
Techniques for Theoretical Prediction of Immunogenic Peptides


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Definition
:1. Background on Immunological Peptides
2. Metrics of Peptide Structure Similarity

3. Peptide Structure Analysis in Immunogenetics
3.1. Significance Levels for TM Score
3.2. Local versus Global Factors of Protein Structure
4. Recognition of Peptides by T Cells
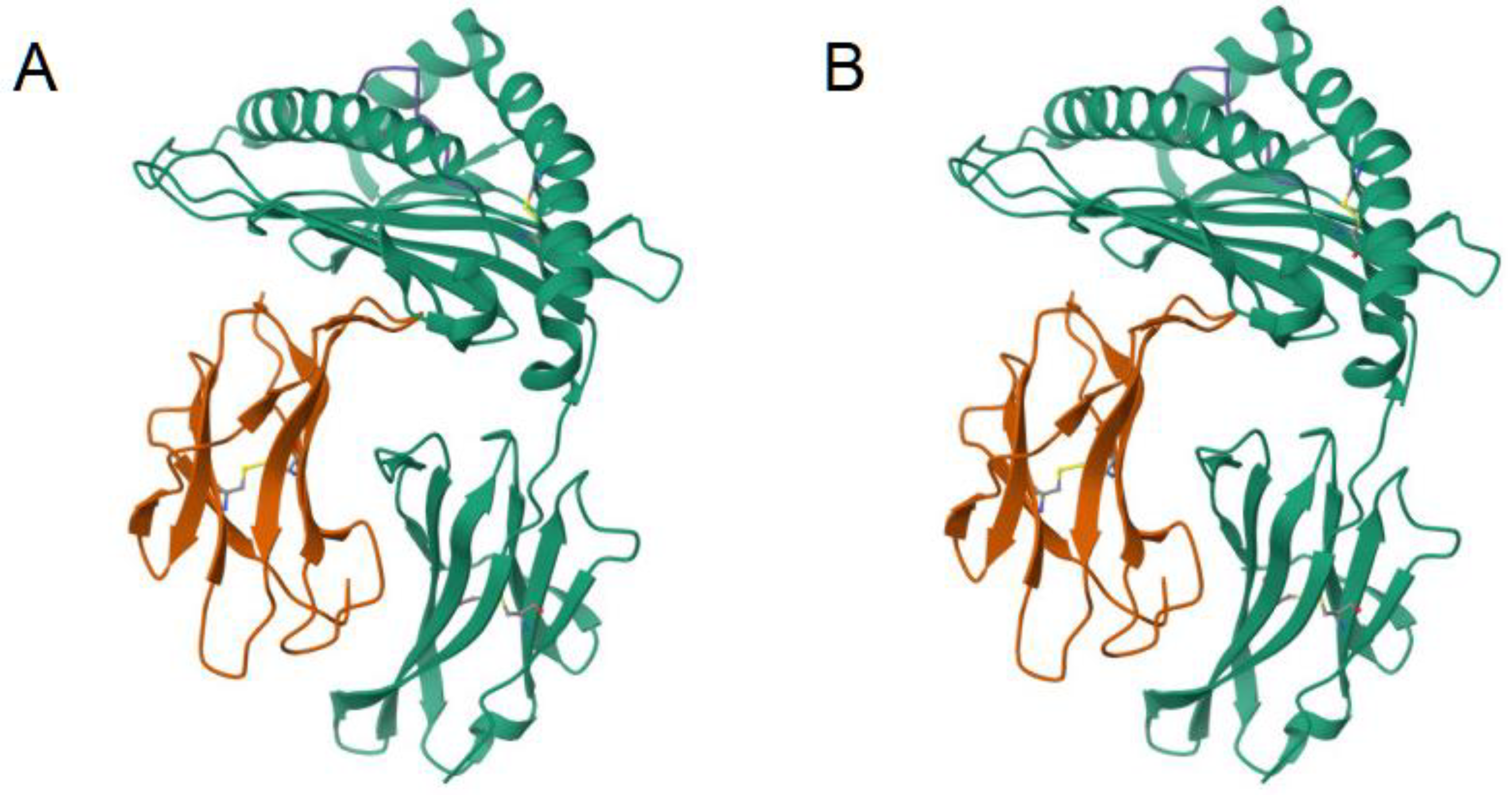
5. Model of T Cell Receptor Structure
5.1. Overview of the ImmuneBuilder Method
5.2. Usage of TCRBuilder2
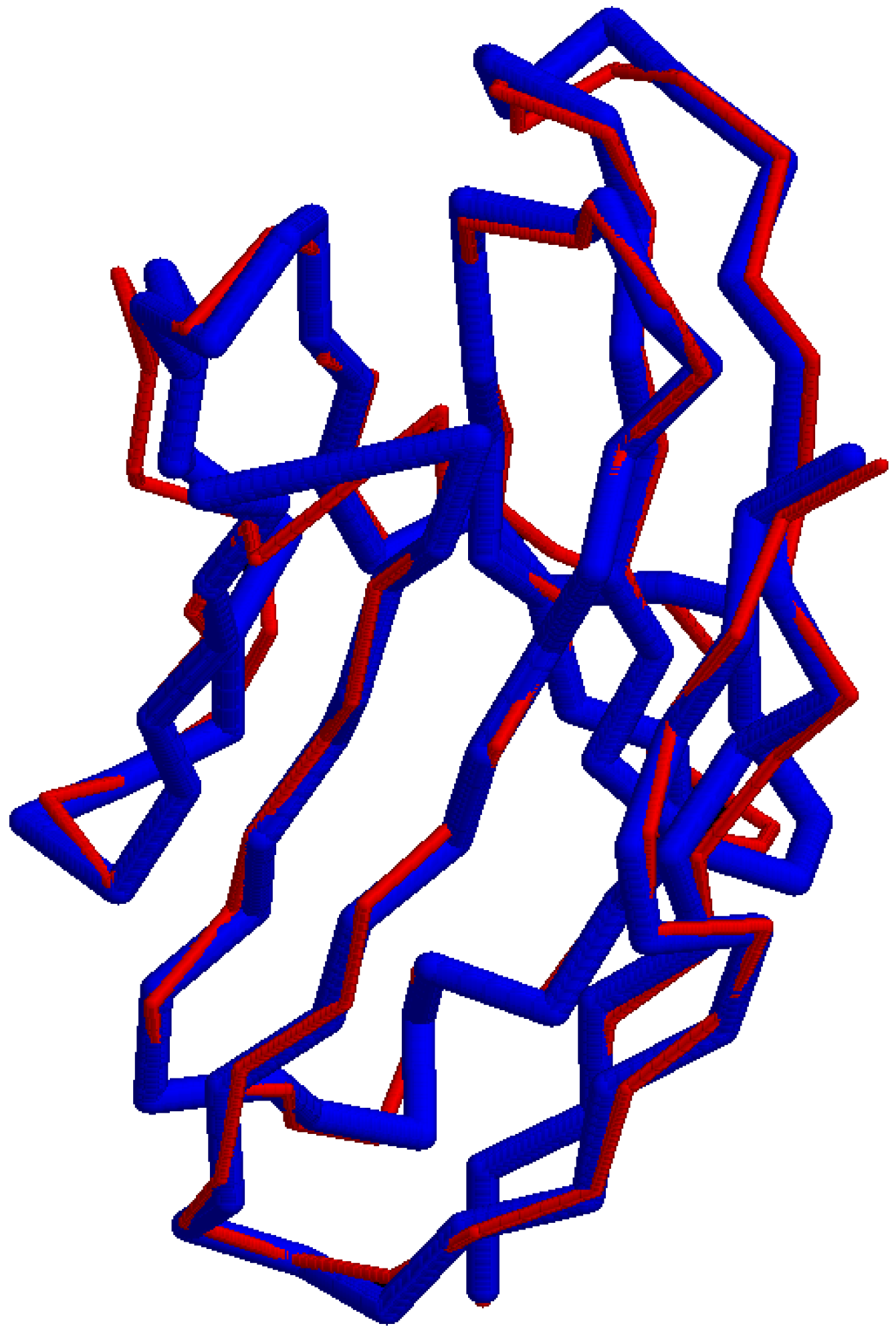
5.3. Verification of the TCRBuilder2 Model
| tmscore 5d2l_prediction_A.pdb 5d2l_ChainA-I.pdb > 5d2l_A_RMSD.out tmscore 5d2l_prediction_B.pdb 5d2l_ChainB-J.pdb > 5d2l_B_RMSD.out |
| tmscore 5d2l_prediction_A.pdb 5d2l_ChainA-I.pdb -o 5d2l_A_SUP tmscore 5d2l_prediction_B.pdb 5d2l_ChainB-J.pdb -o 5d2l_B_SUP |
5.4. Comments on TCR Modeling by Deep Learning
6. Molecular Signature of Peptide Immunogenicity
7. Deep Learning and Immunogenetics
7.1. Deep Learning Architectures
7.2. Meta-Learning Systems
7.3. Interpolation and Extrapolation in Deep Learning
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Peptide Structure Data
| curl –O https://ftp.ebi.ac.uk/pub/databases/alphafold/latest/UP000000589_10090_MOUSE_v4.tar (accessed on 10 March 2024) |
| tar –xvf UP000000589_10090_MOUSE_v4.tar *.pdb.gz |
| gzip –d *.gz |

Appendix A.2. Parsing the PDB Data Files
| https://www.cgl.ucsf.edu/chimera/docs/UsersGuide/tutorials/pdbintro.html (accessed on 10 March 2024) |
Appendix A.3. Format of Data Files for TM Score
Appendix B
| https://www.cgl.ucsf.edu/chimera/docs/UsersGuide/tutorials/pdbintro.html (accessed on 10 March 2024) |
| import glob from Bio.PDB.PDBParser import PDBParser # assign functions parser = PDBParser() # input file for file in glob.glob(‘./*.pdb’): print(“file: “, file) # retrieve PDB structure structure = parser.get_structure(file, file) # iterate over models and chains in file for model in structure: print(“model: “, model) for chain in model: print(“chain: “, chain) |
| import os directory = ‘C:/Peptide3d/data’ files = os.listdir(directory) for file in files: if file.endswith(‘pdb’): print(file) pdb_file = file with open(pdb_file, ‘r’) as f: lines = f.readlines() current_residue = None start_residue = 1 current_residue_number = start_residue − 1 for i, line in enumerate(lines): if line.startswith(‘ATOM’): residue = line[22:26] if residue != current_residue: current_residue = residue current_residue_number += 1 lines[i] = line[:22] + str(current_residue_number).rjust(4) \ + line[26:] if line.startswith(‘TER’): residue = line[22:26] if residue != current_residue: current_residue = residue lines[i] = line[:22] + \ str(current_residue_number).rjust(4) + line[26:] with open(pdb_file, ‘w’) as f: f.writelines(lines) |
Appendix C
- Step 1. Install TCRBuilder2 library dependencies:
