
Akt/mTOR Activation in Lung Cancer Tumorigenic Regulators and Their Potential Value as Biomarkers

Abstract
:Simple Summary
Abstract
1. Introduction
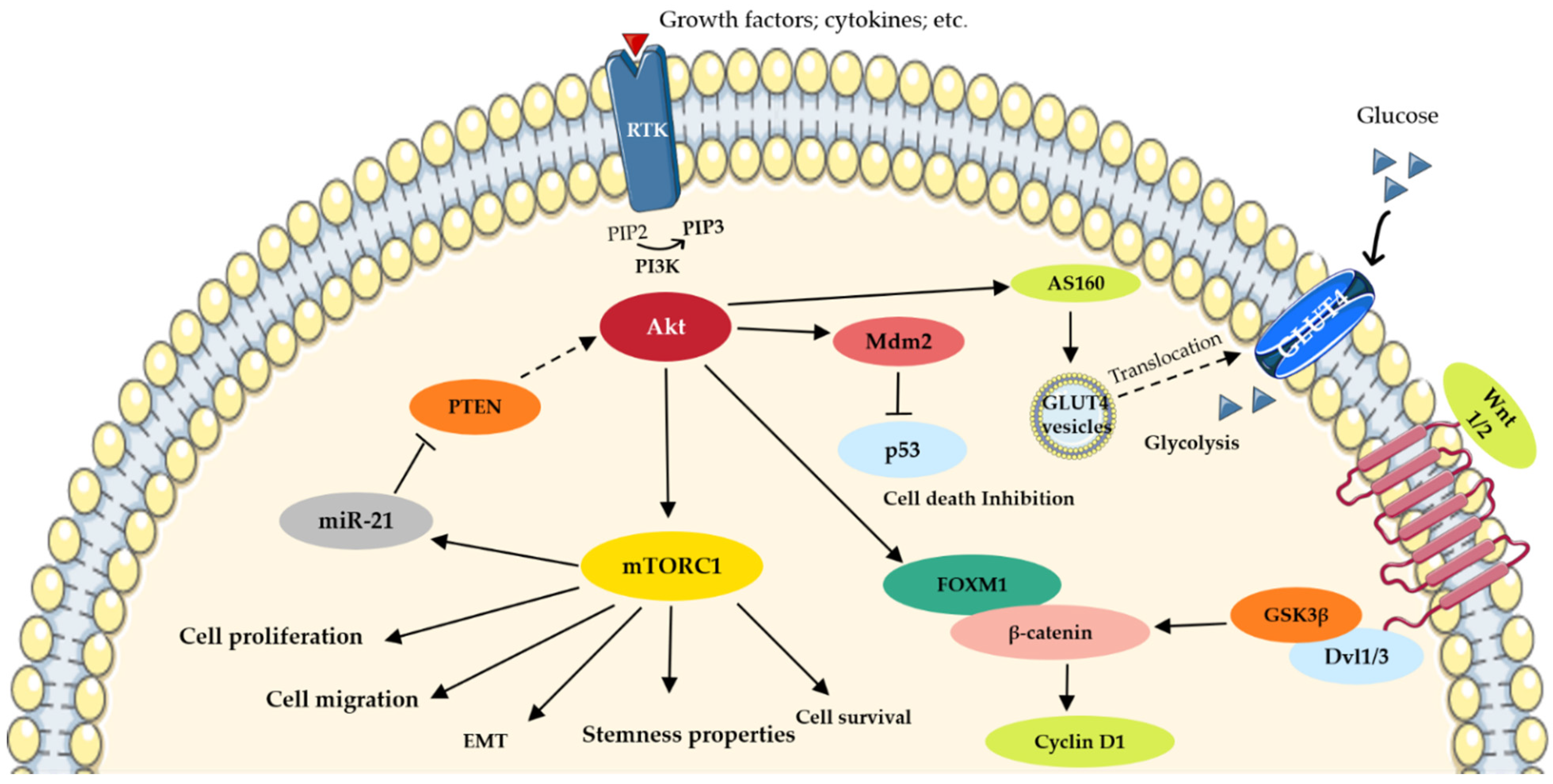
2. PI3K/Akt/mTOR Implication in Lung Cancer Molecular Machinery
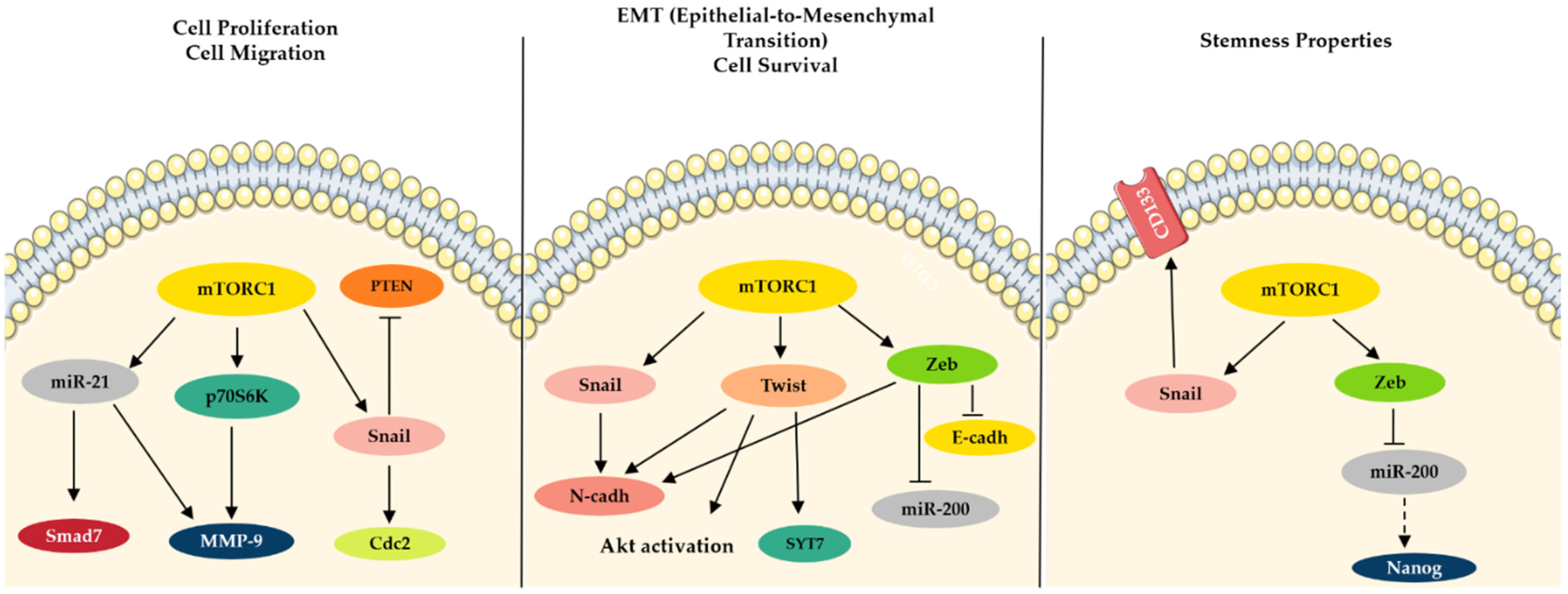
3. Tumor Cells Attain Mesenchymal Traits through Akt/mTOR-Induced EMT Activation
3.1. Twist, Snail and Zeb Transcription Factors at the Crossroads of EMT
3.2. Akt/mTOR/p70S6K Drives Cancer Cell Survival and Proliferation

{kind=link}
{kind=link}
| Cell Regulators | Downstream Target | Upstream Effector | References |
|---|---|---|---|
| Akt | mTOR | PI3K | [8,10,14,15,19] |
| FOXM1 | [104,105,106,107,108] | ||
| mTORC1 | p70S6K | Akt | [20,21] |
| Twist | [42,43] | ||
| Snail | [46,53] | ||
| Zeb (1/2) | [70,71,72,73,74] | ||
| FOXM1 | Cyclin D1 | [109,110,111] | |
| Mdm2 | p53 | [10,112] | |
| AS160 | GLTU4 | [113,114,115,116] | |
| GSK3β | β-catenin | Wnt1/2 | [117,118,119] |
| miR-21 | PTEN | mTORC1 | [120,121] |
| Smad7 | [113,114] | ||
| p70S6K | MMP-9 | [122,123,124] | |
| Snail | N-cadherin | [53,64] | |
| CD133 | [125,126] | ||
| Twist | Akt | [40,41] | |
| N-cadherin | [49,54] | ||
| Zeb (1/2) | E-cadherin | [76,77] | |
| miR-200 | [89,90,91,92,96] | ||
| miR-200 | Nanog | Zeb (1/2) | [77,90] |
3.3. Role of miRNAs as Cell Regulators
4. FOXM1/ β-Catenin/GSK3β Feedback Loop: Intersection between PI3K/Akt and Wnt Signaling Pathways?
5. Membrane Expression of CD133, a Stemness Marker in Lung Cancer
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Word Health Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 13 December 2021).
