
The Use of Interactive Visualizations for Tracking Haplotypic Inheritance in Livestock

Abstract
:1. Introduction
2. Materials and Methods
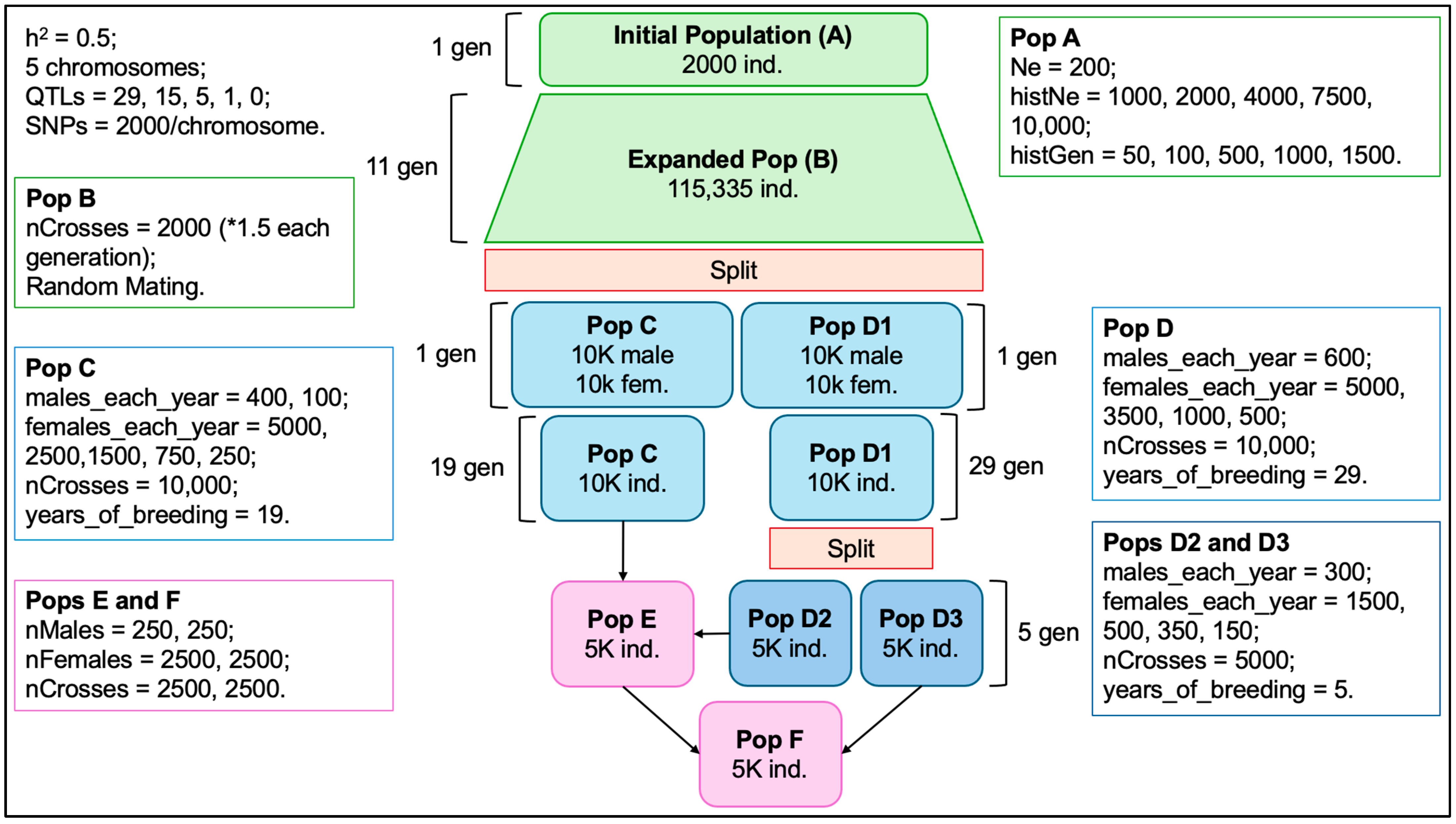
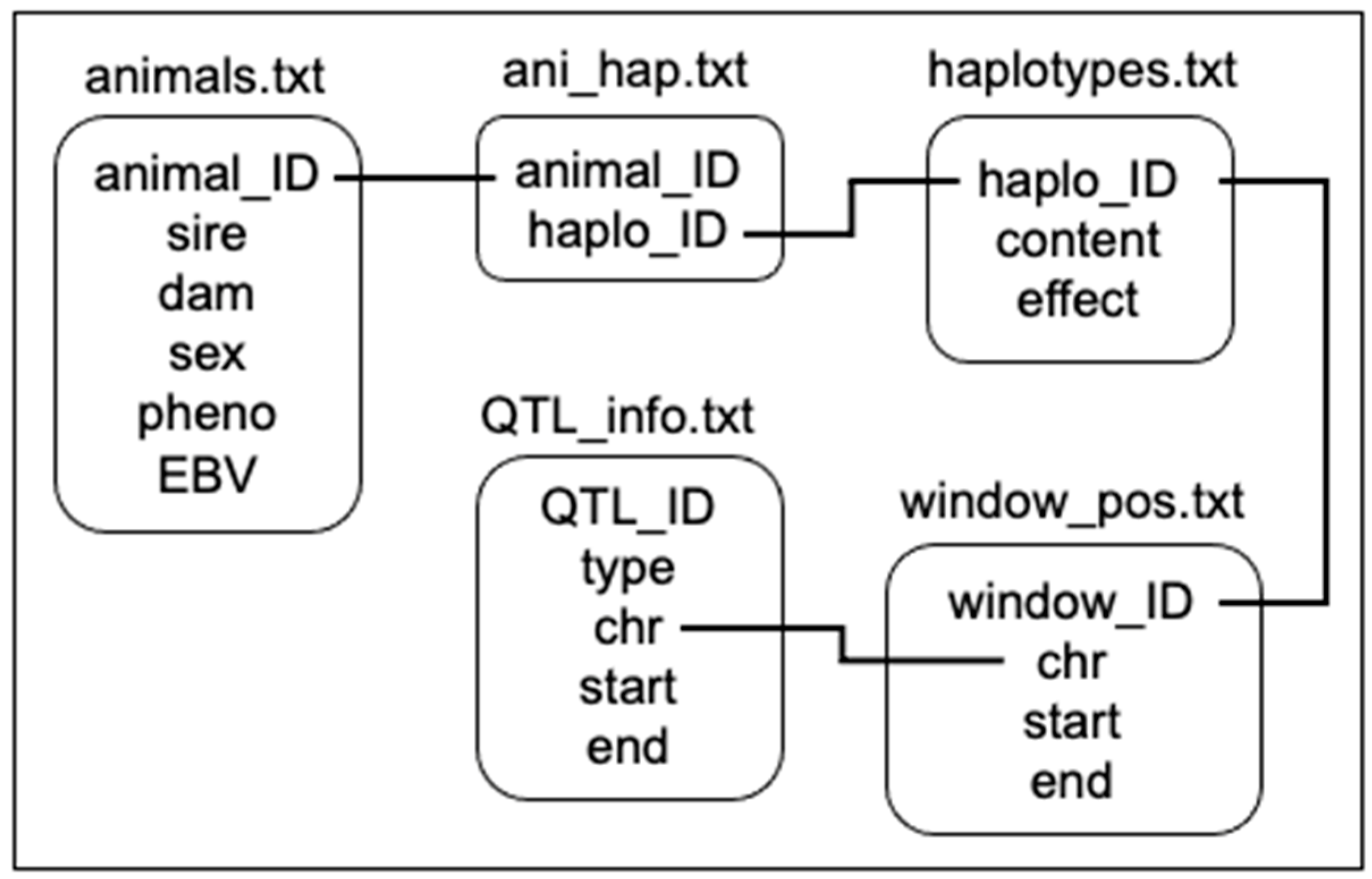
2.1. Data Simulation and Validation
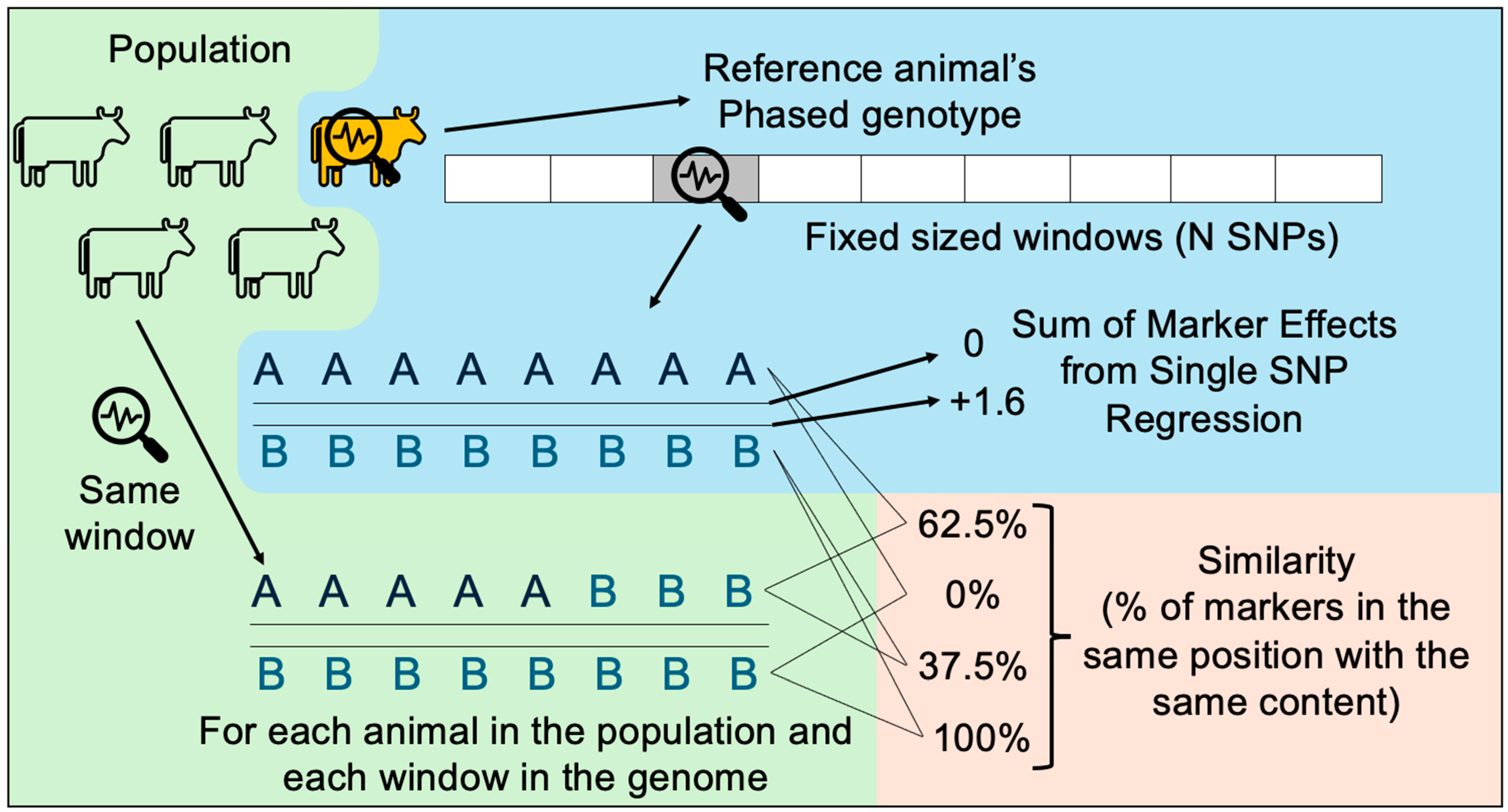
2.2. Single SNP Regression and Preparation of Data
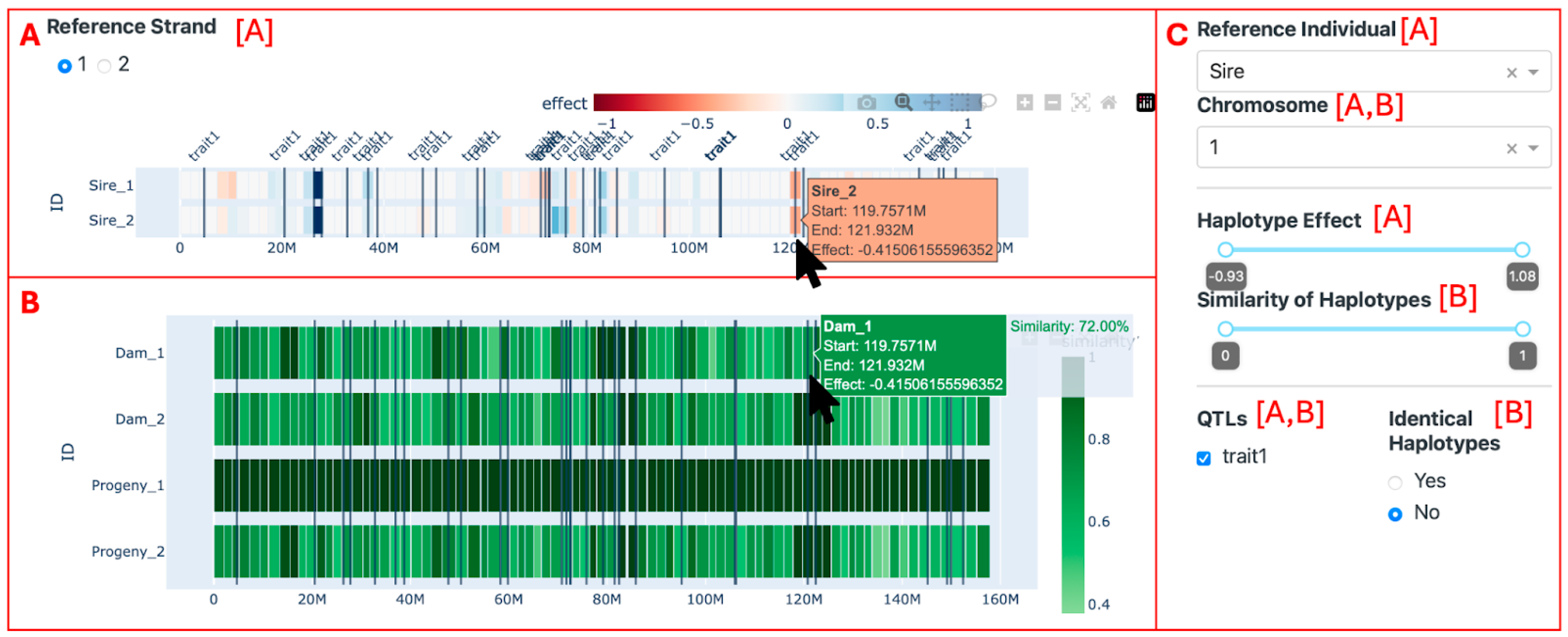
2.3. Visualization Tool
2.4. Scenarios to Illustrate the Visualization Tool
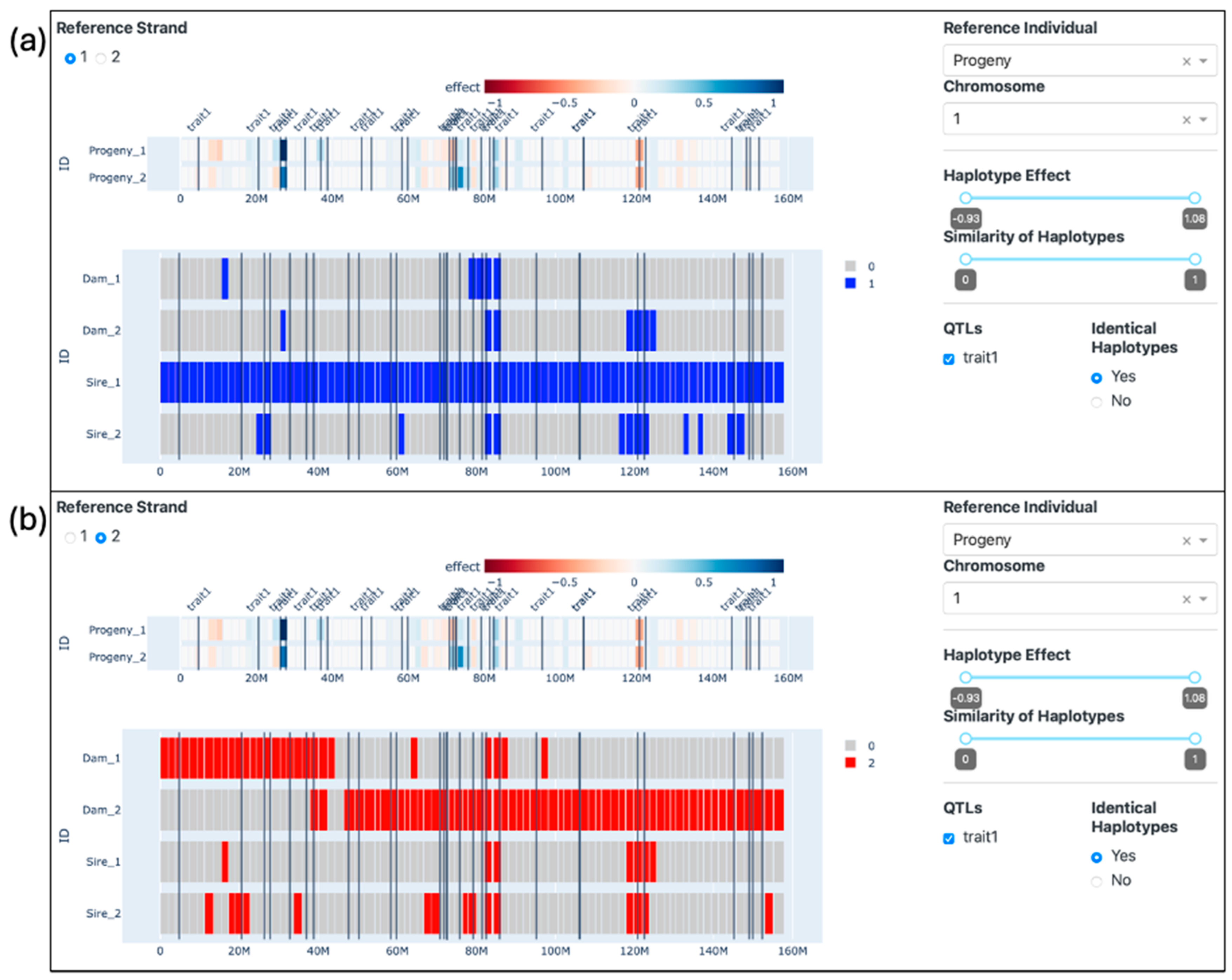
2.4.1. Trio Comparison
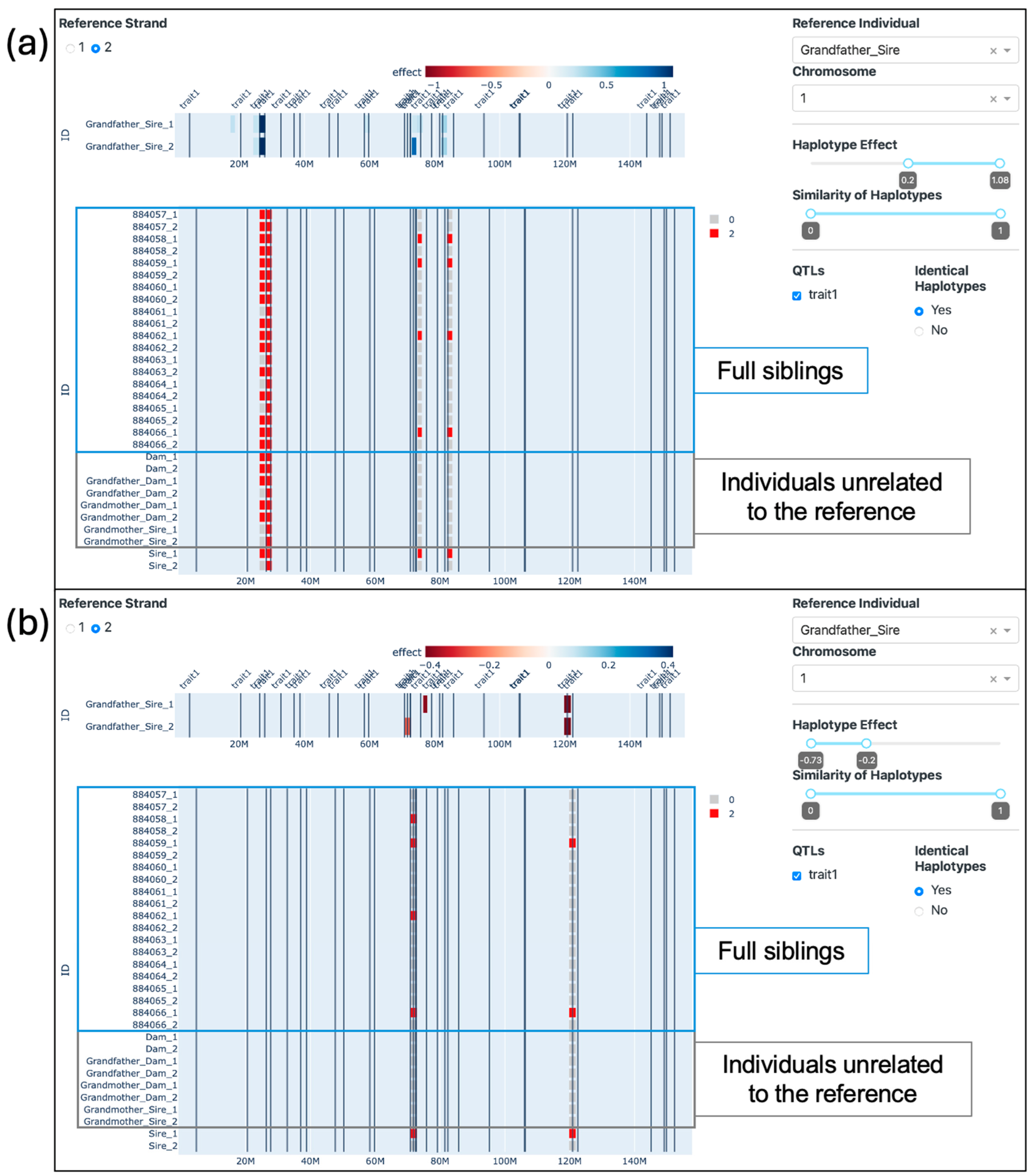
2.4.2. Visualization of Grandparents
2.4.3. Half-Siblings Evaluation
3. Results
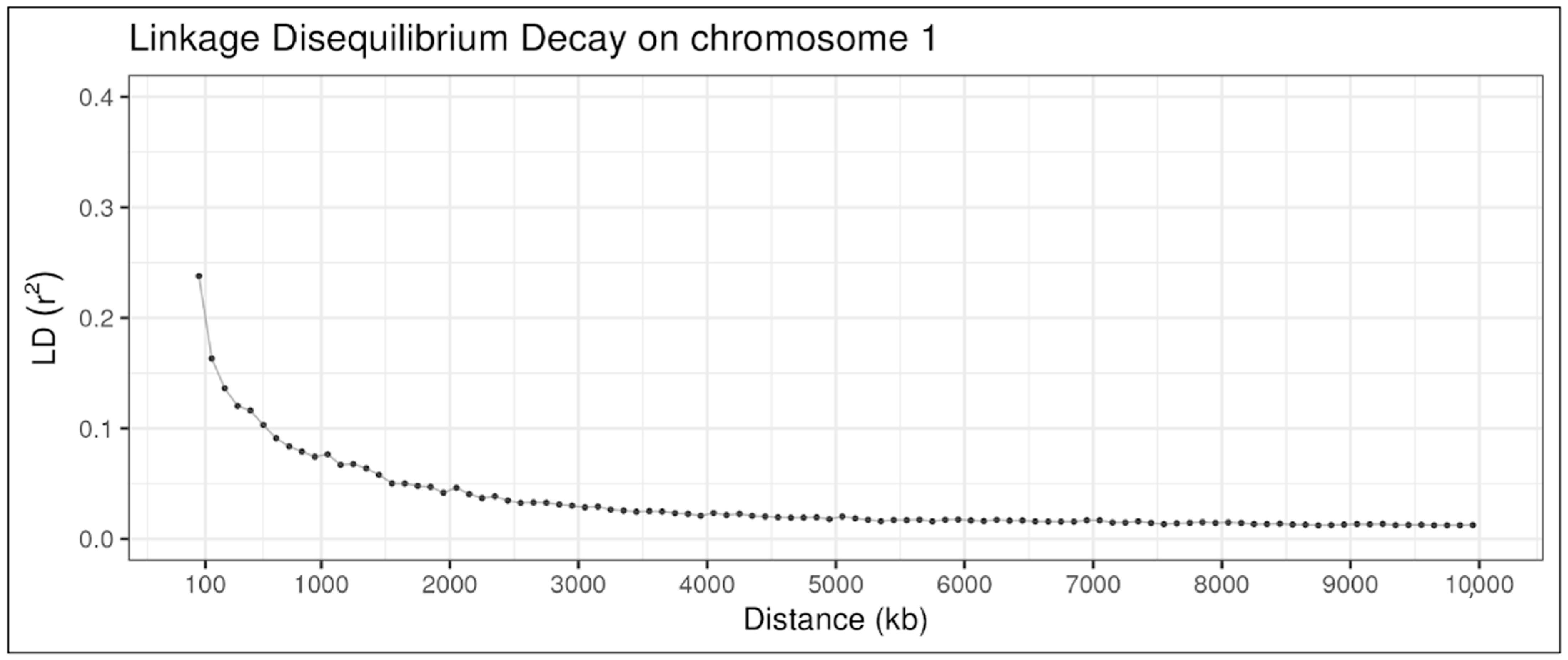