| # Edit sequence_1, sequence_2, filename—the input data for prediction of 3d structure # The Colab runtime may report a crash from an expected restart during installation of a library # Comment out this line to enable verbose output %%capture !pip install ImmuneBuilder # use Python installer to install ImmuneBuilder (TCRBuilder2) !pip install -q condacolab # google colab-compatible access to conda import condacolab, sys # import modules to access their functions condacolab.install_mambaforge() # use of mamba to install conda modules !mamba install openmm # install openmm (toolkit for molecular simulation; refine prediction) !mamba install pdbfixer # install pdbfixer (fix problems in PDB formatted files) !conda install -y -c bioconda anarci # install anarci module from bioconda distribution |
- Step 2. Install TCRBuilder2 and run the model:
| # Delete and restart Colab runtime to avoid unexpected errors in the following code # Comment out this line to enable verbose output %%capture !pip install -q ImmuneBuilder # use Python installer to install ImmuneBuilder (TCRBuilder2) protein_type = “TCR” from anarci import number # github.com/oxpig/ANARCI; aligns sequence to canonical protein from ImmuneBuilder import TCRBuilder2 # prediction of 3d structure # Select model predictor = TCRBuilder2() # “TCRBuilder2” or “ABodyBuilder2” model # Inspect that TCR sequences are annotated as TCR alpha and beta chains # Below is sequence data from www.rcsb.org/structure/5d2l (accessed on 10 March 2024) sequence_1 = ‘MILNVEQSPQSLHVQEGDSTNFTCSFPSSNFYALHWYRWETAKSP\ EALFVMTLNGDEKKKGRISATLNTKEGYSYLYIKGSQPEDSATYLCAFITGNQFYF\ GTGTSLTVIPNIQNPDPAVYQLRDSKSSDKSVCLFTDFDSQTNVSQSKDSDVYITDK\ CVLDMRSMDFKSNSAVAWSNKSDFACANAFNNSIIPEDTFFPSPESS’ sequence_2 = ‘MGAGVSQSPSNKVTEKGKDVELRCDPISGHTALYWYRQRLGQGLE\ FLIYFQGNSAPDKSGLPSDRFSAERTGESVSTLTIQRTQQEDSAVYLCASSQTQLWET\ QYFGPGTRLLVLEDLKNVFPPEVAVFEPSEAEISHTQKATLVCLATGFYPDHVELSW\ WVNGKEVHSGVCTDPQPLKEQPALNDSRYALSSRLRVSATFWQNPRNHFRCQVQF\ YGLSENDEWTQDRAKPVTQIVSAEAWGRAD’ sequence_1 = ““.join(sequence_1.split()) # Remove whitespace sequence_2 = ““.join(sequence_2.split()) # Remove whitespace filename = ‘output.pdb’ # output file name as PDB formatted file (viewable in RasMol) # Anarci will reject the sequence if it is not an expected match to the immunoprotein _, chain1 = number(sequence_1) # set key for chain 1 to input sequence _, chain2 = number(sequence_2) # set key for chain 2 to input sequence input = dict() # initialize hash table of key-value pairs if chain1: input[chain1] = sequence_1 # add sequence value to key for hash table if chain2: input[chain2] = sequence_2 # add sequence value to key for hash table predictor.predict(input).save(filename) # run 3d prediction of TCR, save to file |
References
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Nielsen, M.; Sette, A. T Cell Epitope Predictions. Annu. Rev. Immunol. 2020, 38, 123–145. [Google Scholar] [CrossRef] [PubMed]