- Giustini, N.P.; Jeong, A.R.; Buturla, J.; Bazhenova, L. Advances in Treatment of Locally Advanced or Metastatic Non–Small Cell Lung Cancer: Targeted Therapy. Clin. Chest Med. 2020, 41, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Van Meerbeeck, J.P.; Fennell, D.A.; De Ruysscher, D.K.M. Small-cell lung cancer. Lancet 2011, 378, 1741–1755. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Prim. 2021, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 2020, 383, 640–649. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81–94. [Google Scholar] [CrossRef]
- Kurahara, H.; Takao, S.; Maemura, K.; Mataki, Y.; Kuwahata, T.; Maeda, K.; Ding, Q.; Sakoda, M.; Iino, S.; Ishigami, S.; et al. Epithelial–mesenchymal transition and mesenchymal–epithelial transition via regulation of ZEB-1 and ZEB-2 expression in pancreatic cancer. J. Surg. Oncol. 2012, 105, 655–661. [Google Scholar] [CrossRef]
- Pothongsrisit, S.; Pongrakhananon, V. Targeting the PI3K/AKT/mTOR Signaling Pathway in Lung Cancer: An Update Regarding Potential Drugs and Natural Products. Molecules 2021, 26, 4100. [Google Scholar]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef]
- Chan, C.H.; Jo, U.; Kohrman, A.; Rezaeian, A.H.; Chou, P.C.; Logothetis, C.; Lin, H.K. Posttranslational regulation of Akt in human cancer. Cell Biosci. 2014, 4, 59. [Google Scholar] [CrossRef] [Green Version]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Bacus, S.S.; Altomare, D.A.; Lyass, L.; Chin, D.M.; Farrell, M.P.; Gudkov, A.; Testa, J.R. AKT2 is frequently upregulated in HER-2/neu-positive breast cancers and may contribute to tumor aggressiveness by enhancing cell survival. Oncogene 2002, 21, 3532–3540. [Google Scholar] [CrossRef] [Green Version]
- Gener, P.; Rafael, D.; Seras-franzoso, J.; Perez, A.; Pindado, L.A.; Casas, G.; Arango, D.; Fernández, Y.; Díaz-riascos, Z.V.; Abasolo, I.; et al. Pivotal Role of AKT2 during Dynamic Phenotypic Change of Breast Cancer Stem Cells. Cancers 2019, 11, 1058. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT Kinases in Cancer: Implications for Therapeutic Targeting. Adv. Cancer Res. 2005, 94, 29–86. [Google Scholar]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.J.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.L.; Wu, C.Y.; Wu, J.; Lin, H.K. Regulation of Akt signaling activation by ubiquitination. Cell Cycle 2010, 9, 486–497. [Google Scholar] [CrossRef] [Green Version]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172, 113729. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.; Khursigara, G.; Sun, X.; Franke, T.F.; Chao, M.V. Akt Phosphorylates and Negatively Regulates Apoptosis Signal-Regulating Kinase 1. Mol. Cell. Biol. 2001, 21, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Pan, D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 2001, 15, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.S. Nanotechnology-Based Targeting of mTOR Signaling in Cancer. Int. J. Nanomed. 2020, 15, 5767–5781. [Google Scholar] [CrossRef]
- Mundi, P.S.; Sachdev, J.; McCourt, C.; Kalinsky, K. AKT in cancer: New molecular insights and advances in drug development. Br. J. Clin. Pharmacol. 2016, 82, 943–956. [Google Scholar] [CrossRef] [Green Version]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Oh, M.H.; Lee, H.J.; Yoo, S.B.; Xu, X.; Choi, J.S.; Kim, Y.H.; Lee, S.Y.; Lee, C.T.; Jheon, S.; Chung, J.H. Clinicopathological correlations of mTOR and pAkt expression in non-small cell lung cancer. Virchows Arch. 2012, 460, 601–609. [Google Scholar] [CrossRef]
- Murugan, A.K. mTOR: Role in cancer, metastasis and drug resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef]
- de la Cruz López, K.G.; Toledo Guzmán, M.E.; Sánchez, E.O.; García Carrancá, A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Ogata, H.; Honma, N.; Shibuya, K.; Mikami, T. Expression of mTOR Signaling Pathway Molecules in Triple-Negative Breast Cancer. Pathobiology 2019, 86, 315–321. [Google Scholar] [CrossRef]
- Murayama, T.; Inokuchi, M.; Takagi, Y.; Yamada, H.; Kojima, K.; Kumagai, J.; Kawano, T.; Sugihara, K. Relation between outcomes and localisation of p-mTOR expression in gastric cancer. Br. J. Cancer 2009, 100, 782–788. [Google Scholar] [CrossRef] [Green Version]
- An, J.Y.; Kim, K.M.; Choi, M.G.; Noh, J.H.; Sohn, T.S.; Bae, J.M.; Kim, S. Prognostic role of p-mTOR expression in cancer tissues and metastatic lymph nodes in pT2b gastric cancer. Int. J. Cancer 2010, 126, 2904–2913. [Google Scholar] [CrossRef]
- Karachaliou, N.; Codony-Servat, J.; Teixidó, C.; Pilotto, S.; Drozdowskyj, A.; Codony-Servat, C.; Giménez-Capitán, A.; Molina-Vila, M.A.; Bertrán-Alamillo, J.; Gervais, R.; et al. BIM and mTOR expression levels predict outcome to erlotinib in EGFR-mutant non-small-cell lung cancer. Sci. Rep. 2015, 5, 17499. [Google Scholar] [CrossRef] [Green Version]
- Gremke, N.; Polo, P.; Dort, A.; Schneikert, J.; Elmshäuser, S.; Brehm, C.; Klingmüller, U.; Schmitt, A.; Reinhardt, H.C.; Timofeev, O.; et al. mTOR-mediated cancer drug resistance suppresses autophagy and generates a druggable metabolic vulnerability. Nat. Commun. 2020, 11, 4684. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 143. [Google Scholar] [CrossRef]
- Wang, H.; Duan, L.; Zou, Z.; Li, H.; Yuan, S.; Chen, X.; Zhang, Y.; Li, X.; Sun, H.; Zha, H.; et al. Activation of the PI3K/Akt/mTOR/p70S6K pathway is involved in S100A4-induced viability and migration in colorectal cancer cells. Int. J. Med. Sci. 2014, 11, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W.; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT–mTOR Pathway in Non–Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Schmid, K.; Bago-Horvath, Z.; Berger, W.; Haitel, A.; Cejka, D.; Werzowa, J.; Filipits, M.; Herberger, B.; Hayden, H.; Sieghart, W. Dual inhibition of EGFR and mTOR pathways in small cell lung cancer. Br. J. Cancer 2010, 103, 622–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Tang, H.; Massi, D.; Hemmings, B.A.; Mandalà, M.; Hu, Z.; Wicki, A.; Xue, G. AKT-ions with a TWIST between EMT and MET. Oncotarget 2016, 7, 62767–62777. [Google Scholar] [CrossRef] [Green Version]
- Majc, B.; Sever, T.; Zarić, M.; Breznik, B.; Turk, B.; Lah, T.T. Epithelial-to-mesenchymal transition as the driver of changing carcinoma and glioblastoma microenvironment. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118782. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Wilson, M.M.; Weinberg, R.A.; Lees, J.A.; Guen, V.J. Emerging Mechanisms by which EMT Programs Control Stemness. Trends Cancer 2020, 6, 775–780. [Google Scholar] [CrossRef]
- Pei, H.; Li, Y.; Liu, M.; Chen, Y. Targeting Twist expression with small molecules. Medchemcomm 2017, 8, 268–275. [Google Scholar] [CrossRef]
