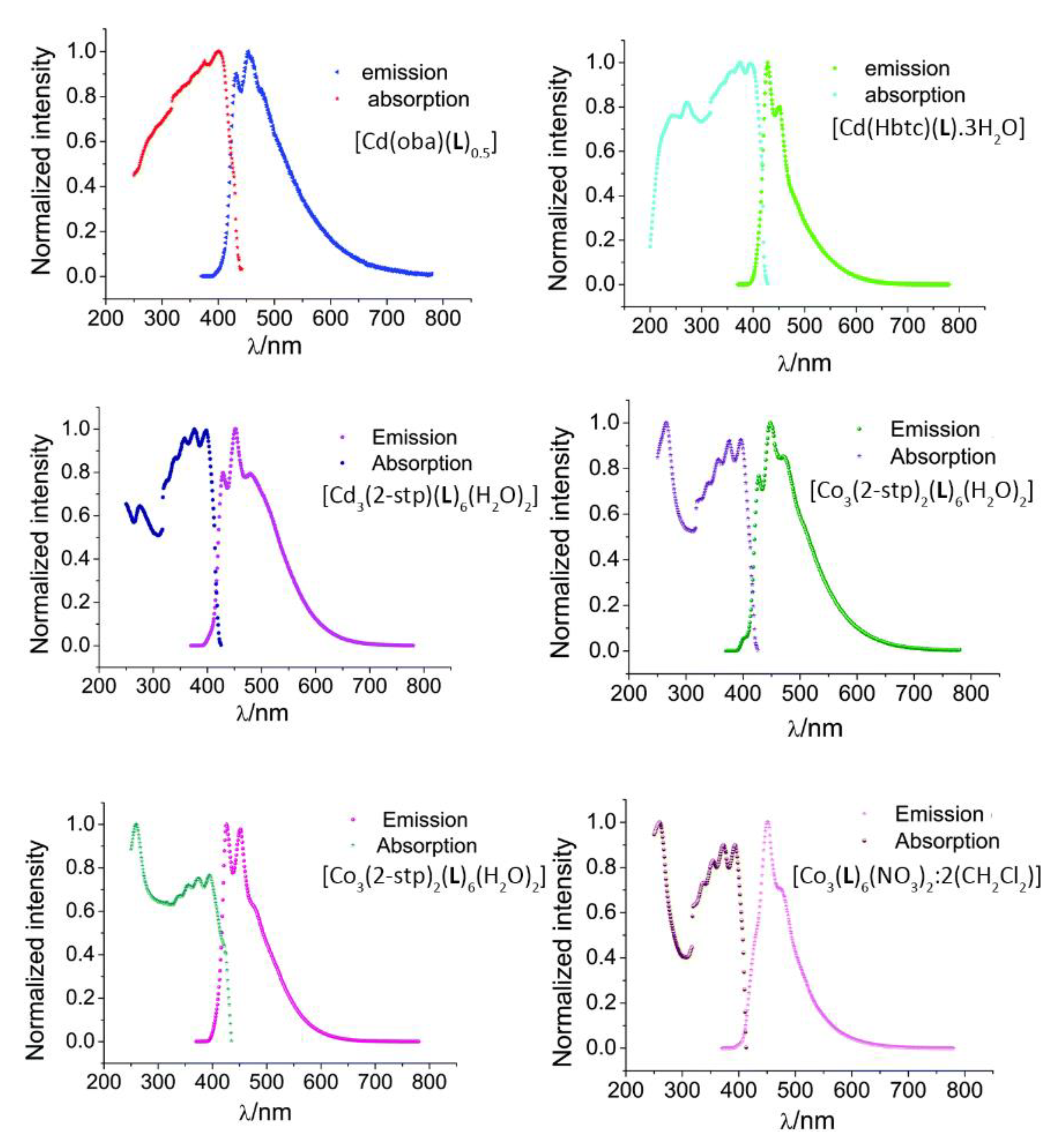
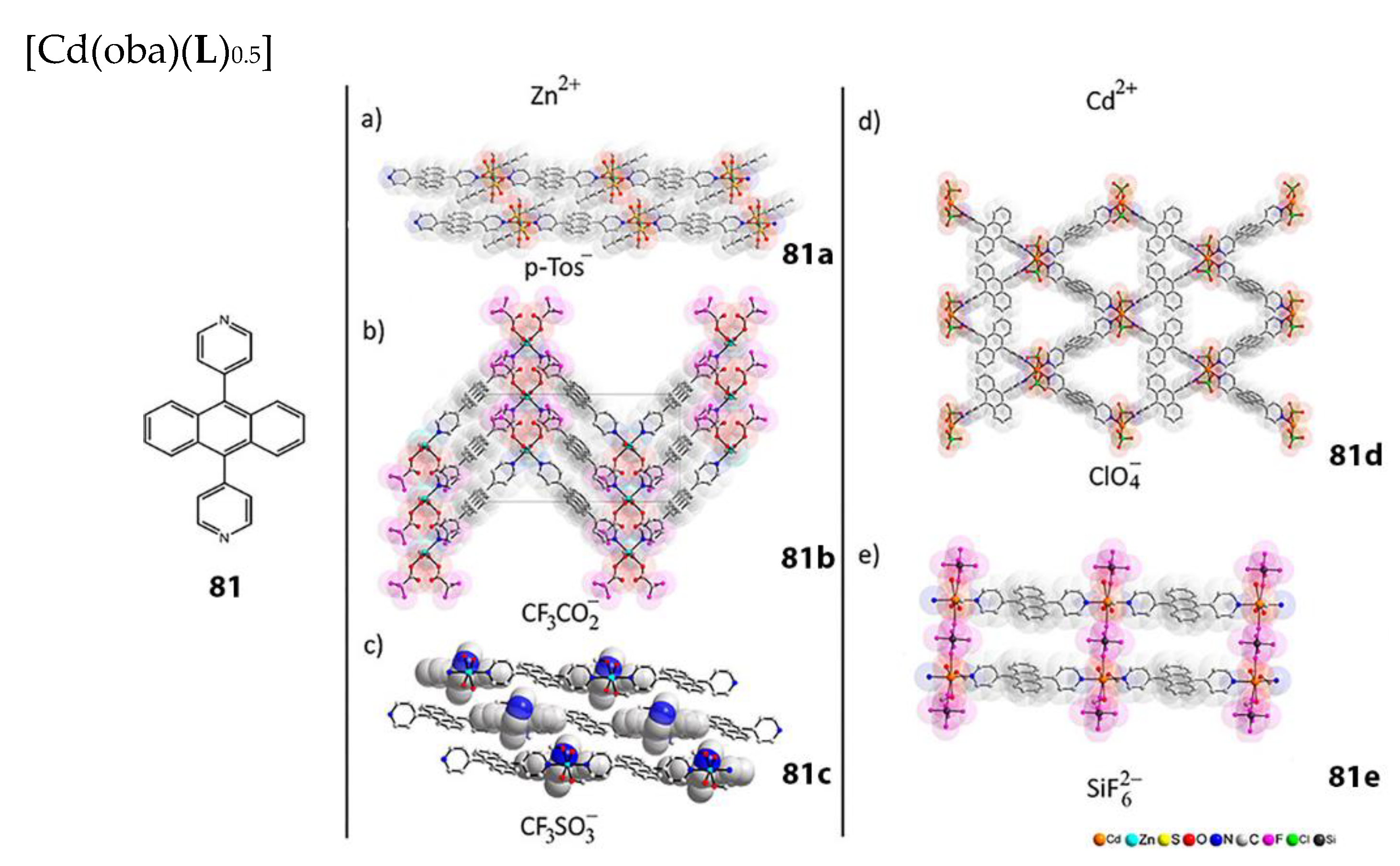
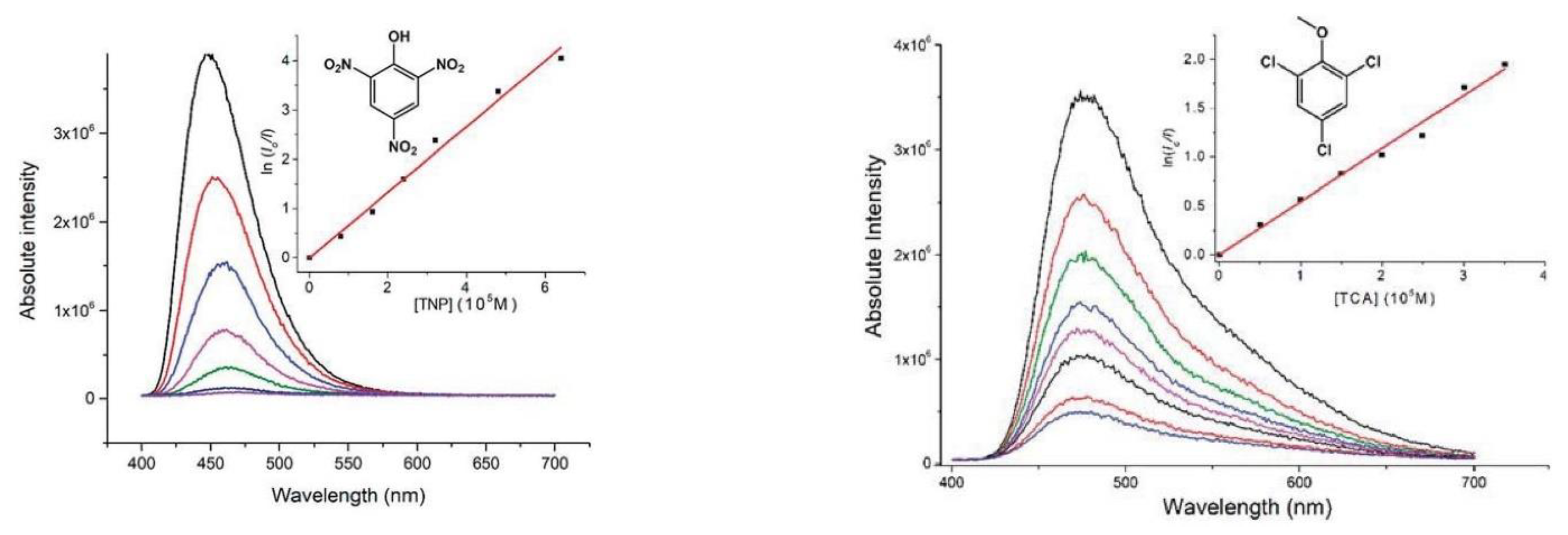
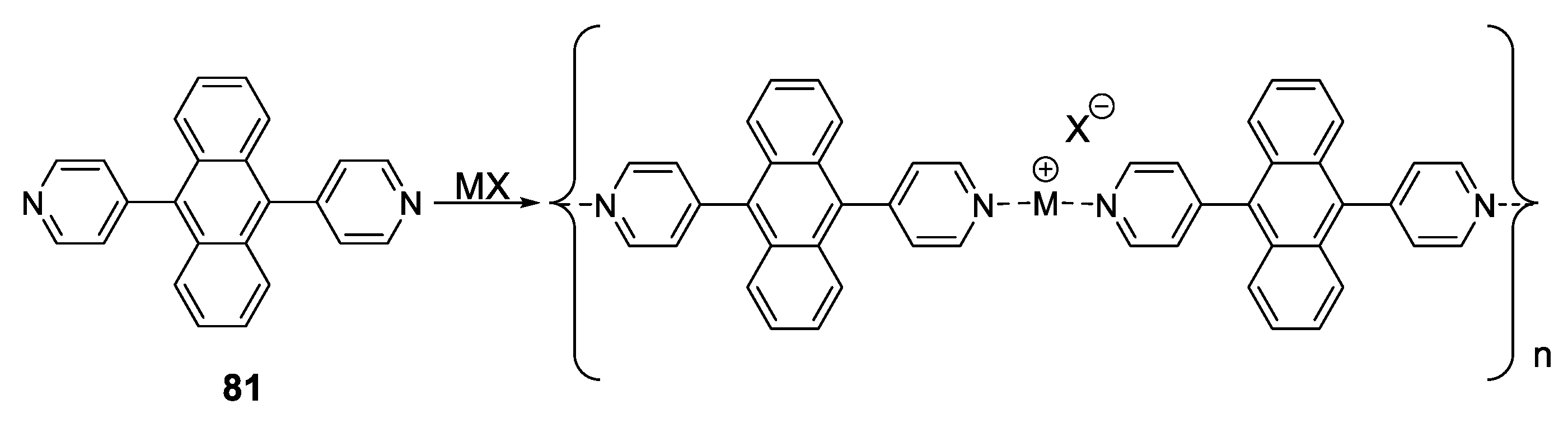
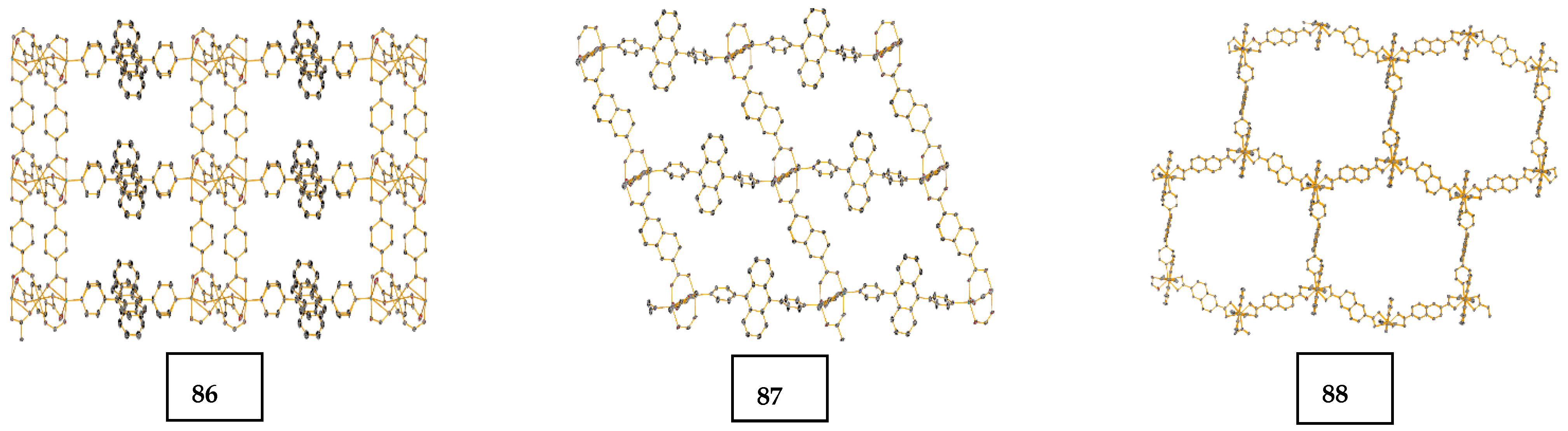
- Dong, J.-L.; Wang, D.-Z.; Jia, Y.-Y.; Wang, D.-H. Three coordination polymers based on 9,10-di(pyridine-4-yl)anthracene ligand: Syntheses, structures and fluorescent properties. J. Mol. Struct. 2017, 1142, 304–310. [Google Scholar] [CrossRef]






















































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