- Engelhard, V.H. Structure of peptides associated with MHC class I molecules. Curr. Opin. Immunol. 1994, 6, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 335, 744. [Google Scholar] [CrossRef]
- Serwold, T.; Gonzalez, F.; Kim, J.; Jacob, R.; Shastri, N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature 2002, 419, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The T Cell Receptor/Cd3 Complex: A Dynamic Protein Ensemble. Annu. Rev. Immunol. 1988, 6, 629–662. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Kono, D.H.; Baccala, R. The multiple pathways to autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef]
- Uemura, Y.; Senju, S.; Maenaka, K.; Iwai, L.K.; Fujii, S.; Tabata, H.; Tsukamoto, H.; Hirata, S.; Chen, Y.Z.; Nishimura, Y.; et al. Systematic Analysis of the Combinatorial Nature of Epitopes Recognized by TCR Leads to Identification of Mimicry Epitopes for Glutamic Acid Decarboxylase 65-Specific TCRs. J. Immunol. 2003, 170, 947–960. [Google Scholar] [CrossRef]
- Borrman, T.; Pierce, B.G.; Vreven, T.; Baker, B.M.; Weng, Z. High-throughput modeling and scoring of TCR-pMHC complexes to predict cross-reactive peptides. Bioinformatics 2020, 36, 5377–5385. [Google Scholar] [CrossRef]
- Prinz, J.C. Immunogenic self-peptides—The great unknowns in autoimmunity: Identifying T-cell epitopes driving the autoimmune response in autoimmune diseases. Front. Immunol. 2023, 13, 1097871. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, Y.; Yoshikai, Y.; Leggett, K.; Clark, S.P.; Aleksander, I.; Mak, T.W. A human T cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature 1984, 308, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, S.M.; Cohen, D.I.; Nielsen, E.A.; Davis, M.M. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature 1984, 308, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Bell, J.J.; Bhandoola, A. T-cell lineage determination. Immunol. Rev. 2010, 238, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Nikolich-Žugich, J.; Slifka, M.K.; Messaoudi, I. The many important facets of T-cell repertoire diversity. Nat. Rev. Immunol. 2004, 4, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Ashby, K.M.; Hogquist, K.A. A guide to thymic selection of T cells. Nat. Rev. Immunol. 2023, 23, 697. [Google Scholar] [CrossRef] [PubMed]
- George, J.T.; Kessler, D.A.; Levine, H. Effects of thymic selection on T cell recognition of foreign and tumor antigenic peptides. Proc. Natl. Acad. Sci USA 2017, 114, E7875–E7881. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Germolec, D.R. Introduction to Immunology and Autoimmunity. Environ. Health Perspect. 1999, 107, 661. [Google Scholar]
- Klein, J.; Figueroa, F. Evolution of the major histocompatibility complex. Crit. Rev. Immunol. 1986, 6, 295–386. [Google Scholar]
- Germain, R.N. MHC-dependent antigen processing and peptide presentation: Providing ligands for T lymphocyte activation. Cell 1994, 76, 287–299. [Google Scholar] [CrossRef]