- Khan, M.A.; Chen, H.C.; Zhang, D.; Fu, J. Twist: A molecular target in cancer therapeutics. Tumor Biol. 2013, 34, 2497–2506. [Google Scholar] [CrossRef]
- Pereira, L.; Horta, S.; Mateus, R.; Videira, M.A. Implications of Akt2/Twist crosstalk on breast cancer metastatic outcome. Drug Discov. Today 2015, 20, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Vesuna, F.; van Diest, P.; Chen, J.H.; Raman, V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem. Biophys. Res. Commun. 2008, 367, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, K.; Natsugoe, S.; Ishigami, S.; Matsumoto, M.; Okumura, H.; Setoyama, T.; Uchikado, Y.; Kita, Y.; Tamotsu, K.; Sakamoto, A.; et al. Significance of Twist expression and its association with E-cadherin in esophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 158. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Wang, X.; Dai, T.; Wu, Y.; Zhang, M.; Cao, R.; Zhang, R.; Wang, G.; Jiang, R.; Zhou, B.P.; et al. Twist promotes tumor metastasis in basal-like breast cancer by transcriptionally upregulating ROR1. Theranostics 2018, 8, 2739–2751. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, O.; Imamura, H.; Shimizu, T.; Kinoshita, J.; Okabe, T.; Hirano, A.; Yoshimatsu, K.; Konno, S.; Aiba, M.; Ogawa, K. Expression of Twist and Wnt in Human Breast Cancer. Anticancer Res. 2004, 24, 3851–3856. [Google Scholar] [PubMed]
- Wang, G.; Ma, W.; Li, Y.; Jiang, Y.; Ma, G.; Zhang, X.; Meng, L.; Du, J. Prognostic value of Twist, Snail and E-cadherin expression in pathological N0 non-small-cell lung cancer: A retrospective cohort study. Eur. J. Cardio-Thorac. Surg. 2018, 54, 237–245. [Google Scholar] [CrossRef]
- Zeng, J.; Zhan, P.; Wu, G.; Yang, W.; Liang, W.; Lv, T.; Song, Y. Prognostic value of twist in lung cancer: Systematic review and meta-analysis. Transl. Lung Cancer Res. 2015, 4, 236–241. [Google Scholar]
- Jiang, W.; Pang, X.G.; Wang, Q.; Shen, Y.X.; Chen, X.K.; Xi, J.J. Prognostic Role of Twist, Slug, and Foxc2 Expression in Stage I Non–Small-Cell Lung Cancer After Curative Resection. Clin. Lung Cancer 2012, 13, 280–287. [Google Scholar] [CrossRef]
- Hui, L.; Zhang, S.; Dong, X.; Tian, D.; Cui, Z.; Qiu, X. Prognostic Significance of Twist and N-Cadherin Expression in NSCLC. PLoS ONE 2013, 8, e62171. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.N.; Zhu, Z.H.; Chang, S.J.; Hang, X.S. Twist expression associated with the epithelial-mesenchymal transition in gastric cancer. Mol. Cell. Biochem. 2012, 367, 195–203. [Google Scholar] [CrossRef]
- Liu, A.; Sun, X.; Xu, J.; Xuan, Y.; Zhao, Y.; Qiu, T.; Hou, F.; Qin, Y.; Wang, Y.; Lu, T.; et al. Relevance and prognostic ability of Twist, Slug and tumor spread through air spaces in lung adenocarcinoma. Cancer Med. 2020, 9, 1986–1998. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Hashimoto, N.; Aoyama, D.; Kohnoh, T.; Sakamoto, K.; Kusunose, M.; Imaizumi, K.; Takeyama, Y.; Sato, M.; Kawabe, T.; et al. Involvement of the transcription factor twist in phenotype alteration through epithelial–mesenchymal transition in lung cancer cells. Mol. Carcinog. 2012, 51, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.H.; Chen, K.F.; Chou, S.H.; Huang, Y.F.; Wu, C.Y.; Cheng, D.E.; Chen, Y.W.; Yang, C.J.; Hung, J.Y.; Huang, M.S. Lung tumor-associated dendritic cell-derived resistin promoted cancer progression by increasing Wolf–Hirschhorn syndrome candidate 1/Twist pathway. Carcinogenesis 2013, 34, 2600–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.Y.; Chang, S.L.; Der Leu, J.; Chang, Y.C.; Hsiao, M.; Lin, L.T.; Lin, H.N.; Lee, Y.J. Comparison of cofilin-1 and Twist-1 protein expression in human non-small cell lung cancer tissues. Oncol. Rep. 2019, 42, 805–816. [Google Scholar] [CrossRef]
- Liu, X.; Li, C.; Yang, Y.; Liu, X.; Li, R.; Zhang, M.; Yin, Y.; Qu, Y. Synaptotagmin 7 in twist-related protein 1-mediated epithelial—Mesenchymal transition of non-small cell lung cancer. EBioMedicine 2019, 46, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shi, L.; Li, J.; Wang, H.; Yang, H. Involvement of twist in NNK exposure-promoted lung cancer cell migration and invasion. Toxicol. Vitr. 2020, 63, 104740. [Google Scholar] [CrossRef]
- Kwok, W.K.; Ling, M.T.; Lee, T.W.; Lau, T.C.M.; Zhou, C.; Zhang, X.; Chua, C.W.; Chan, K.W.; Chan, F.L.; Glackin, C.; et al. Up-Regulation of TWIST in Prostate Cancer and Its Implication as a Therapeutic Target. Cancer Res. 2005, 65, 5153–5162. [Google Scholar] [CrossRef] [Green Version]
- Kaufhold, S.; Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef]
- Skrzypek, K.; Majka, M. Interplay among SNAIL Transcription Factor, MicroRNAs, Long Non-Coding RNAs, and Circular RNAs in the Regulation of Tumor Growth and Metastasis. Cancers 2020, 12, 209. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Li, M.; Wang, X.; Li, L.; Li, Q.; Hou, Z.; Jia, H.; Liu, S. USP37 Promotes Lung Cancer Cell Migration by Stabilizing Snail Protein via Deubiquitination. Front. Genet. 2020, 10, 1324. [Google Scholar] [CrossRef]
- Martin, T.A.; Goyal, A.; Watkins, G.; Jiang, W.G. Expression of the Transcription Factors Snail, Slug, and Twist and Their Clinical Significance in Human Breast Cancer. Ann. Surg. Oncol. 2005, 12, 488–496. [Google Scholar] [CrossRef]
- Pérez-Mancera, P.A.; Pérez-Caro, M.; González-Herrero, I.; Flores, T.; Orfao, A.; Garcia de Herreros, A.; Gutiérrez-Adán, A.; Pintado, B.; Sagrera, A.; Sánchez-Martín, M.; et al. Cancer development induced by graded expression of Snail in mice. Hum. Mol. Genet. 2005, 14, 3449–3461. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Jiang, Y.; Chen, W.; Li, K.; Liu, X.; Gao, S.; Feng, H.; Wang, S.; Jiang, J.; Ma, X.; et al. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget 2015, 6, 6794–6810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. Targeting the EMT transcription factor Snail overcomes resistance to osimertinib in EGFR-mutant non-small cell lung cancer. Thorac. Cancer 2021, 12, 1708–1715. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, M.; Han, H.; Wang, B.; Li, S.; Zhang, Z.; Zhao, W. Silencing Snail suppresses tumor cell proliferation and invasion by reversing epithelial-to-mesenchymal transition and arresting G2/M phase in non-small cell lung cancer. Int. J. Oncol. 2017, 50, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuo, W.; Wang, Y.; Zhuo, X.; Zhang, Y.; Ao, X.; Chen, Z. Knockdown of Snail, a novel zinc finger transcription factor, via RNA interference increases A549 cell sensitivity to cisplatin via JNK/mitochondrial pathway. Lung Cancer 2008, 62, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Osorio, L.A.; Farfán, N.M.; Castellón, E.A.; Contreras, H.R. SNAIL transcription factor increases the motility and invasive capacity of prostate cancer cells. Mol. Med. Rep. 2016, 13, 778–786. [Google Scholar] [CrossRef] [Green Version]