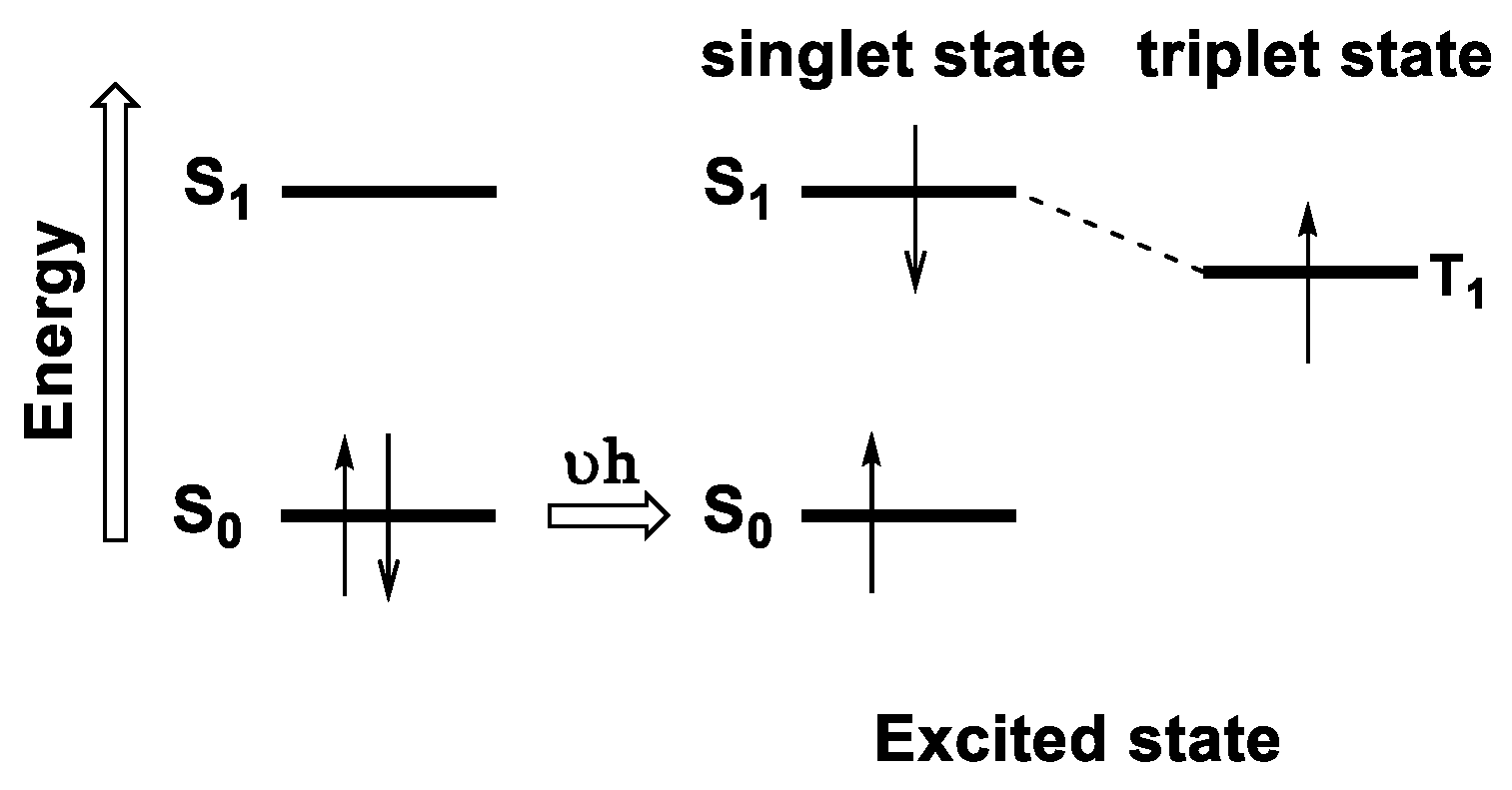{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
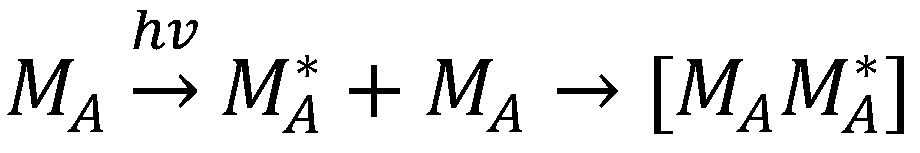{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
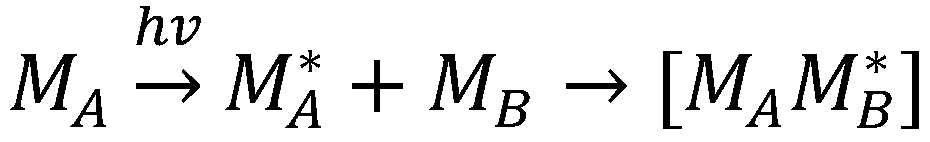{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
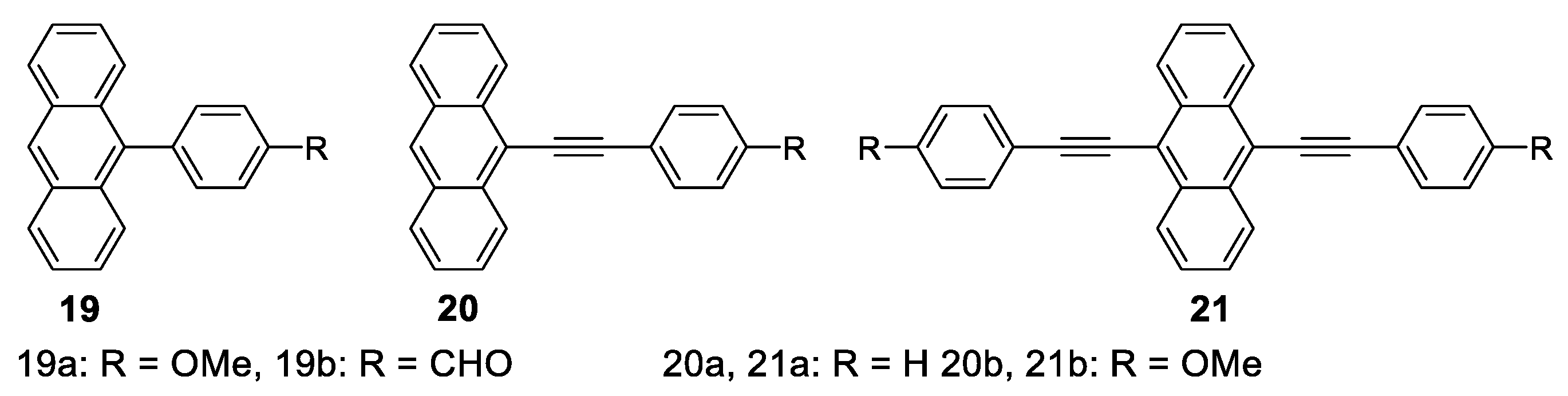
| Compd. | Stokes Shift a/nm | ||||
|---|---|---|---|---|---|
| 19a | 349, 367, 387 | 407, 428 | 61 | 0.41 | 4.59 |
| 19b | 349, 367, 387 | 480 | 113 | 0.20 | 3.10 |
| 20a | 399, 421 | 431, 455 | 32 | 0.54 | 4.77 |
| 20b | 404, 424 | 438, 461 | 34 | 0.75 | 4.26 |