- Nielsen, M.; Andreatta, M.; Peters, B.; Buus, S. Immunoinformatics: Predicting Peptide–MHC Binding. Annu. Rev. Biomed. Data Sci. 2020, 3, 191–215. [Google Scholar] [CrossRef] [PubMed]
- Radwan, J.; Babik, W.; Kaufman, J.; Lenz, T.L.; Winternitz, J. Advances in the Evolutionary Understanding of MHC Polymorphism. Trends Genet. 2020, 36, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Jorde, L.B. Genetic variation and human evolution. Am. Soc. Hum. Genet. 2003, 7, 28–33. [Google Scholar]
- Bjorkman, P.J.; Saper, M.A.; Samraoui, B.; Bennett, W.S.; Strominger, J.L.; Wiley, D.C. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 1987, 329, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Devaurs, D.; Moll, M.; Lizée, G.; Kavraki, L.E. General Prediction of Peptide-MHC Binding Modes Using Incremental Docking: A Proof of Concept. Sci. Rep. 2018, 8, 4327. [Google Scholar] [CrossRef]
- Mei, S.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Giam, K.; Croft, N.P.; Akutsu, T.; Smith, A.I.; Li, J.; Rossjohn, J.; et al. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction. Brief. Bioinform. 2020, 21, 1119–1135. [Google Scholar] [CrossRef] [PubMed]
- Sohail, M.S.; Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. In silico T cell epitope identification for SARS-CoV-2: Progress and perspectives. Adv. Drug Deliv. Rev. 2021, 171, 29–47. [Google Scholar] [CrossRef]
- Raoufi, E.; Hemmati, M.; Eftekhari, S.; Khaksaran, K.; Mahmodi, Z.; Farajollahi, M.M.; Mohsenzadegan, M. Epitope Prediction by Novel Immunoinformatics Approach: A State-of-the-art Review. Int. J. Pept. Res. Ther. 2019, 26, 1155–1163. [Google Scholar] [CrossRef]
- Bradley, P. Structure-based prediction of T cell receptor:peptide-MHC interactions. eLife 2023, 12, e82813. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Zemla, A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003, 31, 3370–3374. [Google Scholar] [CrossRef] [PubMed]
- Leman, J.K.; Szczerbiak, P.; Renfrew, P.D.; Gligorijevic, V.; Berenberg, D.; Vatanen, T.; Taylor, B.C.; Chandler, C.; Janssen, S.; Pataki, A.; et al. Sequence-structure-function relationships in the microbial protein universe. Nat. Commun. 2023, 14, 2351. [Google Scholar] [CrossRef] [PubMed]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2014, 43, D405–D412. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K. Natural history as stamp collecting: A brief history. Arch. Nat. Hist. 2007, 34, 244–258. [Google Scholar] [CrossRef]
- Frede, M. Plato’s Sophist on False Statements. In The Cambridge Companion to Plato; Kraut, R., Ed.; Cambridge University Press: Cambridge, UK, 1992; pp. 397–424. [Google Scholar]