- De Craene, B.; Gilbert, B.; Stove, C.; Bruyneel, E.; Van Roy, F.; Berx, G. The Transcription Factor Snail Induces Tumor Cell Invasion through Modulation of the Epithelial Cell Differentiation Program. Cancer Res. 2005, 65, 6237–6244. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.N.; Burton, L.J.; Henderson, V.; Randle, D.D.; Morton, D.J.; Smith, B.A.; Taliaferro-Smith, L.; Nagappan, P.; Yates, C.; Zayzafoon, M.; et al. Snail Promotes Epithelial Mesenchymal Transition in Breast Cancer Cells in Part via Activation of Nuclear ERK2. PLoS ONE 2014, 9, e104987. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Xiang, L.; Li, Y.; Zhao, Y.; Zhu, H.; Xiao, Y.; Liu, M.; Wu, X.; Wang, Z.; Jiang, P.; et al. Snail/FOXK1/Cyr61 Signaling Axis Regulates the Epithelial–Mesenchymal Transition and Metastasis in Colorectal Cancer. Cell. Physiol. Biochem. 2018, 47, 590–603. [Google Scholar] [CrossRef]
- Ganesan, R.; Mallets, E.; Gomez-Cambronero, J. The transcription factors Slug (SNAI2) and Snail (SNAI1) regulate phospholipase D (PLD) promoter in opposite ways towards cancer cell invasion. Mol. Oncol. 2016, 10, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Faget, J.; Groeneveld, S.; Boivin, G.; Sankar, M.; Zangger, N.; Garcia, M.; Guex, N.; Zlobec, I.; Steiner, L.; Piersigilli, A.; et al. Neutrophils and Snail Orchestrate the Establishment of a Pro-tumor Microenvironment in Lung Cancer. Cell Rep. 2017, 21, 3190–3204. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Lu, X.; Chen, Y.; Rendon, B.; Mitchell, R.A.; Cuatrecasas, M.; Cortés, M.; Postigo, A.; Liu, Y.; Dean, D.C. Zeb1 induces immune checkpoints to form an immunosuppressive envelope around invading cancer cells. Sci. Adv. 2021, 7, eabd7455. [Google Scholar] [CrossRef]
- Singh, A.B.; Sharma, A.; Smith, J.J.; Krishnan, M.; Chen, X.; Eschrich, S.; Washington, M.K.; Yeatman, T.J.; Beauchamp, R.D.; Dhawan, P. Claudin-1 Up-regulates the Repressor ZEB-1 to Inhibit E-Cadherin Expression in Colon Cancer Cells. Gastroenterology 2011, 141, 2140–2153. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Chen, X.; Wang, Y.; Qu, Z.; Lu, Q.; Zhao, J.; Yan, X.; Zhang, H.; Zhou, Y. Notch3 is important for TGF-β-induced epithelial–mesenchymal transition in non-small cell lung cancer bone metastasis by regulating ZEB-1. Cancer Gene Ther. 2014, 21, 364–372. [Google Scholar] [CrossRef]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef]
- Wu, Q.; Cao, J.; Ge, L.; Shi, Y.; Zhu, J.; Ma, D.; Zhang, H.; Yu, D.; Wang, J.; Suo, Z. Increased tumor ZEB1 protein expression is correlated with poor prognosis in patients with non-small cell lung cancers. Int. J. Clin. Exp. Med 2018, 11, 840–847. [Google Scholar]
- Larsen, J.E.; Nathan, V.; Osborne, J.K.; Farrow, R.K.; Deb, D.; Sullivan, J.P.; Dospoy, P.D.; Augustyn, A.; Hight, S.K.; Sato, M.; et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Invest. 2016, 126, 3219–3235. [Google Scholar] [CrossRef] [Green Version]
- Veloso, E.S.; Gonçalves, I.N.N.; Silveira, T.L.; Espirito Santo, J.T.; Figueiredo, L.V.; Varaschin, M.S.; Cassali, G.D.; Del Puerto, H.L.; Ferreira, E. ZEB and Snail expression indicates epithelial-mesenchymal transition in canine melanoma. Res. Vet. Sci. 2020, 131, 7–14. [Google Scholar] [CrossRef]
- Gemmill, R.M.; Roche, J.; Potiron, V.A.; Nasarre, P.; Mitas, M.; Coldren, C.D.; Helfrich, B.A.; Garrett-Mayer, E.; Bunn, P.A.; Drabkin, H.A. ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett. 2011, 300, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Arima, Y.; Hayashi, H.; Sasaki, M.; Hosonaga, M.; Goto, T.M.; Chiyoda, T.; Kuninaka, S.; Shibata, T.; Ohata, H.; Nakagama, H.; et al. Induction of ZEB proteins by inactivation of RB protein is key determinant of mesenchymal phenotype of breast cancer. J. Biol. Chem. 2012, 287, 7896–7906. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Vadhan, A.; Chen, P.H.; Lee, Y.L.; Chao, C.Y.; Cheng, K.H.; Chang, Y.C.; Hu, S.C.S.; Yuan, S.S.F. Cd44 promotes lung cancer cell metastasis through erk–zeb1 signaling. Cancers 2021, 13, 4057. [Google Scholar] [CrossRef]
- Najjary, S.; Mohammadzadeh, R.; Mokhtarzadeh, A.; Mohammadi, A.; Kojabad, A.B.; Baradaran, B. Role of miR-21 as an authentic oncogene in mediating drug resistance in breast cancer. Gene 2020, 738, 144453. [Google Scholar] [CrossRef]
- Bautista-Sánchez, D.; Arriaga-Canon, C.; Pedroza-Torres, A.; De La Rosa-Velázquez, I.A.; González-Barrios, R.; Contreras-Espinosa, L.; Montiel-Manríquez, R.; Castro-Hernández, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. The Promising Role of miR-21 as a Cancer Biomarker and Its Importance in RNA-Based Therapeutics. Mol. Ther.-Nucleic Acids 2020, 20, 409–420. [Google Scholar] [CrossRef]
- Bica-Pop, C.; Cojocneanu-Petric, R.; Magdo, L.; Raduly, L.; Gulei, D.; Berindan-Neagoe, I. Overview upon miR-21 in lung cancer: Focus on NSCLC. Cell. Mol. Life Sci. 2018, 75, 3539–3551. [Google Scholar] [CrossRef]
- Humphries, B.; Yang, C.; Humphries, B.; Yang, C. The microRNA-200 family: Small molecules with novel roles in cancer development, progression and therapy. Oncotarget 2015, 6, 6472–6498. [Google Scholar] [CrossRef] [Green Version]
- Savolainen, K.; Savolainen, K.; Scaravilli, M.; Scaravilli, M.; Scaravilli, M.; Ilvesmäki, A.; Staff, S.; Staff, S.; Staff, S.; Tolonen, T.; et al. Expression of the miR-200 family in tumor tissue, plasma and urine of epithelial ovarian cancer patients in comparison to benign counterparts. BMC Res. Notes 2020, 13, 311. [Google Scholar] [CrossRef]
- Mongroo, P.S.; Rustgi, A.K. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol. Ther. 2010, 10, 219–222. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, L. Members of the microRNA-200 family are promising therapeutic targets in cancer (Review). Exp. Ther. Med. 2017, 14, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Hu, W.; Li, L.L.; Wang, Y.X.; Zhou, Q.; Zhang, F.; Song-Yang, Y.Y.; Zhu, W.; Sun, C.C.; Li, D.J. Roles of miR-200 family members in lung cancer: More than tumor suppressors. Futur. Med. 2018, 14, 2875–2886. [Google Scholar] [CrossRef]
- Hill, L.; Browne, G.; Tulchinsky, E. ZEB/miR-200 feedback loop: At the crossroads of signal transduction in cancer. Int. J. Cancer 2013, 132, 745–754. [Google Scholar] [CrossRef]
- Diaz-Riascos, Z.V.; Ginesta, M.M.; Fabregat, J.; Serrano, T.; Busquets, J.; Buscail, L.; Cordelier, P.; Capellá, G. Expression and Role of MicroRNAs from the miR-200 Family in the Tumor Formation and Metastatic Propensity of Pancreatic Cancer. Mol. Ther.-Nucleic Acids 2019, 17, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.E.; Madhi, H.; Kim, H.; Lee, J.S.; Kim, M.H.; Kim, Y.N.; Goh, S.H. FAM188B Expression Is Critical for Cell Growth via FOXM1 Regulation in Lung Cancer. Biomedicines 2020, 8, 465. [Google Scholar] [CrossRef]