| 21a | 438, 463 | 482, 506 | 19 | 0.59 | 3.57 |
| 21b | 449, 472 | 490 | 18 | 0.57 | 2.61 |
| Compd. | Absorbance | Absorbance Intensity | Emission | Emission Intensity | Crystal Packing |
|---|---|---|---|---|---|
| J-aggregate | Bathochromic (red) shift | High | Sharpening | High | Side by side |
| H-aggregate | Hypsochromic (blue) shift | High | Broadening | Low | Face to face |
| Compd. | Solution | Solid | PL (nm) in THF/H2O (%) | PL (nm) in Different Solvents a | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abs FPL b | PL FPL (%) | 0 | 30 | 50 | 70 | 90 | HEX | TOL | DCM | ACT | Me | |||
| DSA | 410 | 52 | 508 | 14 | 556 | 571 | 568 | 491 | 506 | 545 | 559 | 578 | 586 | 587 |
| 4-BFSA | 410 | 53 | 502 | 18 | 557 | 561 | 576 | 482 | 480 | 545 | 558 | 567 | 585 | 586 |
| 3-BFSA | 410 | 57 | 518 | 49 | 558 | 566 | 567 | 491 | 504 | 545 | 555 | 576 | 582 | 584 |
| BDFSA | 412 | 59 | 448 | 62 | 555 | 566 | 567 | 491 | 505 | 542 | 553 | 560 | 568 | 571 |
| BTFSA | 413 | 56 | 579 | 39 | 509 | 508 | 508 | 508 | 544 | 502 | 506 | 510 | 505 | 501 |
| Compound | F Excimeric | t, ns (Relative Amplitude, %) | ||
|---|---|---|---|---|