- Bero, S.A.; Muda, A.K.; Choo, Y.H.; Muda, N.A.; Pratama, S.F. Similarity Measure for Molecular Structure: A Brief Review. J. Phys. Conf. Ser. 2017, 892, 012015. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Jordan, K.R.; Buhrman, J.D.; Lei, J.; Deng, H.; Marrack, P.; Dai, S.; Kappler, J.W.; Slansky, J.E.; Yin, L.; et al. Structures suggest an approach for converting weak self-peptide tumor antigens into superagonists for CD8 T cells in cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2100588118. [Google Scholar] [CrossRef]
- 6L9M. RCSB Protein Data Bank. Available online: www.rcsb.org/structure/6L9M (accessed on 22 September 2023).
- 6L9N. RCSB Protein Data Bank. Available online: www.rcsb.org/structure/6L9N (accessed on 22 September 2023).
- Python Code to Help Process Files of 3d Protein Structure (PDB Format). Available online: https://github.com/bob-friedman/pdb-file-utilities (accessed on 21 August 2023).
- 1HV4. RCSB Protein Data Bank. Available online: www.rcsb.org/structure/1HV4 (accessed on 6 September 2023).
- Lianga, Y.; Huaa, Z.; Liang, X.; Xu, Q.; Lua, G. The crystal structure of bar-headed goose hemoglobin in deoxy form: The allosteric mechanism of a hemoglobin species with high oxygen affinity. J. Mol. Biol. 2001, 313, 123–137. [Google Scholar] [CrossRef]
- Lin, H.H.; Ray, S.; Tongchusak, S.; Reinherz, E.L.; Brusic, V. Evaluation of MHC class I peptide binding prediction servers: Applications for vaccine research. BMC Immunol. 2008, 9, 8. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemøller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef]
- Chen, G.; Yang, X.; Go, A.; Gao, M.; Zhang, Y.; Shi, A.; Sun, X.; Mariuzza, R.A.; Weng, N.-P. Sequence and structural analyses reveal distinct and highly diverse human CD8+ TCR repertoires to immunodominant viral antigens. Cell Rep. 2017, 19, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Szeto, C.; Lobos, C.A.; Nguyen, A.T.; Gras, S. TCR Recognition of Peptide–MHC-I: Rule Makers and Breakers. Int. J. Mol. Sci. 2020, 22, 68. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, F.; Mösch, A.; Machart, P.; Li, K.; Alqassem, I.; O’Donnell, T.J.; Min, M.R. On TCR binding predictors failing to generalize to unseen peptides. Front. Immunol. 2022, 13, 1014256. [Google Scholar] [CrossRef]
- Paul, S.; Croft, N.P.; Purcell, A.W.; Tscharke, D.C.; Sette, A.; Nielsen, M.; Peters, B. Benchmarking predictions of MHC class I restricted T cell epitopes in a comprehensively studied model system. PLOS Comput. Biol. 2020, 16, e1007757. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Bennink, J.R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 1999, 17, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Gao, Y.; Fan, Y.; Zhu, C.; Wei, Z.; Zhou, C.; Chuai, G.; Chen, Q.; Zhang, H.; Liu, Q.; et al. Pan-Peptide Meta Learning for T-cell receptor–antigen binding recognition. Nat. Mach. Intell. 2023, 5, 236–249. [Google Scholar] [CrossRef]
- PanPep: Pan-Peptide Meta Learning for T-Cell Receptor-Antigen Binding Recognition. Available online: https://github.com/bm2-lab/PanPep (accessed on 18 September 2023).