- Xiao, H.; Jiang, Z.; Fu, X.; Kuang, Y.; Lin, S.; Cai, Y.; Zhang, Q.; Zheng, F. High expression of forkhead box M1 (FOXM1) is a poor prognostic biomarker in lung adenocarcinoma. Transl. Cancer Res. 2020, 9, 6331–6343. [Google Scholar] [CrossRef]
- Xu, N.; Zhang, X.; Wang, X.; Ge, H.Y.; Wang, X.Y.; Garfield, D.; Yang, P.; Song, Y.L.; Bai, C.X. FoxM1 mediated resistance to gefitinib in non-smallcell lung cancer cells. Acta Pharmacol. Sin. 2012, 33, 675–681. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Li, J.; Li, B.; Wang, Y.; Li, M.; Ma, D.; Wang, X. Genistein exhibits anti-cancer effects via down-regulating FoxM1 in H446 small-cell lung cancer cells. Tumor Biol. 2013, 35, 4137–4145. [Google Scholar] [CrossRef]
- Wei, P.; Zhang, N.; Wang, Y.; Li, D.; Wang, L.; Sun, X.; Shen, C.; Yang, Y.; Zhou, X.; Du, X. FOXM1 promotes lung adenocarcinoma invasion and metastasis by upregulating SNAIL. Int. J. Biol. Sci. 2015, 11, 186–198. [Google Scholar] [CrossRef] [Green Version]
- Bao, B.; Wang, Z.; Ali, S.; Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Over-expression of FoxM1 leads to epithelial–mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J. Cell. Biochem. 2011, 112, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.F.; Qu, Z.Q.; Yuan, H.H.; Wang, J.Y.; Zhao, M.; Guo, Y.H.; Shi, J.; Gong, X.-D.; Zhu, Y.L.; Liu, F.; et al. Overexpression of FOXM1 is associated with EMT and is a predictor of poor prognosis in non-small cell lung cancer. Oncol. Rep. 2014, 31, 2660–2668. [Google Scholar] [CrossRef] [Green Version]
- Glibo, M.; Serman, A.; Karin-Kujundzic, V.; Vlatkovic, I.B.; Miskovic, B.; Vranic, S.; Serman, L. The role of glycogen synthase kinase 3 (GSK3) in cancer with emphasis on ovarian cancer development and progression: A comprehensive review. Bosn. J. Basic Med. Sci. 2021, 21, 5–18. [Google Scholar] [CrossRef]
- Wang, H.; Tan, Z.; Hu, H.; Liu, H.; Wu, T.; Zheng, C.; Wang, X.; Luo, Z.; Wang, J.; Liu, S.; et al. MicroRNA-21 promotes breast cancer proliferation and metastasis by targeting LZTFL1. BMC Cancer 2019, 19, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wu, X. MiR-21-5p promotes the progression of non-small-cell lung cancer by regulating the expression of SMAD7. Onco. Targets. Ther. 2018, 11, 8445–8454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Wu, Y.; Awadasseid, A.; Tanaka, Y.; Zhang, W. New Advances in Canonical Wnt/beta-Catenin Signaling in Cancer. Cancer Manag. Res. 2020, 12, 6987–6998. [Google Scholar] [CrossRef]
- Zhu, G.X.; Gao, D.; Shao, Z.Z.; Chen, L.; Ding, W.J.; Yu, Q.F. Wnt/β-catenin signaling: Causes and treatment targets of drug resistance in colorectal cancer (Review). Mol. Med. Rep. 2021, 23, 105. [Google Scholar] [CrossRef] [PubMed]
- Jackstadt, R.; Hodder, M.C.; Sansom, O.J. WNT and β-Catenin in Cancer: Genes and Therapy. Annu. Rev. Cancer Biol. 2020, 4, 177–196. [Google Scholar] [CrossRef] [Green Version]
- Vincent, E.E.; Elder, D.J.E.; O’Flaherty, L.; Pardo, O.E.; Dzien, P.; Phillips, L.; Morgan, C.; Pawade, J.; May, M.T.; Sohail, M.; et al. Glycogen Synthase Kinase 3 Protein Kinase Activity Is Frequently Elevated in Human Non-Small Cell Lung Carcinoma and Supports Tumour Cell Proliferation. PLoS ONE 2014, 9, e114725. [Google Scholar] [CrossRef] [Green Version]
- Vijay, G.V.; Zhao, N.; Den Hollander, P.; Toneff, M.J.; Joseph, R.; Pietila, M.; Taube, J.H.; Sarkar, T.R.; Ramirez-Pena, E.; Werden, S.J.; et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 37. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Liu, D.; Qiu, Z.; Huang, Y.; Chen, B.; Wang, L.; Xu, H.; Huang, N.; Liu, L.; Li, W. GSK3β Overexpression Indicates Poor Prognosis and Its Inhibition Reduces Cell Proliferation and Survival of Non-Small Cell Lung Cancer Cells. PLoS ONE 2014, 9, e91231. [Google Scholar] [CrossRef]
- Nishijima, N.; Seike, M.; Soeno, C.; Chiba, M.; Miyanaga, A.; Noro, R.; Sugano, T.; Matsumoto, M.; Kubota, K.; Gemma, A. miR-200/ZEB axis regulates sensitivity to nintedanib in non-small cell lung cancer cells. Int. J. Oncol. 2016, 48, 937–944. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Meng, L.; Ye, J.; Wei, X.; Shang, Y.; Tian, Y.; He, Y.; Peng, Z.; Chen, L.; Chen, W.; et al. Transcriptional repression of miR-200 family members by Nanog in colon cancer cells induces epithelial–mesenchymal transition (EMT). Cancer Lett. 2017, 392, 26–38. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, D.; Sun, G.; Mei, F.; Cui, Y.; Xu, H. Effect of miR-21 on Apoptosis in Lung Cancer Cell Through Inhibiting the PI3K/ Akt/NF-κB Signaling Pathway in Vitro and in Vivo. Cell. Physiol. Biochem. 2018, 46, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xia, H.; Liu, Y.; Li, M. Silencing miR-21 sensitizes non-small cell lung cancer A549 cells to ionizing radiation through inhibition of PI3K/Akt. Biomed Res. Int. 2014, 2014, 617868. [Google Scholar] [CrossRef] [PubMed]
- Ni, K.; Wang, D.; Xu, H.; Mei, F.; Wu, C.; Liu, Z.; Zhou, B. MiR-21 promotes non-small cell lung cancer cells growth by regulating fatty acid metabolism. Cancer Cell Int. 2019, 19, 219. [Google Scholar] [CrossRef] [Green Version]
- Fakiruddin, K.S.; Lim, M.N.; Nordin, N.; Rosli, R.; Zakaria, Z.; Abdullah, S. Targeting of CD133+ Cancer Stem Cells by Mesenchymal Stem Cell Expressing TRAIL Reveals a Prospective Role of Apoptotic Gene Regulation in Non-Small Cell Lung Cancer. Cancers 2019, 11, 1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C.; Hsu, H.S.; Chen, Y.W.; Tsai, T.H.; How, C.K.; Wang, C.Y.; Hung, S.C.; Chang, Y.L.; Tsai, M.L.; Lee, Y.Y.; et al. Oct-4 Expression Maintained Cancer Stem-Like Properties in Lung Cancer-Derived CD133-Positive Cells. PLoS ONE 2008, 3, e2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Title, A.C.; Hong, S.J.; Pires, N.D.; Hasenöhrl, L.; Godbersen, S.; Stokar-Regenscheit, N.; Bartel, D.P.; Stoffel, M. Genetic dissection of the miR-200–Zeb1 axis reveals its importance in tumor differentiation and invasion. Nat. Commun. 2018, 9, 4671. [Google Scholar] [CrossRef]
- Bendoraite, A.; Knouf, E.C.; Garg, K.S.; Parkin, R.K.; Kroh, E.M.; O’Briant, K.C.; Ventura, A.P.; Godwin, A.K.; Karlan, B.Y.; Drescher, C.W.; et al. Regulation of miR-200 family microRNAs and ZEB transcription factors in ovarian cancer: Evidence supporting a mesothelial-to-epithelial transition. Gynecol. Oncol. 2010, 116, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Liu, Y.; Wang, Y.; Zhao, M.; Tu, L.; Luo, F. Decitabine reverses TGF-beta1-induced epithelial-mesenchymal transition in non-small-cell lung cancer by regulating miR-200/ZEB axis. Drug Des. Devel. Ther. 2017, 11, 969–983. [Google Scholar] [CrossRef] [Green Version]
