1. Introduction
Biogas production through anaerobic digestion (AD) and energy recovery from methane (CH
4) are some of the ways that have been studied to generate renewable energy and reduce greenhouse gas (GHG) emissions [
1]. The literature shows that the use of different wastes can be used as substrates for AD, such as restaurant food waste, agro-industrial waste, animal manure, lignocellulosic waste, and domestic sewage [
2,
3,
4]. Among several agro-industry residues, the use of by-products from ethanol production, such as vinasse, stands out in biogas production. The vinasse is a residue with a high amount of organic matter and provides a way for integrating ethanol plants in biorefinery concepts, as it converts waste into energy [
5,
6].
First-generation (1G) ethanol production distilleries generate residues, in addition to vinasse, such as filter cake and sugarcane straw, which also have the huge potential to produce methane (CH
4) within anaerobic digestion [
7]. This fact makes sugarcane plants become biorefineries, since, in addition to producing ethanol as a biofuel, they can produce other types of bioenergy through the use of their residues. An additional strategy to produce biogas is from the residues of second-generation ethanol (2G) production from lignocellulosic residues, such as sugarcane straw [
1]. To produce 2G ethanol, several pretreatments are applied to lignocellulosic materials, and Brenelli et al. [
8] demonstrated the efficiency of acetic group removal through the alkaline pretreatment of sugarcane straw to produce 2G ethanol. From this process, a residue rich in acetate emerged, called deacetylation liquor, that had a high biochemical methane potential (BMP) [
7]. In the work of Volpi et al. [
7], in-depth investigation comparing the mono-digestion of vinasse 1G, mono-digestion of filter cake, and the addition of 2G ethanol residue were performed, and the results clearly showed that the deacetylation liquor increased the production of CH
4 when placed in co-digestion with the filter cake and vinasse.
To promote the integration of 1G2G ethanol biorefineries, we previously demonstrated that the anaerobic co-digestion of vinasse and filter cake, both from the production of 1G ethanol and the deacetylation liquor (2G ethanol) in a continuous reactor, allows for the recovery of 234 NmLCH
4 gVS
−1 [
9]. In addition, by converting the biogas produced into electricity, an installed capacity of 58 MW can be obtained for a 1G2G ethanol plant, and if this biogas is purified using biomethane, the entire truck fleet of the largest ethanol plant in Brazil can be supplied during the harvest period [
10]. The co-digestion was important for the balance of nutrients in the process, mainly as the C:N ratio of the residues used and phosphorus (P) contents. For anaerobic digestion, the recommended C:N ratio can range from 10 to 40 [
11]. The vinasse used in the tests had a C:N ratio, ranging from 28:1–40:1, fitting within the requirements for AD; however, the deacetylation liquor had a C:N ratio of 90:1–137:1, quite exceeding the condition considered ideal for AD [
9]. On the other hand, the filter cake had a C:N ratio close to 18:1 [
7], well below the recommended, reinforcing the importance of co-digestion and evidencing that the use of these three residues was beneficial to the process of obtaining CH
4. The P content for vinasse and the deacetylation liquor was 9 mg L
−1 and 2 mg L
−1, respectively, both being at low concentrations, and thus, not interfering with the accumulation of this nutrient and consequent nutritional imbalance in the process. In order to optimize biogas production, our research group carried out another operation in a continuous reactor for the co-digestion of the same residues (vinasse 1G, filter cake, and deacetylation liquor 2G), adding Fe
3O
4 nanoparticles (NP). The literature has shown the good performance of reactors, introducing micronutrients using NP [
12,
13,
14]. The choice of Fe
3O
4 NP was due to the stimulating properties that iron ions (Fe) have on methanogenic archaea. Fe is a micronutrient that stimulates the formation of cytochromes and ferredoxins, important for cellular energy metabolism, especially of archaea. The hydrolysis and acidification phase of AD is also benefited by Fe as a growth factor, since Fe supplementation can accelerate these steps [
12,
13]. The results obtained from this reactor operation showed a 91% increase in CH
4 production, compared to the previous operation, and also allowed for an increase in the organic load rate (OLR), which consequently allowed for an increase in the volume of waste to be treated, improving AD in general [
15].
Despite all the operational discussions of the co-digestion of residues from ethanol production for CH
4 recuperation, biochemical aspects of the process need to be better understood. The AD process is quite complex from a microbiological point of view. The bacterial consortium responsible for the metabolism of waste to the final product comprises several species of bacteria and archaea, and they may prefer different metabolic routes, depending on the experimental conditions and the substrates used, as there are numerous metabolic routes within the process [
16]. We have demonstrated that, in the microbial consortium of co-digestion of vinasse, filter cake, and deacetylation liquor, in both reactor operations (with and without NP), the predominant archaea group was
Methanoculleus. This methanogenic group is characterized by the metabolic route syntrophic acetate oxidation (SAO), coupled with hydrogenotrophic methanogenesis [
9], which indicates this metabolic route was possibly the main one to produce CH
4 from these residues. However, just by identifying the existing microorganisms, it is difficult to distinguish which possible route is being preferred in the process according to the experimental conditions. Little is known about the functional activities of the various abundant groups of anaerobic sludges from AD bioreactors [
16]. Therefore, it is important to gain insights into the biochemistry of the bioprocess to ensure a better performance and optimization of sugarcane residue co-digestion.