- Nahm, F.S. Receiver operating characteristic curve: Overview and practical use for clinicians. Korean J. Anesthesiol. 2022, 75, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Parra, Z.E.; Baker, M.L.; Schwarz, R.S.; Deakin, J.E.; Lindblad-Toh, K.; Miller, R.D. A unique T cell receptor discovered in marsupials. Proc. Natl. Acad. Sci. USA 2007, 104, 9776–9781. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Alt, F.W.; Hughes, M.M.; D’Auteuil, M.; Wehrly, T.D.; Woodman, B.B.; Gärtner, F.; White, J.M.; Davidson, L.; Sleckman, B.P. Recombination signal sequences restrict chromosomal V (D) J recombination beyond the 12/23 rule. Nature 2000, 405, 583–586. [Google Scholar] [CrossRef]
- Max, E.E.; Seidman, J.G.; Leder, P. Sequences of five potential recombination sites encoded close to an immunoglobulin kappa constant region gene. Proc. Natl. Acad. Sci. USA 1979, 76, 3450–3454. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.R.; Sheriff, S.; Padlan, E.A. Antibody-Antigen Complexes. Annu. Rev. Biochem. 1990, 59, 439–473. [Google Scholar] [CrossRef] [PubMed]
- Abanades, B.; Wong, W.K.; Boyles, F.; Georges, G.; Bujotzek, A.; Deane, C.M. ImmuneBuilder: Deep-Learning models for predicting the structures of immune proteins. Commun. Biol. 2023, 6, 575. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv 2021. bioRxiv:2021.10.04.463034. [Google Scholar]
- Leem, J.; de Oliveira, S.H.P.; Krawczyk, K.; Deane, C.M. STCRDab: The structural T-cell receptor database. Nucleic Acids Res. 2017, 46, D406–D412. [Google Scholar] [CrossRef]
- Carugo, O.; Pongor, S. A normalized root-mean-square distance for comparing protein three-dimensional structures. Protein Sci. 2008, 10, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, J.; Fuchs, A.; Shi, J.; Deane, C.M. ABangle: Characterising the VH-VL orientation in antibodies. Protein Eng. Des. Sel. 2013, 26, 611–620. [Google Scholar] [CrossRef]
- Leem, J.; Georges, G.; Shi, J.; Deane, C.M. Antibody side chain conformations are position-dependent. Proteins Struct. Funct. Bioinform. 2018, 86, 383–392. [Google Scholar] [CrossRef]
- ImmuneBuilder. GitHub. Available online: https://github.com/oxpig/ImmuneBuilder (accessed on 2 November 2023).
- Bisong, E. Google Colaboratory. In Building Machine Learning and Deep Learning Models on Google Cloud Platform; APress: Berkeley, CA, USA, 2019. [Google Scholar]
- Sayle, R.A.; Milner-White, E.J. RASMOL: Biomolecular graphics for all. Trends Biochem. Sci. 1995, 20, 374–376. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef]
- Yang, X.; Gao, M.; Chen, G.; Pierce, B.G.; Lu, J.; Weng, N.-P.; Mariuzza, R.A. Structural Basis for Clonal Diversity of the Public T Cell Response to a Dominant Human Cytomegalovirus Epitope. J. Biol. Chem. 2015, 290, 29106–29119. [Google Scholar] [CrossRef] [PubMed]
- ClustalW. Available online: www.genome.jp/tools-bin/clustalw (accessed on 2 November 2023).