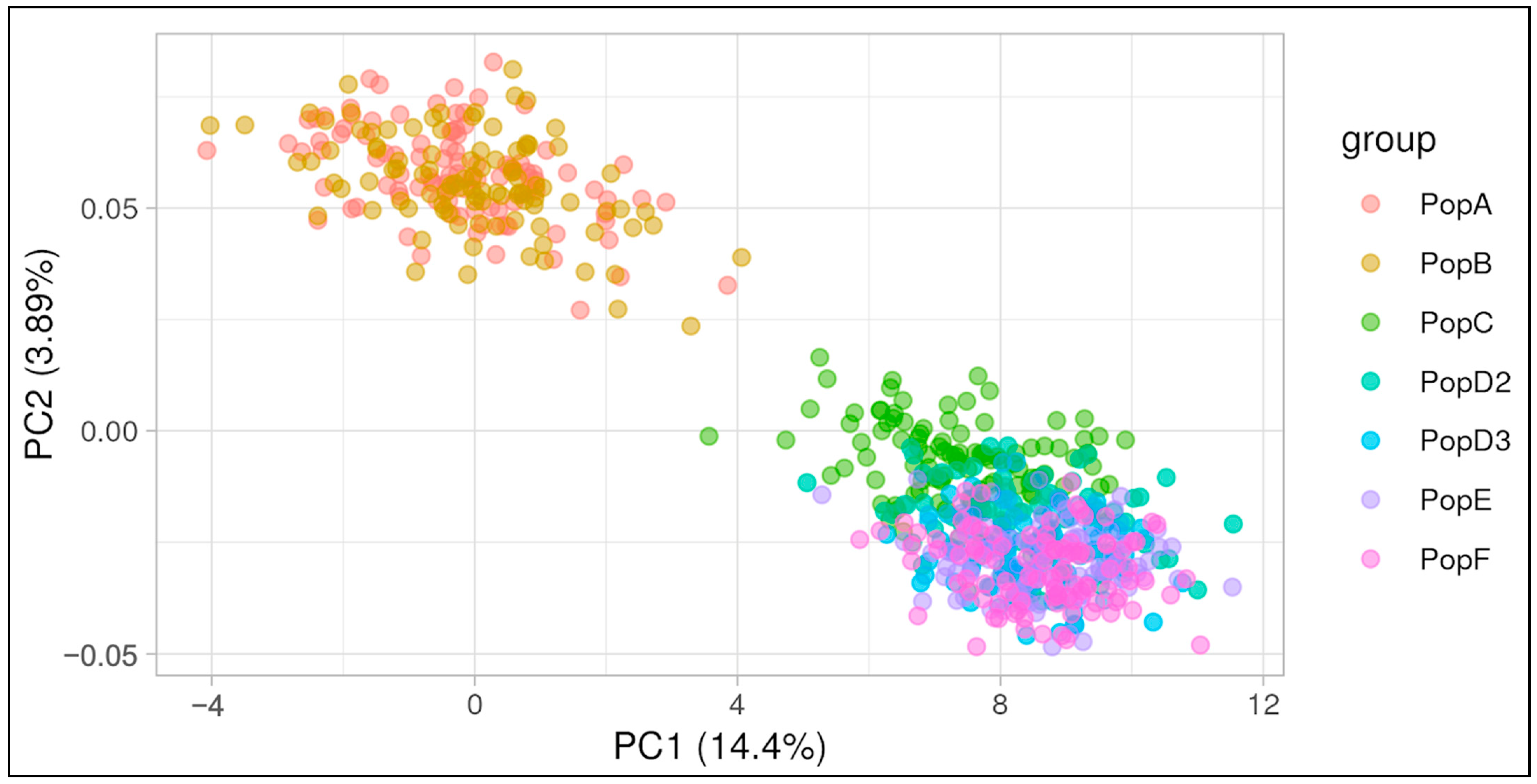
3.1. Data Simulation and Validation
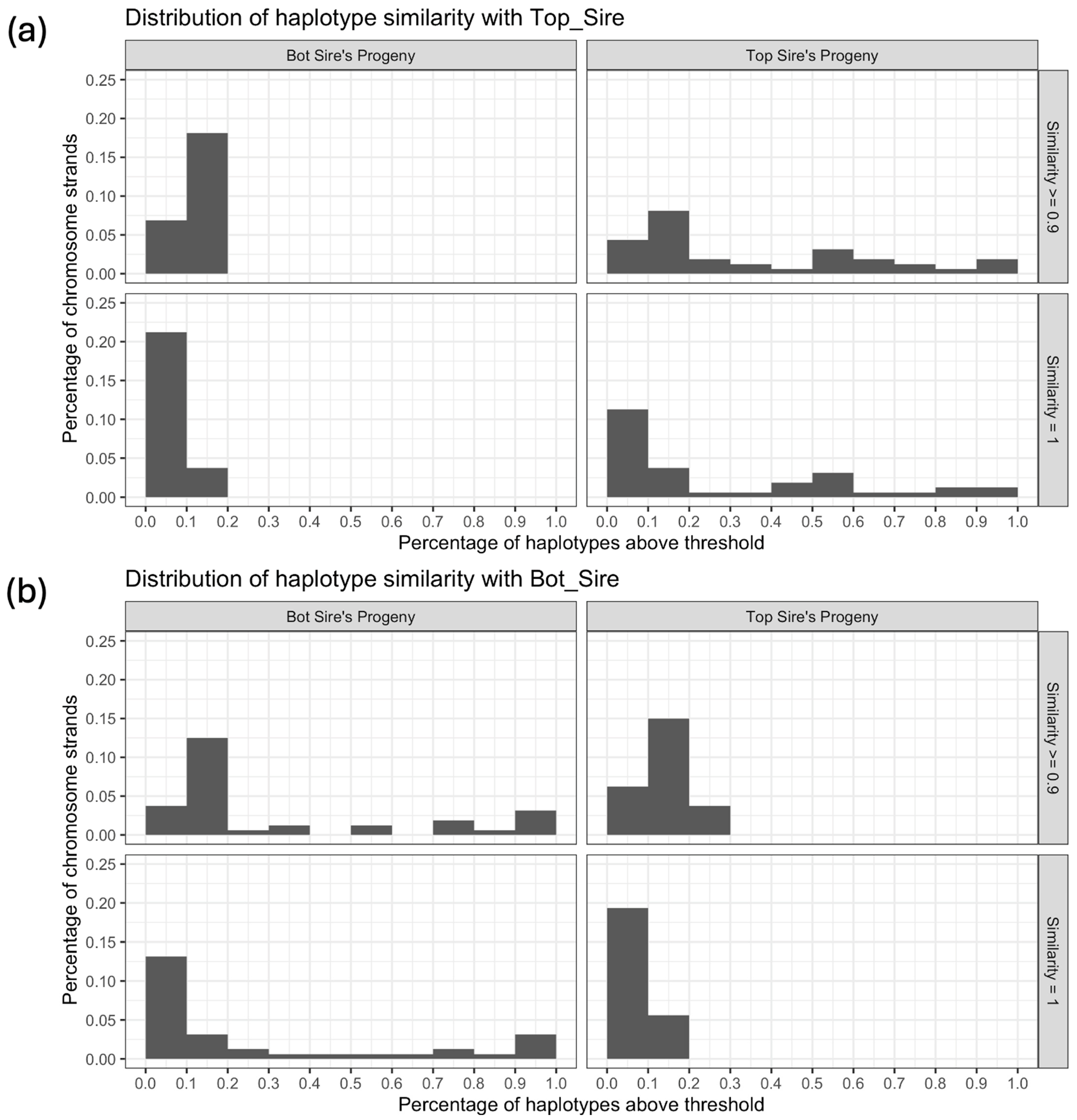
3.2. Visualization of Trios
3.3. Visualization of Grandparents
3.4. Visualization of Half-Siblings
4. Discussion
4.1. Data Simulation and Validation
4.2. Visualization of Trios
4.3. Visualization of Grandparents
4.4. Visualization of Half-Siblings
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oldenbroek, K.; van der Waaij, L. Textbook Animal Breeding: Animal Breeding and Genetics for BSc Students; Centre for Genetic Resources and Animal Breeding and Genomics Group, Wageningen University and Research Centre: Wageningen, The Netherlands, 2014. [Google Scholar]