The literature is quite scarce regarding the proteomics analysis of anaerobic reactors that use sugarcane residues, mainly 2G ethanol residues. Even less information is found on the proteomics analysis of the residue’s anaerobic co-digestion from the sugar-energy industry. According to studies, the metaproteomics approach was successfully used to examine the expression of essential microbial functions in a variety of environments, such as enhanced biological phosphorus removal reactors, activated sludge, and local mine acid drainage, as well as beneath local soil and seascapes [
17].
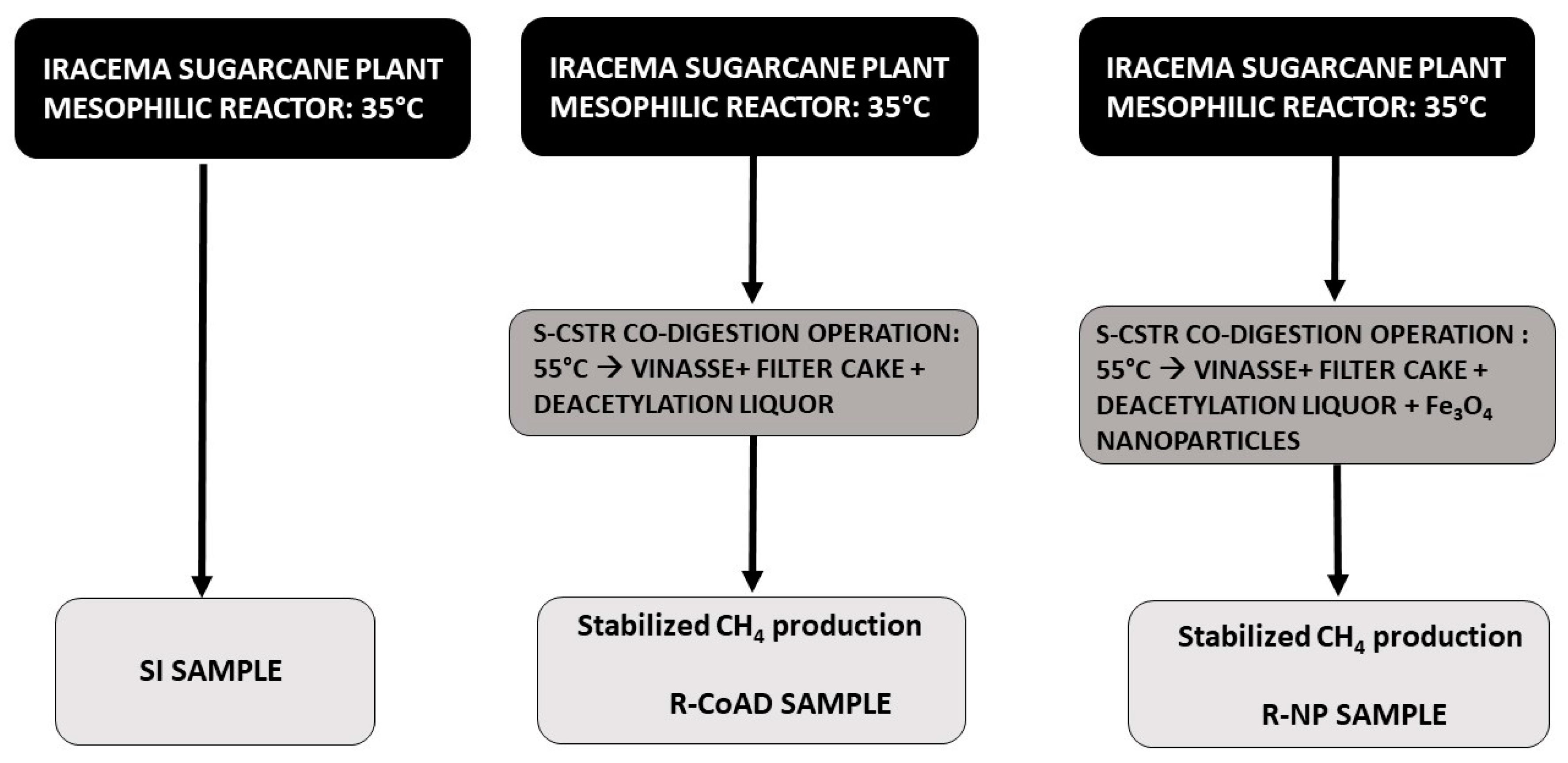
Therefore, the objective of the present work was to carry out a metaproteomic analysis of samples from anaerobic reactors that contained residues from 1G ethanol production and 2G ethanol production in co-digestion for biogas production. Metaproteomic analyses were performed for three types of samples: the first type is the anaerobic sludge alone, before adding substrate, that is, the seed inoculum; the second type is from a reactor with the co-digestion of filter cake, vinasse, and deacetylation liquor; and the last sample type is from another reactor with the same previous co-digestion condition adding Fe3O4 NP. The goal was to analyze whether there would be changes in the abundance of the proteins between the different sample types and if there could be a change or preference in the metabolic routes detected in these complex microorganism communities.