- Ma, Y.J.; Liang, W.; Wang, G.; Huang, D.-A.; Bastani, O.; Jayaraman, D.; Zhu, Y.; Fan, L.; Anandkumar, A. Eureka: Human-Level Reward Design via Coding Large Language Models. arXiv 2023, arXiv:2310.12931. [Google Scholar]
- Bickle, J. The first two decades of CREB-memory research: Data for philosophy of neuroscience. AIMS Neurosci. 2021, 8, 322. [Google Scholar] [CrossRef]
- Li, W.-H. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 1993, 36, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Scharloo, W. Canalization: Genetic and Developmental Aspects. Annu. Rev. Ecol. Syst. 1991, 22, 65–93. [Google Scholar] [CrossRef]
- Waddington, C.H. Canalization of Development and the Inheritance of Acquired Characters. Nature 1942, 150, 563–565. [Google Scholar] [CrossRef]
- Meyer, A.; Zardoya, R. Recent Advances in the (Molecular) Phylogeny of Vertebrates. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 311–338. [Google Scholar] [CrossRef]
- Bengio, Y.; Lecun, Y.; Hinton, G. Deep learning for AI. Commun. ACM 2021, 64, 58–65. [Google Scholar] [CrossRef]
- Park, M.; Seo, S.-W.; Park, E.; Kim, J. EpiBERTope: A sequence-based pre-trained BERT model improves linear and structural epitope prediction by learning long-distance protein interactions effectively. bioRxiv 2022. bioRxiv:2022.02.27.481241. [Google Scholar]
- Friedman, R. Tokenization in the Theory of Knowledge. Encyclopedia 2023, 3, 380–386. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- François-Lavet, V.; Peter, P.; Islam, R.; Bellemare, M.G.; Pineau, J. An Introduction to Deep Reinforcement Learning. Found. Trends Mach. Learn. 2018, 11, 219–354. [Google Scholar] [CrossRef]
- Fawzi, A.; Balog, M.; Huang, A.; Hubert, T.; Romera-Paredes, B.; Barekatain, M.; Novikov, A.; Ruiz, F.J.R.; Schrittwieser, J.; Swirszcz, G.; et al. Discovering faster matrix multiplication algorithms with reinforcement learning. Nature 2022, 610, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R. A Hierarchy of Interactions between Pathogenic Virus and Vertebrate Host. Symmetry 2022, 14, 2274. [Google Scholar] [CrossRef]
- Wei, J.; Wang, X.; Schuurmans, D.; Bosma, M.; Chi, E.; Le, Q.; Zhou, D. Chain-of-thought prompting elicits reasoning in large language models. Adv. Neural Inf. Process. Syst. 2022, 35, 24824–24837. [Google Scholar]
- Zhuge, M.; Liu, H.; Faccio, F.; Ashley, D.R.; Csordás, R.; Gopalakrishnan, A.; Hamdi, A.; Hammoud, H.A.A.K.; Herrmann, V.; Irie, K.; et al. Mindstorms in Natural Language-Based Societies of Mind. arXiv 2023, arXiv:2305.17066. [Google Scholar]
- Zhou, W.; Jiang, Y.E.; Li, L.; Wu, J.; Wang, T.; Qiu, S.; Zhang, J.; Chen, J.; Wu, R.; Wang, S.; et al. Sachan Agents: An Open-source Framework for Autonomous Language Agents. arXiv 2023, arXiv:2309.07870. [Google Scholar]
- Masoudnia, S.; Ebrahimpour, R. Mixture of experts: A literature survey. Artif. Intell. Rev. 2012, 42, 275–293. [Google Scholar] [CrossRef]
- Open AI. GPT-4 Technical Report. arXiv 2023, arXiv:2303.08774. [Google Scholar]
- Anil, R.; Dai, A.M.; Firat, O.; Johnson, M.; Lepikhin, D.; Passos, A.; Shakeri, S.; Taropa, E.; Bailey, P.; Chen, Z.; et al. PaLM 2 Technical Report. arXiv 2023, arXiv:2305.10403. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- Asanovic, K.; Bodik, R.; Demmel, J.; Keaveny, T.; Keutzer, K.; Kubiatowicz, J.; Morgan, N.; Patterson, D.; Sen, K.; Wawrzynek, J.; et al. A view of the parallel computing landscape. Commun. ACM 2009, 52, 56–67. [Google Scholar] [CrossRef]
- Touvron, H.; Martin, L.; Stone, K.; Albert, P.; Almahairi, A.; Babaei, Y.; Bashlykov, N.; Batra, S.; Bhargava, P.; Bhosale, S.; et al. Llama 2: Open Foundation and Fine-Tuned Chat Models. arXiv 2023, arXiv:2307.09288. [Google Scholar]
- Taylor, R.; Kardas, M.; Cucurull, G.; Scialom, T.; Hartshorn, A.; Saravia, E.; Poulton, A.; Kerkez, V.; Stojnic, R. Galactica: A Large Language Model for Science. arXiv 2022, arXiv:2211.09085. [Google Scholar]
- Wang, X.; Wei, J.; Schuurmans, D.; Le, Q.; Chi, E.; Zhou, D. Self-Consistency Improves Chain of Thought Reasoning in Language Models. arXiv 2022, arXiv:2203.11171. [Google Scholar]