- Bohmanova, J.; Sargolzaei, M.; Schenkel, F.S. Characteristics of Linkage Disequilibrium in North American Holsteins. BMC Genom. 2010, 11, 421. [Google Scholar] [CrossRef]
- Brito, F.V.; Neto, J.B.; Sargolzaei, M.; Cobuci, J.A.; Schenkel, F.S. Accuracy of Genomic Selection in Simulated Populations Mimicking the Extent of Linkage Disequilibrium in Beef Cattle. BMC Genet. 2011, 12, 80. [Google Scholar] [CrossRef]
- Cook, M.P. Visual Representations in Science Education: The Influence of Prior Knowledge and Cognitive Load Theory on Instructional Design Principles. Sci. Educ. 2006, 90, 1073–1091. [Google Scholar] [CrossRef]
- Selli, A.; Ventura, R.V.; Fonseca, P.A.S.; Buzanskas, M.E.; Andrietta, L.T.; Balieiro, J.C.C.; Brito, L.F. Detection and Visualization of Heterozygosity-Rich Regions and Runs of Homozygosity in Worldwide Sheep Populations. Animals 2021, 11, 2696. [Google Scholar] [CrossRef] [PubMed]
- Van Essen, D.C.; Anderson, C.H.; Felleman, D.J. Information Processing in the Primate Visual System: An Integrated Systems Perspective. Science 1992, 255, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Dosher, B.; Lu, Z.-L. Visual Perceptual Learning and Models. Annu. Rev. Vis. Sci. 2017, 3, 343–363. [Google Scholar] [CrossRef]
- Rougier, N.P.; Droettboom, M.; Bourne, P.E. Ten Simple Rules for Better Figures. PLoS Comput. Biol. 2014, 10, e1003833. [Google Scholar] [CrossRef]