3. Results
In general, a total of 439 proteins were found, which included 319 proteins for the SI sample, 293 proteins for the R-CoAD sample, and 299 proteins for the R-NP sample. From this data, contaminating proteins and proteins from other organisms were excluded, but considering proteins of the same suggested function attributed to different microbial species.
Tables S1–S3 in the SM show the identified proteins that were differentially abundant among the samples. These tables also contain the NCBI-Uniprot identifier for each protein.
Figure 2 shows the relative abundance of the proteins found in each of the samples.
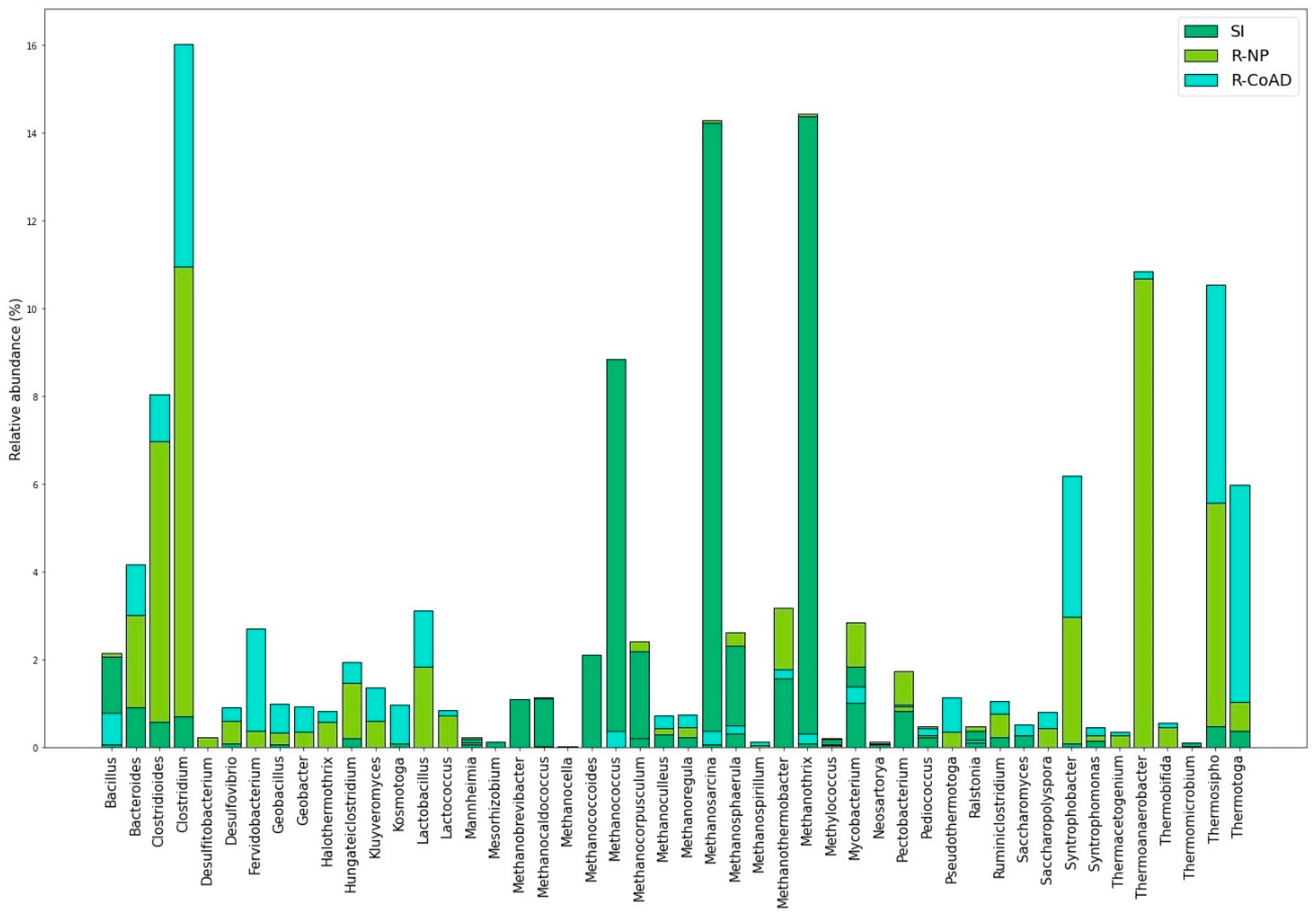
Figure 3 shows the relative abundance of proteins of the main microorganism genus. The relationship between the protein abundance and the possible microorganism that synthesized it resulted from the LC-MS analysis, as the database in which the cross was performed (NCBI non-redundant database) also had these data on the microbial community. The three samples have already been carried out and were published in studies on reactor operation [
9,
15].