| 51 | 262, 357 365, 384 | 420, 442, 472 | 0.26 | τ1 = 3.52 (22.34) τ2 = 7.46 (77.66) |
| 52 | 264, 355 366, 384 | 420, 444, 474 | 0.22 | τ1 = 2.78 (33.79) τ2 = 5.56 (04.76) τ3 = 11.10 (61.44) |
| 53 | 264, 355 365, 382 | 418, 442, 471 | 0.16 | τ1 = 2.72 (19.12) τ2 = 7.28 (80.88) |
| 54 | 263, 356 364, 384 | 420, 443, 473 | 0.33 | τ1 = 2.31 (32.89) τ2 = 7.07 (67.11) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kastrati, A.; Oswald, F.; Scalabre, A.; Fromm, K.M. Photophysical Properties of Anthracene Derivatives. Photochem 2023, 3, 227-273. https://doi.org/10.3390/photochem3020015
Kastrati A, Oswald F, Scalabre A, Fromm KM. Photophysical Properties of Anthracene Derivatives. Photochem. 2023; 3(2):227-273. https://doi.org/10.3390/photochem3020015
Chicago/Turabian StyleKastrati, Agonist, Franck Oswald, Antoine Scalabre, and Katharina M. Fromm. 2023. "Photophysical Properties of Anthracene Derivatives" Photochem 3, no. 2: 227-273. https://doi.org/10.3390/photochem3020015
APA StyleKastrati, A., Oswald, F., Scalabre, A., & Fromm, K. M. (2023). Photophysical Properties of Anthracene Derivatives. Photochem, 3(2), 227-273. https://doi.org/10.3390/photochem3020015