- Bateman, S.; Mandryk, R.L.; Gutwin, C.; Genest, A.; McDine, D.; Brooks, C. Useful Junk? The Effects of Visual Embellishment on Comprehension and Memorability of Charts. In Proceedings of the SIGCHI Conference on Human Factors in Computing Systems, Atlanta, GA, USA, 10 April 2010; pp. 2573–2582. [Google Scholar]
- Hullman, J.; Diakopoulos, N. Visualization Rhetoric: Framing Effects in Narrative Visualization. IEEE Trans. Vis. Comput. Graph. 2011, 17, 2231–2240. [Google Scholar] [CrossRef]
- Zhou, L.; Hansen, C.D. A Survey of Colormaps in Visualization. IEEE Trans. Vis. Comput. Graph. 2016, 22, 2051–2069. [Google Scholar] [CrossRef] [PubMed]
- Zacks, J.; Tversky, B. Bars and Lines: A Study of Graphic Communication. Mem. Cognit. 1999, 27, 1073–1079. [Google Scholar] [CrossRef]
- Ali, S.M.; Gupta, N.; Nayak, G.K.; Lenka, R.K. Big Data Visualization: Tools and Challenges. In Proceedings of the 2016 2nd International Conference on Contemporary Computing and Informatics, Greater Noida, India, 14–17 December 2016; pp. 656–660. [Google Scholar]
- Morota, G.; Cheng, H.; Cook, D.; Tanaka, E. ASAS-NANP Symposium: Prospects for Interactive and Dynamic Graphics in the Era of Data-Rich Animal Science1. J. Anim. Sci. 2021, 99, skaa402. [Google Scholar] [CrossRef]
- Plotly. Dash Documentation & User Guide. Available online: https://dash.plotly.com/ (accessed on 10 March 2022).