Table S4 brings the results of all identified microorganism genus, with the respective relative abundance of identified proteins. The R-NP and R-CoAD samples also had proteins that were produced by methanogenic archaea and also by groups of bacteria that are important for AD, such as
Thermoanaerobacter,
Thermobifida,
Thermomicrobium,
Thermosipho,
Thermotoga,
Syntrophomonas,
Ruminiclostridium, and
Pseudomonas. Figure 4A shows the heatmap of the results obtained from clustering the protein groups of the three samples with the three repetitions performed. This graph shows that the samples were clustered into two groups, one in purple and the other two in red. As it is not possible to read the IDs of the proteins in
Figure 4A and
Figure S2 in the SM, a larger figure with the ID of each of the proteins can be found.
Figure 5 shows the metabolic route that was proposed for the three samples together, according to the identification of proteins. To propose this route, KEGG’s specific metabolic routes (
http://www.kegg.jp/kegg/1) accessed on 25 July 2021 were followed.
4. Discussion
4.1. Identified Proteins
The samples R-NP and R-CoAD were the ones with the highest numbers of differentially abundant proteins, as these samples were collected from different reactors. However, this does not mean that they have followed different metabolic routes for CH
4 production, but that the addition of Fe
3O
4 NP in the reactor may have influenced a higher or lower abundance of a certain protein, as NP acts as an archaea growth stimulant and acts as a protein cofactor [
24,
25] improving the AD process.
It can be observed that sample SI has a greater diversification of proteins than the other samples, as was identified in
Figure 2. This could be due to the fact that this sample came from an external reactor—Iracemapólis plant (as described in the methodology)—and not from the reactors that were operated for the present study, presenting greater operational variability and being able to generate greater diversity of proteins, compared to samples R-CoAD and R-NP. However, it is worth pointing out that this inoculum (SI sample) was introduced in the start operation reactors, where the R-CoAD and R-NP samples came from. This could lead to differences in the abundance of the proteins, once there were changes in the initial metabolic routes, until the given moment of sampling. The same observation was seem by Volpi et al. [
9] on the identification of the microbial community in the samples of SI and R-CoAD. The sample from the reactor operation showed a greater diversification of microorganisms, in relation to the seed inoculum sample, indicating that the substrates and conditions of the reactor directed to the co-digestion process selected new bacteria from the microbial community in a way that some members of the community increased their relative abundance, while others decreased.
The main proteins found in sample R-CoAD were classified in the following functional classes: (~10%) 60 kDa chaperonin, (~24%) enolase, (~0.72%) xylose isomerase, (~4%) argininosuccinate lyase, (~38%) ornithine carbamoyltransferase. For sample, R-NP, the main proteins identified, belonged to the functional classes: (~22%) chaperone protein DnaK, (~30%) enolase, (~13%) xylose Isomerase, and (~3.4%) pyruvate phosphate dikinase.
In both samples, the presence of the enolase enzyme was highly represented, and carbohydrate appeared to be the main initial carbon source. This enzyme was associated with glucose metabolism, and as the reactors were treating substrates with a high cellulose content, this may explain their high relative abundance. When degraded, cellulose can follow both the glycolysis metabolic pathway and the pentose phosphate pathway [
16]. The pyruvate phosphate dikinase enzyme could work at the end of the glycolytic pathway, metabolizing phosphoenolpyruvate into pyruvate [
16]. In addition to enolase, another enzyme associated with cellulose metabolism is xylose isomerase, indicating that the metabolic route of xylulose can also be involved in the cellulose catabolism that takes place under AD. The enzyme xylose isomerase appeared in greater abundance in the R-NP sample than in the R-CoAD sample (13% vs. 0.7%). The presence of Fe
3O
4 NPs may have stimulated acidogenic and fermentative bacteria [
26], causing them to express greater amounts of enzymes responsible for breaking down carbohydrates, such as xylose isomerase.