- Kundu, S.T.; Byers, L.A.; Peng, D.H.; Roybal, J.D.; Diao, L.; Wang, J.; Tong, P.; Creighton, C.J.; Gibbons, D.L. The miR-200 family and the miR-183~96~182 cluster target Foxf2 to inhibit invasion and metastasis in lung cancers. Oncogene 2015, 35, 173–186. [Google Scholar] [CrossRef]
- Jin, H.F.; Wang, J.F.; Song, T.T.; Zhang, J.; Wang, L. MiR-200b Inhibits Tumor Growth and Chemoresistance via Targeting p70S6K1 in Lung Cancer. Front. Oncol. 2020, 10, 643. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, J.; Yang, P.; Lu, Q.; Zhang, T.; Yang, Y. MicroRNA-200 promotes lung cancer cell growth through FOG2-independent AKT activation. IUBMB Life 2015, 67, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulos, D.; Polytarchou, C.; Hatziapostolou, M.; Kottakis, F.; Maroulakou, I.G.; Struhl, K.; Tsichlis, P.N. MicroRNAs Differentially Regulated by Akt Isoforms Control EMT and Stem Cell Renewal in Cancer Cells. Sci. Signal. 2009, 2, ra62. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.L.; Sun, J.; Lu, Y.; Liu, Y.; Cao, H.; Zhang, H.; Calin, G.A. MiR-200 family and cancer: From a meta-analysis view. Mol. Aspects Med. 2019, 70, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; Barbano, R.; Dama, E.; Pasculli, B.; Rendina, M.; Morritti, M.G.; Melocchi, V.; Castelvetere, M.; Valori, V.M.; Ravaioli, S.; et al. Combined analysis of miR-200 family and its significance for breast cancer. Sci. Rep. 2021, 11, 2980. [Google Scholar] [CrossRef] [PubMed]
- Javid, H.; Soltani, A.; Mohammadi, F.; Hashemy, S.I. Emerging roles of microRNAs in regulating the mTOR signaling pathway during tumorigenesis. J. Cell. Biochem. 2019, 120, 10874–10883. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.H.; Tsao, C.J. Emerging role of microRNA-21 in cancer (Review). Biomed. Rep. 2016, 5, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Zhao, J.J.; Tao, Y.; Guo, M.; Ya, Z.; Chen, C.; Qin, N.; Zheng, J.; Luo, J.; Xu, L. MicroRNA-21: A promising biomarker for the prognosis and diagnosis of non-small cell lung cancer. Oncol. Lett. 2018, 16, 2777–2782. [Google Scholar] [CrossRef]
- Pop-Bica, C.; Pintea, S.; Magdo, L.; Cojocneanu, R.; Gulei, D.; Ferracin, M.; Berindan-Neagoe, I. The Clinical Utility of miR-21 and let-7 in Non-small Cell Lung Cancer (NSCLC). A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 2210. [Google Scholar] [CrossRef]
- Ourô, S.; Mourato, C.; Velho, S.; Cardador, A.; Ferreira, M.P.; Albergaria, D.; Castro, R.E.; Maio, R.; Rodrigues, C.M.P. Potential of miR-21 to Predict Incomplete Response to Chemoradiotherapy in Rectal Adenocarcinoma. Front. Oncol. 2020, 10, 2212. [Google Scholar] [CrossRef]
- Liu, X.G.; Zhu, W.Y.; Huang, Y.Y.; Ma, L.N.; Zhou, S.Q.; Wang, Y.K.; Zeng, F.; Zhou, J.H.; Zhang, Y.K. High expression of serum miR-21 and tumor miR-200c associated with poor prognosis in patients with lung cancer. Med. Oncol. 2011, 29, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Sahraei, M.; Chaube, B.; Liu, Y.; Sun, J.; Kaplan, A.; Price, N.L.; Ding, W.; Oyaghire, S.; García-Milian, R.; Mehta, S.; et al. Suppressing miR-21 activity in tumor-associated macrophages promotes an antitumor immune response. J. Clin. Invest. 2019, 129, 5518–5536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunita, A.; Morita, S.; Irisa, T.U.; Goto, A.; Niki, T.; Takai, D.; Nakajima, J.; Fukayama, M. MicroRNA-21 in cancer-associated fibroblasts supports lung adenocarcinoma progression. Sci. Rep. 2018, 8, 8838. [Google Scholar] [CrossRef] [PubMed]
- Marin, I.; Ofek, E.; Bar, J.; Prisant, N.; Perelman, M.; Avivi, C.; Lavy-Shahaf, G.; Onn, A.; Katz, R.; Barshack, I. MiR-21, EGFR and PTEN in non-small cell lung cancer: An in-situ hybridisation and immunohistochemistry study. J. Clin. Pathol. 2020, 73, 636–641. [Google Scholar] [CrossRef]
- Sun, L.H.; Tian, D.; Yang, Z.C.; Li, J.L. Exosomal miR-21 promotes proliferation, invasion and therapy resistance of colon adenocarcinoma cells through its target PDCD4. Sci. Rep. 2020, 10, 8271. [Google Scholar] [CrossRef]
- Li, B.; Ren, S.; Li, X.; Wang, Y.; Garfield, D.; Zhou, S.; Chen, X.; Su, C.; Chen, M.; Kuang, P.; et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer 2014, 83, 146–153. [Google Scholar] [CrossRef]
- Seike, M.; Goto, A.; Okano, T.; Bowman, E.D.; Schetter, A.J.; Horikawa, I.; Mathe, E.A.; Jen, J.; Yang, P.; Sugimura, H.; et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc. Natl. Acad. Sci. USA 2009, 106, 12085–12090. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Li, X.; Cheng, T.; Wu, J. miR-21-5p/SMAD7 axis promotes the progress of lung cancer. Thorac. Cancer 2021, 12, 2307–2313. [Google Scholar] [CrossRef]
- Shang, S.; Hua, F.; Hu, Z.-W.; Shang, S.; Hua, F.; Hu, Z.-W. The regulation of β-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [Green Version]
- Rapp, J.; Jaromi, L.; Kvell, K.; Miskei, G.; Pongracz, J.E. WNT signaling-lung cancer is no exception. Respir. Res. 2017, 18, 167. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D.J. Wnt Signaling Pathway in Non–Small Cell Lung Cancer. J. Natl. Cancer Inst. 2014, 106, djt356. [Google Scholar] [CrossRef]
- Yu, W.; Li, L.; Zheng, F.; Yang, W.; Zhao, S.; Tian, C.; Yin, W.; Chen, Y.; Guo, W.; Zou, L.; et al. β-Catenin Cooperates with CREB Binding Protein to Promote the Growth of Tumor Cells. Cell. Physiol. Biochem. 2017, 44, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, K.; Ochiai, A.; Akimoto, S.; Nakamura, S.; Baba, S.; Moriya, Y.; Hirohashi, S. Cytoplasmic Beta-Catenin Accumulation as a Predictor of Hematogenous Metastasis in Human Colorectal Cancer. Oncology 2000, 59, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhan, P.; Katoh, M.; Kobayashi, S.S.; Phan, K.; Qian, H.; Li, H.; Wang, X.; Wang, X.; Song, Y. Prognostic significance of β-catenin expression in patients with non-small cell lung cancer: A meta-analysis. Transl. Lung Cancer Res. 2017, 6, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, T.; Shiozaki, H.; Shibamoto, S.; Oka, H.; Kimura, Y.; Tamura, S.; Inoue, M.; Monden, T.; Ito, F.; Monden, M. Beta-catenin expression in human cancers. Am. J. Pathol. 1996, 148, 39. [Google Scholar] [PubMed]