- Yao, S.; Yu, D.; Zhao, J.; Shafran, I.; Griffiths, T.L.; Cao, Y.; Narasimhan, K. Tree of Thoughts: Deliberate Problem Solving with Large Language Models. arXiv 2023, arXiv:2305.10601. [Google Scholar]
- Li, C.; Liang, J.; Zeng, A.; Chen, X.; Hausman, K.; Sadigh, D.; Levine, S.; Fei-Fei, L.; Xia, F.; Ichter, B. Chain of Code: Reasoning with a Language Model-Augmented Code Emulator. arXiv 2023, arXiv:2312.04474. [Google Scholar]
- Friedman, R. Higher Cognition: A Mechanical Perspective. Encyclopedia 2022, 2, 1503–1516. [Google Scholar] [CrossRef]
- Merchant, A.; Batzner, S.; Schoenholz, S.S.; Aykol, M.; Cheon, G.; Cubuk, E.D. Scaling deep learning for materials discovery. Nature 2023, 624, 80–85. [Google Scholar] [CrossRef]
- Schrittwieser, J.; Antonoglou, I.; Hubert, T.; Simonyan, K.; Sifre, L.; Schmitt, S.; Guez, A.; Lockhart, E.; Hassabis, D.; Graepel, T.; et al. Mastering Atari, Go, chess and shogi by planning with a learned model. Nature 2020, 588, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Zebari, R.; Abdulazeez, A.; Zeebaree, D.; Zebari, D.; Saeed, J. A Comprehensive Review of Dimensionality Reduction Techniques for Feature Selection and Feature Extraction. J. Appl. Sci. Technol. Trends 2020, 1, 56–70. [Google Scholar] [CrossRef]
- Pang, R.; Lansdell, B.J.; Fairhall, A.L. Dimensionality reduction in neuroscience. Curr. Biol. 2016, 26, R656–R660. [Google Scholar] [CrossRef] [PubMed]
- Fusi, S.; Miller, E.K.; Rigotti, M. Why neurons mix: High dimensionality for higher cognition. Curr. Opin. Neurobiol. 2016, 37, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Pujara, J.; Ren, X.; Chen, X.; Cheng, H.T.; Le, Q.V.; Chi, E.H.; Zhou, D.; Mishra, S.; Zheng, H.S. Self-discover: Large language models self-compose reasoning structures. arXiv 2024, arXiv:2402.03620. [Google Scholar]
- Friedman, R. Large Language Models and Logical Reasoning. Encyclopedia 2023, 3, 687–697. [Google Scholar] [CrossRef]
- Mitra, A.; Corro, L.D.; Mahajan, S.; Codas, A.; Simoes, C.; Agarwal, S.; Chen, X.; Razdaibiedina, A.; Jones, E.; Aggarwal, K.; et al. Orca 2: Teaching Small Language Models How to Reason. arXiv 2023, arXiv:2311.11045. [Google Scholar]
- Wang, X.; Zhou, D. Chain-of-Thought Reasoning without Prompting. arXiv 2024, arXiv:2402.10200. [Google Scholar]
- Balestriero, R.; Pesenti, J.; LeCun, Y. Learning in High Dimension Always Amounts to Extrapolation. arXiv 2021, arXiv:2110.09485. [Google Scholar]
- Zou, X.; Dou, Z.-Y.; Yang, J.; Gan, Z.; Li, L.; Li, C.; Dai, X.; Behl, H.; Wang, J.; Yuan, L.; et al. Generalized Decoding for Pixel, Image, and Language; Institute of Electrical and Electronics Engineers (IEEE): Piscataway, NJ, USA, 2023; pp. 15116–15127. [Google Scholar]
- Nakajima, K. The mechanism of antigenic shift and drift of human influenza virus. Nihon Rinsho Jpn. J. Clin. Med. 2003, 61, 1897–1903. [Google Scholar]
- Xiao, C.; Ren, Z.; Zhang, B.; Mao, L.; Zhu, G.; Gao, L.; Su, J.; Ye, J.; Long, Z.; Zhu, Y.; et al. Insufficient epitope-specific T cell clones are responsible for impaired cellular immunity to inactivated SARS-CoV-2 vaccine in older adults. Nat. Aging 2023, 3, 418–435. [Google Scholar] [CrossRef]
- Qi, Q.; Liu, Y.; Cheng, Y.; Glanville, J.; Zhang, D.; Lee, J.-Y.; Olshen, R.A.; Weyand, C.M.; Boyd, S.D.; Goronzy, J.J. Diversity and clonal selection in the human T-cell repertoire. Proc. Natl. Acad. Sci. USA 2014, 111, 13139–13144. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedman, R. Techniques for Theoretical Prediction of Immunogenic Peptides. Encyclopedia 2024, 4, 600-621. https://doi.org/10.3390/encyclopedia4010038
Friedman R. Techniques for Theoretical Prediction of Immunogenic Peptides. Encyclopedia. 2024; 4(1):600-621. https://doi.org/10.3390/encyclopedia4010038
Chicago/Turabian StyleFriedman, Robert. 2024. "Techniques for Theoretical Prediction of Immunogenic Peptides" Encyclopedia 4, no. 1: 600-621. https://doi.org/10.3390/encyclopedia4010038
APA StyleFriedman, R. (2024). Techniques for Theoretical Prediction of Immunogenic Peptides. Encyclopedia, 4(1), 600-621. https://doi.org/10.3390/encyclopedia4010038