The presence of proteins such as chaperones and chaperonins are usually related to stress responses, due to environmental conditions and survival challenges in extreme or changing conditions, and are not directly related to the metabolic pathways involved in the AD of polysaccharides and biogas production. However, these proteins may be common in anaerobic reactor sample analyses, as reported by Lam et al. [
27], and may be important to ensure the proper cellular response and protein folding under AD [
28]. The samples from reactors present different types of chaperones, the main ones being 60 kDa chaperonine and DnaK chaperones. As they have practically the same function, it is possible to indicate that the microorganisms were experiencing some level of stress within the bioreactors at the time of sampling, as it is a complex process, with different microorganisms and different metabolites being generated and consumed. However, the literature shows that GroEL chaperonins can be overexpressed in the presence of acetic acid [
28], and an abundance of this protein could be interpreted as a response to environmental stress exposure [
29]. The presence of different fermentative products in the R-CoAD sample, such as VFA (acetic acid, latic acid, propionic acid), may have led to a greater presence of this type of chaperonin, concerning the R-NP sample (10% vs. 2%). This expression of different types of chaperones is likely to be related to the different VFA found in the samples.
Among the main proteins related to the metabolic process of CH
4 production within anaerobic digestion, methyl coenzyme-M reductase is a key enzyme at the end of CH
4 production [
16]. This enzyme was found in the three samples, (~0.5% in sample SI, ~1.6% in sample R-NP, and 0.2% in sample R-CoAD), indicating that the CH
4 production route was probably active in the sludge.
The acetate kinase enzyme was also found (~1.5%) in sample R-NP and (~0.5%) in sample R-CoAD. This enzyme is responsible for converting acetyl phosphate into acetate (and vice versa) within the metabolic pathway to produce CH
4. The acetate is the main precursor of CH
4 production [
30] and the presence of this protein, which is widespread in bacteria fermentation consortiums, suggesting that complex organic matter is degraded to acetate, as well in these AD conditions, to produce CH
4.
Acetyl-coenzyme A synthetase, acyl-CoA dehydrogenase, as related to the acetoclastic pathway in CH4 production, were also identified in the three samples. The SI sample showed the highest number of these proteins (e.g., ~7% acetyl-CoA decarbonylase/synthase) in the presence of archaea Methanosarcina and Methanothrix, possibly indicating that the acetoclastic route was predominant in the inoculum before being inserted into the reactor. The fact that these enzymes were not found in greater amounts in the R-CoAD and R-NP samples did not indicate that the acetoclastic route was not present inside the reactor, but perhaps that, as the CH4 production was already stable, only proteins related to the final steps of methanogenesis, as was the case with methyl-CoM reductase, were detected.
4.2. Relationship of Proteins and the Microbial Community
Our proteomics results revealed a high number of proteins identified and annotated to the microorganisms of the
Thermotoga genus, which is characteristic of thermophilic processes [
9]. The abundance of these proteins was higher in the samples from the two reactors, which operated at 55 °C, than in the SI sample that comes from a mesophilic reactor.
These same samples showed proteins that are abundant in the acetogenic and hydrolysis phases of the organisms from genus
Clostridium and
Thermoanaerobacter, which are part of the first stages of AD and are important for driving the entire metabolic process [
31]. Proteins from the bacteria of the genus Bacteroidetes were also detected in the samples, which are characteristic of the hydrolysis and acidogenesis phases of anaerobic digestion, mainly in reactors with the presence of straw and Fe
3O
4 NP, as was the case of the R-NP sample [
32]. Microorganisms of the genus
Lactobacillus for example, whose proteins were identified in samples R-NP and R-CoAD, are responsible for converting pyruvate into lactic acid, which can then be converted to acetate [
27]. Species of the genus
Clostridium are involved in the degradation of pyruvate to butyrate [
27], and this butyrate was possibly converted to acetate. All these microorganisms from the early stages of AD are important for preparing substrates that will be reduced to CH
4, such as acetate and CO, which will be used by the methanogenic archaea species.
Another factor that draws attention is that most of the proteins synthesized by methanogenic archaea (
Figure 3) had a greater abundance in the SI sample, indicating that this sample, even being from the inoculum at the beginning of the studied process, already had activity in the methanogenic pathway. This inoculum, coming from a reactor for treating vinasse and producing biogas and having been in operation for some years, suggests that its methanogenesis was better stabilized than the reactors that operated in this study. However, the difference in the substrate and operating conditions confirm the possibility of changes in metabolic pathways in SI sample to R-CoAD and R-NP sample.