- Van Rossum, G.; Drake, F.L., Jr. Python Reference Manual; Centrum voor Wiskunde en Informatica: Amsterdam, The Netherlands, 1995. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Tableau. Business Intelligence and Analytics Software. Available online: https://www.tableau.com/ (accessed on 26 October 2023).
- Microsoft Power BI. Data Visualisation. Available online: https://powerbi.microsoft.com/en-gb/ (accessed on 26 October 2023).
- Curti, P.D.F.; Selli, A.; Pinto, D.L.; Merlos-Ruiz, A.; Balieiro, J.C.D.C.; Ventura, R.V. Applications of Livestock Monitoring Devices and Machine Learning Algorithms in Animal Production and Reproduction: An Overview. Anim. Reprod. 2023, 20, e20230077. [Google Scholar] [CrossRef]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. JBrowse: A Dynamic Web Platform for Genome Visualization and Analysis. Genome Biol. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- L’Yi, S.; Gehlenborg, N. Multi-View Design Patterns and Responsive Visualization for Genomics Data. IEEE Trans. Vis. Comput. Graph. 2023, 29, 559–569. [Google Scholar] [CrossRef]
- Deeb, J.; Juan, R.P.; Kendall, D.; Castellani, D.; Heuer, C.; Utsunomiya, Y.T.; Su, H.; Westberry, S.; Utsunomiya, A. Chromosomal Mating: A Data-Driven Approach to Improving Dairy Cattle Breeding Decisions. In Proceedings of the Plant and Animal Genome XXXI, San Diego, CA, USA, 12–17 January 2024. [Google Scholar]
- Larkin, D.M.; Daetwyler, H.D.; Hernandez, A.G.; Wright, C.L.; Hetrick, L.A.; Boucek, L.; Bachman, S.L.; Band, M.R.; Akraiko, T.V.; Cohen-Zinder, M.; et al. Whole-Genome Resequencing of Two Elite Sires for the Detection of Haplotypes under Selection in Dairy Cattle. Proc. Natl. Acad. Sci. USA 2012, 109, 7693–7698. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, R.C.; Gorjanc, G.; Hickey, J.M. AlphaSimR: An R Package for Breeding Program Simulations. G3 GenesGenomesGenetics 2021, 11, jkaa017. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Qiu, Y.; Inouye, M. FlashPCA2: Principal Component Analysis of Biobank-Scale Genotype Datasets. Bioinformatics 2017, 33, 2776–2778. [Google Scholar] [CrossRef] [PubMed]
- Gondro, C. Primer to Analysis of Genomic Data Using R.—Use R! Springer International Publishing: Cham, Switzerland, 2015; ISBN 978-3-319-14474-0. [Google Scholar]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 56–61. [Google Scholar]
- Sargolzaei, M.; Schenkel, F.S. QMSim: A Large-Scale Genome Simulator for Livestock. Bioinformatics 2009, 25, 680–681. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Sargolzaei, M.; Kelly, M.; Li, C.; Voort, G.V.; Wang, Z.; Plastow, G.; Moore, S.; Miller, S.P. Linkage Disequilibrium in Angus, Charolais, and Crossbred Beef Cattle. Front. Genet. 2012, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pérez O’Brien, A.M.; Mészáros, G.; Utsunomiya, Y.T.; Sonstegard, T.S.; Garcia, J.F.; Van Tassell, C.P.; Carvalheiro, R.; da Silva, M.V.B.; Sölkner, J. Linkage Disequilibrium Levels in Bos Indicus and Bos Taurus Cattle Using Medium and High Density SNP Chip Data and Different Minor Allele Frequency Distributions. Livest. Sci. 2014, 166, 121–132. [Google Scholar] [CrossRef]
- Buzanskas, M.E.; Ventura, R.V.; Chud, T.C.S.; Bernardes, P.A.; De Abreu Santos, D.J.; De Almeida Regitano, L.C.; De Alencar, M.M.; De Alvarenga Mudadu, M.; Zanella, R.; Da Silva, M.V.G.B.; et al. Study on the Introgression of Beef Breeds in Canchim Cattle Using Single Nucleotide Polymorphism Markers. PLoS ONE 2017, 12, 1–16. [Google Scholar] [CrossRef]
- Roach, J.C.; Glusman, G.; Smit, A.F.A.; Huff, C.D.; Hubley, R.; Shannon, P.T.; Rowen, L.; Pant, K.P.; Goodman, N.; Bamshad, M.; et al. Analysis of Genetic Inheritance in a Family Quartet by Whole-Genome Sequencing. Science 2010, 328, 636–639. [Google Scholar] [CrossRef]
- Gibson, J.; Morton, N.E.; Collins, A. Extended Tracts of Homozygosity in Outbred Human Populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef]