- Retera, J.M.A.M.; Leers, M.P.G.; Sulzer, M.A.; Theunissen, P.H.M.H. The expression of beta-catenin in non-small-cell lung cancer: A clinicopathological study. J. Clin. Pathol. 1998, 51, 891–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourboulia, D.; Han, H.; Jensen-Taubman, S.; Gavil, N.; Isaac, B.; Wei, B.; Neckers, L.; Stetler-Stevenson, W.; Bourboulia, D.; Han, H.; et al. TIMP-2 modulates cancer cell transcriptional profile and enhances E-cadherin/beta-catenin complex expression in A549 lung cancer cells. Oncotarget 2013, 4, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, S.; Sng, N.; Carretero, J.; Welner, R.; Hayashi, Y.; Yamamoto, M.; Tan, A.J.; Yamaguchi, N.; Yasuda, H.; Li, D.; et al. β-Catenin Contributes to Lung Tumor Development Induced by EGFR Mutations. Cancer Res. 2014, 74, 5891–5902. [Google Scholar] [CrossRef] [Green Version]
- Kafka, A.; Tomas, D.; Beroš, V.; Pećina, H.I.; Zeljko, M.; Pećina-Šlaus, N. Brain Metastases from Lung Cancer Show Increased Expression of DVL1, DVL3 and Beta-Catenin and Down-Regulation of E-Cadherin. Int. J. Mol. Sci. 2014, 15, 10635–10651. [Google Scholar] [CrossRef] [Green Version]
- Uematsu, K.; He, B.; You, L.; Xu, Z.; Mccormick, F.; Jablons, D.M. Activation of the Wnt pathway in non small cell lung cancer: Evidence of dishevelled overexpression. Oncogene 2003, 22, 7218–7221. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Sun, P.L.; Li, J.Z.; Jheon, S.; Lee, C.T.; Chung, J.H. Aberrant Wnt1/β-Catenin Expression is an Independent Poor Prognostic Marker of Non-small Cell Lung Cancer After Surgery. J. Thorac. Oncol. 2011, 6, 716–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Miller, S.A.; Wang, H.Y.; Xia, W.; Wen, Y.; Zhou, B.P.; Li, Y.; Lin, S.Y.; Hung, M.C. β-catenin interacts with and inhibits NF-κB in human colon and breast cancer. Cancer Cell 2002, 2, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Kalathil, D.; John, S.; Nair, A.S. FOXM1 and Cancer: Faulty Cellular Signaling Derails Homeostasis. Front. Oncol. 2021, 10, 3472. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, F.; Tan, Q.; Guo, M.; Ma, P.; Wang, X.; Zhang, S.; Xu, J.; Luo, P.; Jin, Y. The multifaceted roles of FOXM1 in pulmonary disease. Cell Commun. Signal. 2019, 17, 35. [Google Scholar] [CrossRef] [Green Version]
- Liao, G.-B.; Li, X.Z.; Zeng, S.; Liu, C.; Yang, S.M.; Yang, L.; Hu, C.J.; Bai, J.Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Qiao, W.-B.; Shan, L. Expression and functional characterization of FOXM1 in non-small cell lung cancer. Onco. Targets. Ther. 2018, 11, 3385–3393. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Jia, D.; Chen, W.; Wang, H.; Liu, F.; Ge, H.; Zhu, X.; Song, Y.; Zhang, X.; Zhang, D.; et al. FoxM1 Is Associated with Poor Prognosis of Non-Small Cell Lung Cancer Patients through Promoting Tumor Metastasis. PLoS ONE 2013, 8, e59412. [Google Scholar] [CrossRef]
- Liang, S.K.; Hsu, C.C.; Song, H.L.; Huang, Y.C.; Kuo, C.W.; Yao, X.; Li, C.C.; Yang, H.C.; Hung, Y.L.; Chao, S.Y.; et al. FOXM1 is required for small cell lung cancer tumorigenesis and associated with poor clinical prognosis. Oncogene 2021, 40, 4847–4858. [Google Scholar] [CrossRef]
- Xiu, G.; Sui, X.; Wang, Y.; Zhang, Z. FOXM1 regulates radiosensitivity of lung cancer cell partly by upregulating KIF20A. Eur. J. Pharmacol. 2018, 833, 79–85. [Google Scholar] [CrossRef]
- Kwok, J.M.M.; Peck, B.; Monteiro, L.J.; Schwenen, H.D.C.; Millour, J.; Coombes, R.C.; Myatt, S.S.; Lam, E.W.F. FOXM1 Confers Acquired Cisplatin Resistance in Breast Cancer Cells. Mol. Cancer Res. 2010, 8, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wen, L.; Zhao, S.-H.; Ai, Z.H.; Guo, J.-Z.; Liu, W.-C. FoxM1 expression is significantly associated with cisplatin-based chemotherapy resistance and poor prognosis in advanced non-small cell lung cancer patients. Lung Cancer 2013, 79, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, J.; Zhou, W.; Ren, Y.; Wang, X.; Chen, H.; Zhang, J.; Chen, J.; Sun, Y.; Cui, L.; et al. Activation of an AKT/FOXM1/STMN1 pathway drives resistance to tyrosine kinase inhibitors in lung cancer. Br. J. Cancer 2017, 117, 974–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domoto, T.; Uehara, M.; Bolidong, D.; Minamoto, T. Glycogen Synthase Kinase 3β in Cancer Biology and Treatment. Cells 2020, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Chiarini, F.; Paganelli, F.; Marmiroli, S.; Martelli, A.M. Crosstalks of GSK3 signaling with the mTOR network and effects on targeted therapy of cancer. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118635. [Google Scholar] [CrossRef] [Green Version]
- Sahin, I.; Eturi, A.; De Souza, A.; Pamarthy, S.; Tavora, F.; Giles, F.J.; Carneiro, B.A. Glycogen synthase kinase-3 beta inhibitors as novel cancer treatments and modulators of antitumor immune responses. Cancer Biol. Ther. 2019, 20, 1047–1056. [Google Scholar] [CrossRef]
- Alves, M.; de Paula Borges, D.; Kimberly, A.; Martins Neto, F.; Oliveira, A.C.; de Sousa, J.C.; Nogueira, C.D.; Carneiro, B.A.; Tavora, F. Glycogen Synthase Kinase-3 Beta Expression Correlates With Worse Overall Survival in Non-Small Cell Lung Cancer—A Clinicopathological Series. Front. Oncol. 2021, 11, 343. [Google Scholar] [CrossRef]
- Ren, J.; Liu, T.; Han, Y.; Wang, Q.; Chen, Y.; Li, G.; Jiang, L. GSK-3β inhibits autophagy and enhances radiosensitivity in non-small cell lung cancer. Diagn. Pathol. 2018, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Remsing Rix, L.L.; Kuenzi, B.M.; Luo, Y.; Remily-Wood, E.; Kinose, F.; Wright, G.; Li, J.; Koomen, J.M.; Haura, E.B.; Lawrence, H.R.; et al. GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem. Biol. 2014, 9, 353–358. [Google Scholar] [CrossRef]
- Marchand, B.; Tremblay, I.; Cagnol, S.; Boucher, M.J. Inhibition of glycogen synthase kinase-3 activity triggers an apoptotic response in pancreatic cancer cells through JNK-dependent mechanisms. Carcinogenesis 2012, 33, 529–537. [Google Scholar] [CrossRef]
- Kao, S.H.; Wang, W.L.; Chen, C.Y.; Chang, Y.L.; Wu, Y.Y.; Wang, Y.T.; Wang, S.P.; Nesvizhskii, A.I.; Chen, Y.J.; Hong, T.M.; et al. GSK3β controls epithelial–mesenchymal transition and tumor metastasis by CHIP-mediated degradation of Slug. Oncogene 2013, 33, 3172–3182. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K.; Takeuchi, S.; Arai, S.; Kita, K.; Tanimoto, A.; Nishiyama, A.; Yano, S. Glycogen synthase kinase-3 inhibition overcomes epithelial-mesenchymal transition-associated resistance to osimertinib in EGFR-mutant lung cancer. Cancer Sci. 2020, 111, 2374–2384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, C.; Sun, B.; Lv, J.; Liu, Z.; Liu, S.; Li, H. Integrated High Throughput Analysis Identifies GSK3 as a Crucial Determinant of p53-Mediated Apoptosis in Lung Cancer Cells. Cell. Physiol. Biochem. 2017, 42, 1177–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.Y.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates β-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yu, D.; Yu, Z.; Gao, Q.; Chen, R.; Zhou, L.; Wang, R.; Li, Y.; Qian, Y.; Zhao, J.; et al. TIPE3 promotes non-small cell lung cancer progression via the protein kinase B/extracellular signal-regulated kinase 1/2-glycogen synthase kinase 3β-β-catenin/Snail axis. Transl. Lung Cancer Res. 2021, 10, 936–954. [Google Scholar] [CrossRef]