It is noteworthy that all samples have proteins from methanogenic archaea annotated in the acetoclastic pathway, such as
Methanosarcin, as well as from the hydrogenotrophic pathway, such as
Methanoculleus. We previously identified the main archaea genus found in the samples of AD as
Methanoculleus in the co-digestion of vinasse, filter cake, and deacetylation liquor, indicating that the probable metabolic route with these substrates would be a syntrophic acetate oxidation (SAO) process coupled with hydrogenotrophic methanogenesis [
9]. The fact that the two reactors under AD have the presence of enzymes from both the acetoclastic metanogenesis route (e.g., ~0.5% and ~1% of acetate kinase, for R-CoAD and R-NP, respectively) and the hydrogenotrophic methanogenesis route (e.g., ~0.16% and ~0.94% of acetyl CoA descarbonylase/synthase for R-CoAD and R-NP, respectively) confirms the possibility that SAO was coupled to the hydrogenotrophic route and may be the most likely within the waste co-digestion of the ethanol production industry.
4.3. Protein Functional Analysis
In the first cluster on
Figure 4, it can be seen, in general, that the R-CoAD samples and the R-NP sample have similar patterns of protein abundance, while in the second cluster, most proteins that were present in the SI sample were not in the other two samples. To assess the biological function of the set of proteins of the two clusters identified, we performed an analysis using Blastkoala, which is shown in
Figure 4B.
Cluster 1, which has the proteins from the R-CoAD and R-NP samples with high abundance (
Figure 4B), had an intense carbohydrate metabolism activity, in addition to amino acid metabolism. Cluster 2 had the highest abundance of proteins in the SI sample, and the greatest functions of the detected proteins were the metabolism of other amino acids, the energy metabolism, and the cellular process. In general, these protein functions, even though none are directly related to the CH