- Fariello, M.I.; Boitard, S.; Naya, H.; SanCristobal, M.; Servin, B. Detecting Signatures of Selection Through Haplotype Differentiation Among Hierarchically Structured Populations. Genetics 2013, 193, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.F.; Kijas, J.W.; Ventura, R.V.; Sargolzaei, M.; Porto-Neto, L.R.; Cánovas, A.; Feng, Z.; Jafarikia, M.; Schenkel, F.S. Genetic Diversity and Signatures of Selection in Various Goat Breeds Revealed by Genome-Wide SNP Markers. BMC Genomics 2017, 18, 229. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-Wide Analysis of the World’s Sheep Breeds Reveals High Levels of Historic Mixture and Strong Recent Selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.F.; McEwan, J.C.; Miller, S.P.; Pickering, N.K.; Bain, W.E.; Dodds, K.G.; Schenkel, F.S.; Clarke, S.M. Genetic Diversity of a New Zealand Multi-Breed Sheep Population and Composite Breeds’ History Revealed by a High-Density SNP Chip. BMC Genet. 2017, 18, 25. [Google Scholar] [CrossRef] [PubMed]
- Olenski, K.; Kamiński, S.; Szyda, J.; Cieslinska, A. Polymorphism of the Beta-Casein Gene and Its Associations with Breeding Value for Production Traits of Holstein–Friesian Bulls. Livest. Sci. 2010, 131, 137–140. [Google Scholar] [CrossRef]
- Malher, X.; Beaudeau, F.; Philipot, J.M. Effects of Sire and Dam Genotype for Complex Vertebral Malformation (CVM) on Risk of Return-to-Service in Holstein Dairy Cows and Heifers. Theriogenology 2006, 65, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Charlier, C.; Agerholm, J.S.; Coppieters, W.; Karlskov-Mortensen, P.; Li, W.; De Jong, G.; Fasquelle, C.; Karim, L.; Cirera, S.; Cambisano, N.; et al. A Deletion in the Bovine FANCI Gene Compromises Fertility by Causing Fetal Death and Brachyspina. PLoS ONE 2012, 7, e43085. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M.; Null, D.J.; Olson, K.M.; Hutchison, J.L. Reporting of Haplotypes with Recessive Effects on Fertility. Interbull Bull. 2011, 44, 117–121. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance (kb) | Number of SNP Pairs | Mean r2 | Standard Deviation r2 | Median r2 | Percentage of r2 ≥ 0.3 |
|---|---|---|---|---|---|
| 0–50 | 5786 | 0.26 | 0.34 | 0.09 | 0.3 |
| 50–100 | 5557 | 0.19 | 0.26 | 0.07 | 0.21 |
| 100–200 | 10,943 | 0.15 | 0.22 | 0.05 | 0.17 |
| 200–300 | 11,191 | 0.12 | 0.18 | 0.04 | 0.12 |
| 300–400 | 11,127 | 0.11 | 0.17 | 0.04 | 0.11 |
| 400–500 | 11,070 | 0.1 | 0.15 | 0.04 | 0.09 |
| 500–1000 | 55,122 | 0.08 | 0.12 | 0.03 | 0.06 |
| Group | Reference Strand | N | Similarity = 1 | Similarity ≥ 0.9 | ||||
|---|---|---|---|---|---|---|---|---|
| Mean | SD | Median | Mean | SD | Median | |||
| Bot Progeny | 1 | 20 | 5.3 | 2.27 | 5.5 | 10.7 | 2.41 | 11.5 |
| 2 | 20 | 5.55 | 2.86 | 5 | 10.55 | 3.90 | 9.5 | |
| Bot Sire | 1 | 2 | 4.5 | 2.12 | 4.5 | 10.5 | 2.12 | 10.5 |
| 2 | 2 | 6 | 4.24 | 6 | 11 | 7.07 | 11 | |
| Dams | 1 | 20 | 6.55 | 3.69 | 6 | 11.05 | 4.21 | 11 |
| 2 | 20 | 4.6 | 2.68 | 4.5 | 9.55 | 3.98 | 10 | |
| Top Progeny | 1 | 20 | 28.25 | 27.37 | 17.5 | 32.9 | 26.11 | 23 |
| 2 | 20 | 18.2 | 19.60 | 7.5 | 24.25 | 19.01 | 16.5 | |
| Group | Reference Strand | N | Similarity = 1 | Similarity ≥ 0.9 | ||||
|---|---|---|---|---|---|---|---|---|
| Mean | SD | Median | Mean | SD | Median | |||
| Bot Progeny | 1 | 20 | 18.3 | 22.09 | 8 | 21.55 | 21.09 | 13 |
| 2 | 20 | 30.6 | 30.88 | 9 | 33.4 | 29.28 | 15 | |
| Dams | 1 | 20 | 7.3 | 3.06 | 6.5 | 11.55 | 3.38 | 10.5 |
| 2 | 20 | 7.1 | 2.86 | 6.5 | 12.1 | 4.44 | 12 | |
| Top Progeny | 1 | 20 | 5.35 | 3.51 | 4 | 9.65 | 3.47 | 9 |
| 2 | 20 | 7 | 3.26 | 6.5 | 13.15 | 4.36 | 13 | |
| Top Sire | 1 | 2 | 3 | 0.00 | 3 | 7.5 | 2.12 | 7.5 |
| 2 | 2 | 7.5 | 2.12 | 7.5 | 14 | 2.83 | 14 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selli, A.; Miller, S.P.; Ventura, R.V. The Use of Interactive Visualizations for Tracking Haplotypic Inheritance in Livestock. Ruminants 2024, 4, 90-111. https://doi.org/10.3390/ruminants4010006
Selli A, Miller SP, Ventura RV. The Use of Interactive Visualizations for Tracking Haplotypic Inheritance in Livestock. Ruminants. 2024; 4(1):90-111. https://doi.org/10.3390/ruminants4010006
Chicago/Turabian StyleSelli, Alana, Stephen P. Miller, and Ricardo V. Ventura. 2024. "The Use of Interactive Visualizations for Tracking Haplotypic Inheritance in Livestock" Ruminants 4, no. 1: 90-111. https://doi.org/10.3390/ruminants4010006
APA StyleSelli, A., Miller, S. P., & Ventura, R. V. (2024). The Use of Interactive Visualizations for Tracking Haplotypic Inheritance in Livestock. Ruminants, 4(1), 90-111. https://doi.org/10.3390/ruminants4010006