- Basu, A.; Lambring, C.B. Akt Isoforms: A Family Affair in Breast Cancer. Cancers 2021, 13, 3445. [Google Scholar] [CrossRef]
- Liu, P.; Begley, M.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef]
- Moore, S.M.; Rintoul, R.C.; Walker, T.R.; Chilvers, E.R.; Haslett, C.; Sethi, T. The Presence of a Constitutively Active Phosphoinositide 3-Kinase in Small Cell Lung Cancer Cells Mediates Anchorage-independent Proliferation via a Protein Kinase B and p70s6k-dependent Pathway. Cancer Res. 1998, 58, 5239–5247. [Google Scholar]
- Kwon, H.K.; Bae, G.U.; Yoon, J.W.; Yong, K.K.; Lee, H.Y.; Lee, H.W.; Han, J.W. Constitutive activation of p70S6k in cancer cells. Arch. Pharm. Res. 2002, 25, 685–690. [Google Scholar] [CrossRef]
- Wang, X.; Yao, J.; Wang, J.; Zhang, Q.; Brady, S.W.; Arun, B.; Seewaldt, V.L.; Yu, D. Targeting Aberrant p70S6K Activation for Estrogen Receptor–Negative Breast Cancer Prevention. Cancer Prev. Res. 2017, 10, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Chung, K.S.; Lee, H.H.; Ko, D.; Kang, M.; Yoo, H.; Ahn, J.H.; Lee, J.Y.; Lee, K.T. Improved tumor-suppressive effect of OZ-001 combined with cisplatin mediated by mTOR/p70S6K and STAT3 inactivation in A549 human lung cancer cells. Biomed. Pharmacother. 2021, 142, 111961. [Google Scholar] [CrossRef]
- Li, M.; Chen, H.; Sun, T.; Ma, Z.; Chen, X.; Wu, D.; Huang, W.; Wang, X. p70S6K Promotes Acquired Resistance of Erlotinib Through Induction of Epithelial-Mesenchymal Transition in Non-Small Cell Lung Carcinoma. Onco. Targets. Ther. 2020, 13, 5257–5270. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Wong, A.S.T. Activation of p70S6K Induces Expression of Matrix Metalloproteinase 9 Associated with Hepatocyte Growth Factor-Mediated Invasion in Human Ovarian Cancer Cells. Endocrinology 2006, 147, 2557–2566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pothongsrisit, S.; Arunrungvichian, K.; Hayakawa, Y.; Sritularak, B.; Mangmool, S.; Pongrakhananon, V. Erianthridin suppresses non-small-cell lung cancer cell metastasis through inhibition of Akt/mTOR/p70S6K signaling pathway. Sci. Rep. 2021, 11, 6618. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, H.; Nieminen, A.; Saarela, M.; Kallioniemi, A.; Klefström, J.; Hautaniemi, S.; Monni, O. Deciphering downstream gene targets of PI3K/mTOR/p70S6K pathway in breast cancer. BMC Genomics 2008, 9, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Z.X.; Sun, R.F.; Mo, X.M.; Li, W.M. The p70S6K Specific Inhibitor PF-4708671 Impedes Non-Small Cell Lung Cancer Growth. PLoS ONE 2016, 11, e0147185. [Google Scholar] [CrossRef] [Green Version]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef]
- Sakamoto, K.; Holman, G.D. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol.-Endocrinol. Metab. 2008, 295, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.T.; Pessin, J.E. Bridging the GAP between insulin signaling and GLUT4 translocation. Trends Biochem. Sci. 2006, 31, 215–222. [Google Scholar] [CrossRef]
- Thong, F.S.L.; Bilan, P.J.; Klip, A. The Rab GTPase-Activating Protein AS160 Integrates Akt, Protein Kinase C, and AMP-Activated Protein Kinase Signals Regulating GLUT4 Traffic. Diabetes 2007, 56, 414–423. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Chen, Y.; Xue, P.; Fan, Y.; Deng, Y.; Peng, G.; Yang, F.; Xu, T. RUVBL2, a novel AS160-binding protein, regulates insulin-stimulated GLUT4 translocation. Cell Res. 2009, 19, 1090–1097. [Google Scholar] [CrossRef] [Green Version]
- Gongpan, P.; Lu, Y.; Wang, F.; Xu, Y.; Xiong, W. AS160 controls eukaryotic cell cycle and proliferation by regulating the CDK inhibitor p21. Cell Cycle 2016, 15, 1733–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.H.; Sun, J.W.; Xu, M.; Jiang, X.F.; Liu, C.F.; Lu, Y. Frequent hyperphosphorylation of AS160 in breast cancer. Cancer Biol. Ther. 2010, 10, 362–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Liou, G.Y. CD133 as a regulator of cancer metastasis through the cancer stem cells. Int. J. Biochem. Cell Biol. 2019, 106, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brugnoli, F.; Grassilli, S.; Al-Qassab, Y.; Capitani, S.; Bertagnolo, V. CD133 in Breast Cancer Cells: More than a Stem Cell Marker. J. Oncol. 2019, 2019, 7512632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Zhu, H.; Feng, J.; Ni, S.; Huang, J. High CD133 expression in the nucleus and cytoplasm predicts poor prognosis in non-small cell lung cancer. Dis. Markers 2015, 2015, 986095. [Google Scholar] [CrossRef]
- Wu, H.; Qi, X.W.; Yan, G.N.; Zhang, Q.B.; Xu, C.; Bian, X.W. Is CD133 Expression a Prognostic Biomarker of Non-Small-Cell Lung Cancer? A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e100168. [Google Scholar] [CrossRef] [Green Version]
- Hilbe, W.; Dirnhofer, S.; Oberwasserlechner, F.; Schmid, T.; Gunsilius, E.; Hilbe, G.; Wöll, E.; Kähler, C.M. CD133 positive endothelial progenitor cells contribute to the tumour vasculature in non-small cell lung cancer. J. Clin. Pathol. 2004, 57, 965–969. [Google Scholar] [CrossRef] [Green Version]
- Salnikov, A.V.; Gladkich, J.; Moldenhauer, G.; Volm, M.; Mattern, J.; Herr, I. CD133 is indicative for a resistance phenotype but does not represent a prognostic marker for survival of non-small cell lung cancer patients. Int. J. Cancer 2010, 126, 950–958. [Google Scholar] [CrossRef]
- Yang, Y.C.; Chiou, P.C.; Chen, P.C.; Liu, P.Y.; Huang, W.C.; Chao, C.C.; Tang, C.H. Melatonin reduces lung cancer stemness through inhibiting of PLC, ERK, p38, β-catenin, and Twist pathways. Environ. Toxicol. 2019, 34, 203–209. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, C.; Silva-Lima, B.; Videira, M. Akt/mTOR Activation in Lung Cancer Tumorigenic Regulators and Their Potential Value as Biomarkers. Onco 2022, 2, 36-55. https://doi.org/10.3390/onco2010004
Sousa C, Silva-Lima B, Videira M. Akt/mTOR Activation in Lung Cancer Tumorigenic Regulators and Their Potential Value as Biomarkers. Onco. 2022; 2(1):36-55. https://doi.org/10.3390/onco2010004
Chicago/Turabian StyleSousa, Carolina, Beatriz Silva-Lima, and Mafalda Videira. 2022. "Akt/mTOR Activation in Lung Cancer Tumorigenic Regulators and Their Potential Value as Biomarkers" Onco 2, no. 1: 36-55. https://doi.org/10.3390/onco2010004
APA StyleSousa, C., Silva-Lima, B., & Videira, M. (2022). Akt/mTOR Activation in Lung Cancer Tumorigenic Regulators and Their Potential Value as Biomarkers. Onco, 2(1), 36-55. https://doi.org/10.3390/onco2010004