4 route, were already shown to have different metabolic pathways, and the R-CoAD and R-NP samples were classified under the same functions. It is worth remembering that these samples come from the anaerobic co-digestion operation of the same residues, with stabilized CH
4 production.
According to the functions represented in the BlastKOALA (
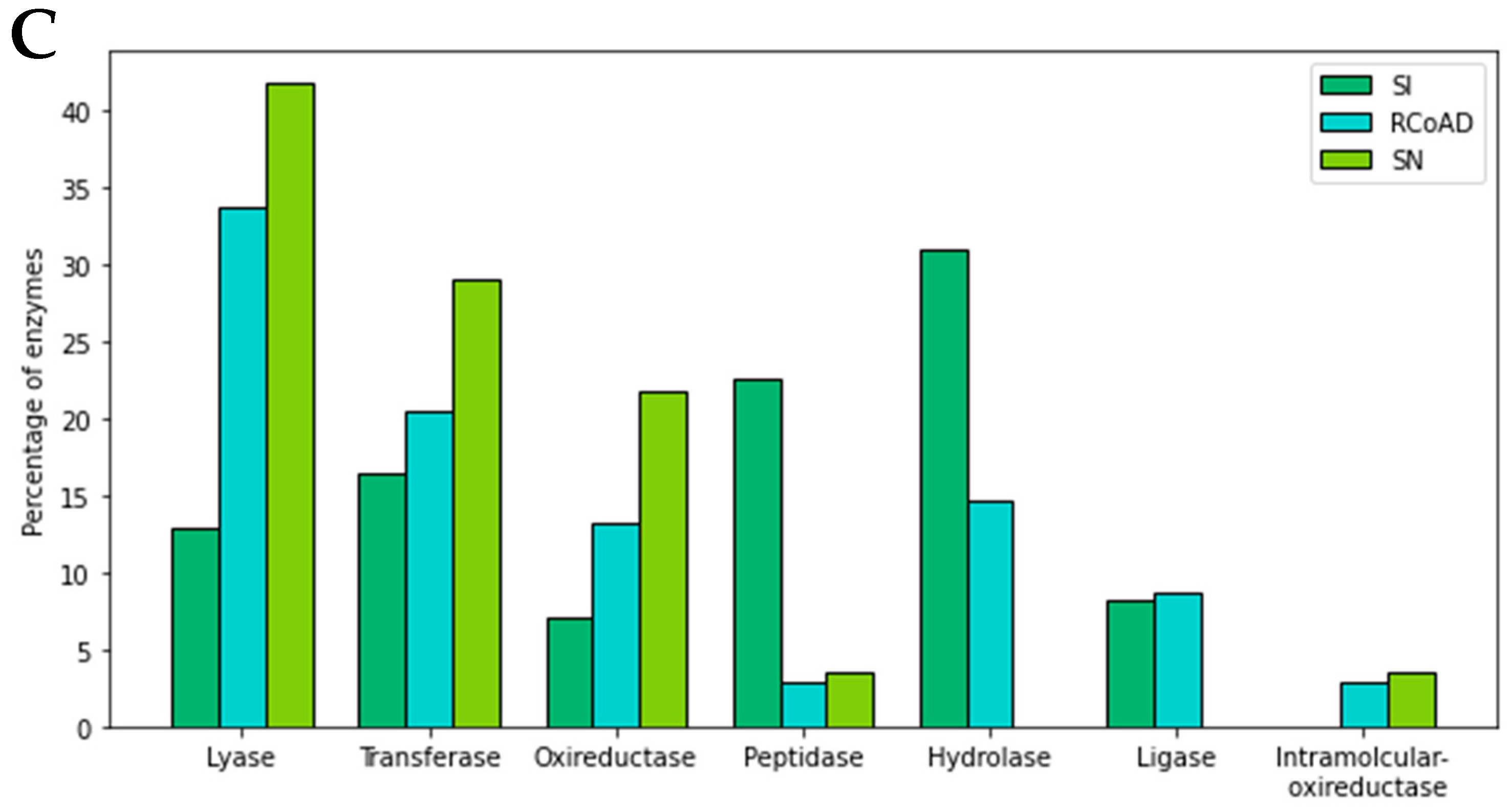
Figure 4B), there was a higher abundance of proteins that act in the first AD phases, which are the hydrolysis and acidogenic phases, for all samples. Even samples R-NP and R-CoAD were removed from the reactor in the methanogenesis phase. This can be confirmed in
Figure 4C, which shows the frequency of each of the enzymes identified in the samples, according to the analysis of the gene ontology (GO) of UniProtKB, in relation to the molecular function of these proteins. In all samples, the active carbohydrate and protein metabolism enzymes that were detected, such as hydrolase, lyase, peptidase, and some other auxiliary enzymes [
33], were part of the hydrolysis steps. Moreover, these enzymes are more frequent in SI samples than in the other two from methanogenesis. These enzymes are important in the fatty acid biosynthesis process, which is an essential step for biogas production [
34]. What probably occurred is that smaller amounts of proteins related to methanogenesis were identified, which were not detected in the analysis carried out in the blastkoala because of low quantification. Even with this difficulty, proteins related to the CH
4 metabolic route could be detected, as described in section above.
4.4. Metabolic Pathway
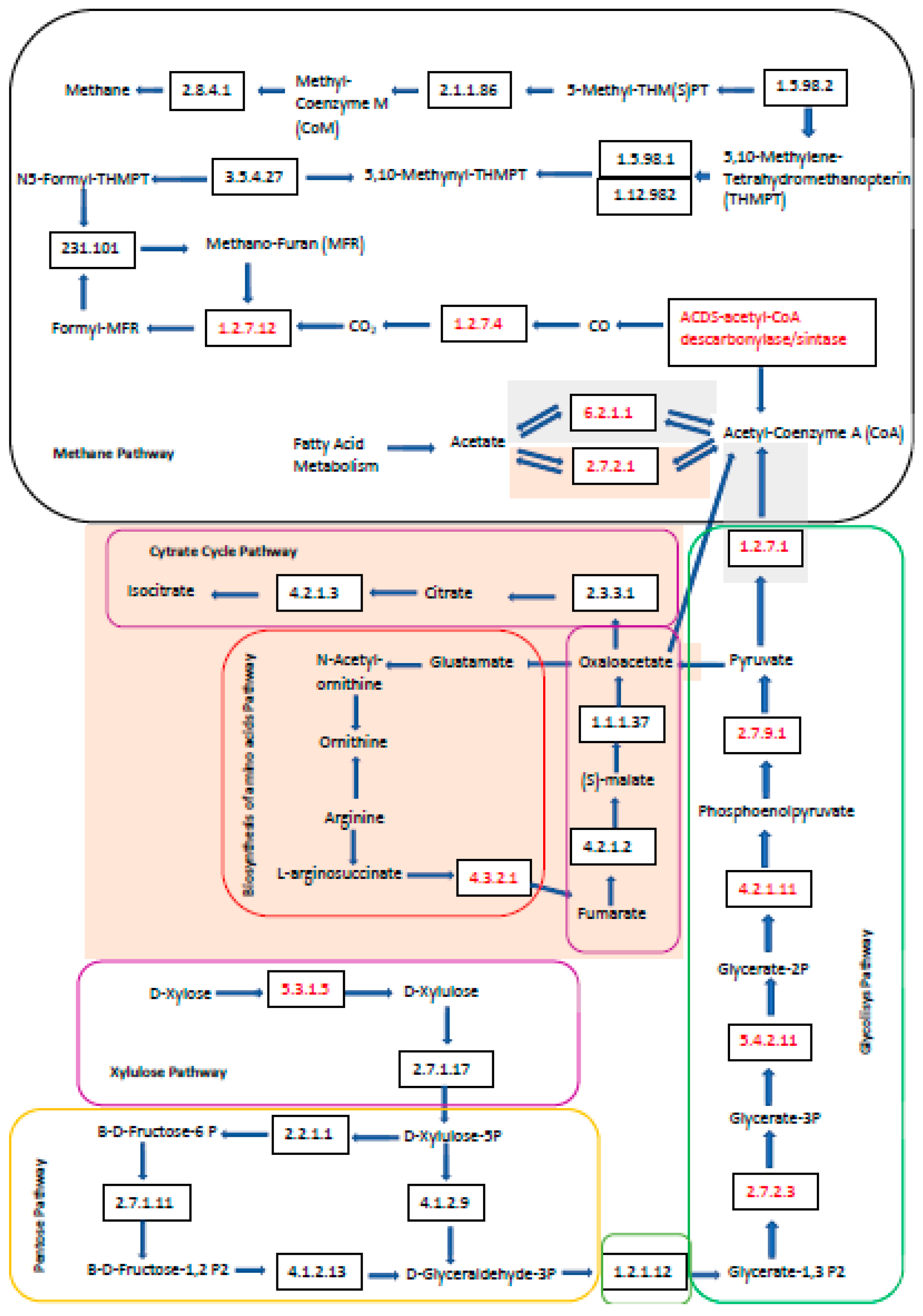
According to
Figure 5, the main differences between the three samples occurred in the early stages of AD, such as hydrolysis and acetogenesis, which is a process where macromolecules, such as cellulose, lignin, and xylose, are degraded into volatile fatty acids by different types of microorganisms. These acids enter the acetogenesis and methanogenesis phase.
It is likely that methanogenesis was not different for the three samples, because the same residues were used as substrates, with the same compositional characteristics [
7].
At the beginning of the metabolic route, the xylose, pentose, and glycolysis pathways probably occurred in the three samples as the presence of the Xylose isomerase enzyme (EC: 5.3.1.5) was detected. Then, the proteins phosphoglycerate kinase (EC: 2.7.2.3) enolase (EC: 4.2.1.11) and pyruvate phosphate dikinase (EC: 2.7.9.1) were detected, which were all involved in the glycolysis pathway, probably for the pyruvate synthesis.
Based on this, there were some differences in the SI sample routes with the R-CoAD and R-NP samples. The R-CoAD and R-NP samples showed several proteins related to the metabolism of amino acids, such as arginine succinate lyase and ornithine carbometyltransferase. The presence of these proteins allowed the route of the biosynthesis of amino acids, arginine biosynthesis, and citrate cycle (shown in pink in the map in
Figure 5) to be explored. However, these routes are not extremely important for producing CH
4, as they are part of the initial stages of the process.
In the SI sample, acetyl-CoA may have followed an acetoclastic methanogenic metabolic route, as the acetyl-coenzyme A synthase protein (EC: 6.2.1.1) was detected. In the R-CoA and R-NP sample, the acetoclastic pathway can also be identified as the acetate kinase protein (EC: 2.7.2.1) was detected in greater proportions in both samples. In general, all samples followed this route of acetyl-CoA generation, from metabolites such as the acetate, glucose, citrate cycle, and fatty acid metabolism pathways.
In the end, it is likely that acetate could be converted to CO, and later to CO
2, by the protein acetyl-CoA decarbonylase/synthase complex subunit alpha (EC: 1.2.7.4) inside of the acetyl-CoA pathway (M00422-methane metabolic route-KEGG) [
27]. This protein is part of the ACDS complex that catalyzes the reversible cleavage of acetyl-CoA, allowing for autotrophic growth from CO
2. This CO
2 could then be used by the hydrogenotrophic methanogens, as reported by Lametal [
27], following the degradation processes until the formation of CH
4 as the methyl-CoM enzyme was detected.
In this study, proteins that are part of the metabolic pathway of the acetoclastic methanogenic (M00357 methane metabolic route KEGG) and proteins that participate in the hydrogenotrophic pathway (M00567 methane metabolic route KEGG) were identified in all samples. This is a common situation for bioreactors that are fed with glucose, as reported by Lam et al. [
27]. CH
4 production from both CO
2 and acetate correlated with the observation of a temporally increasing ratio (2.1–3.3 times) of hydrogenotrophic to acetoclastic methanogenic activity.
The initial proposal of previous studies carried out by our research group (9) reported that the predominant metabolic route in the process would be the SAO coupled to hydrogenotrophic methanogenesis (SAO-HM), since, by the identified proteins, the acetate can be oxidized and converted into CO
2, and this, together with the hydrogen, would form CH
4 (
Figure 5). As this reaction generates H
2 and is thermodynamically unfavorable (reaction 1), the hydrogenotrophic methanogenic archeae consumed the present H
2 and generated CH
4 (reaction 2) [
30]. Therefore, as proteins from the two methanogenic routes and microorganisms classified as participants in the SAO-HM were identified [
9], the two reactions may be coupled, and it was probably the predominant one in co-digestion with residues from ethanol production 1G2G.
Syntrophic acetate oxidizing reaction
Hydrogenotrophic methanogenesis


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}