1. Introduction
Nickel is ubiquitous in our daily live s and is also found in trace amounts in environmental water [
1]. According to the WHO guidelines, the nickel concentration of drinking water should be 0.07 mg/L. The International Agency for Research on Cancer concluded that inhaled nickel compounds are carcinogenic to humans (Group 1) [
2]. Nickel is used in a wide range of compounds, including stainless steel and nickel alloys [
2]. Moreover, nickel is used in nickel-cadmium and nickel-metal hydride batteries as well as lithium-ion batteries. These batteries are used in electric vehicles and their demand is expected to rise with their widespread use in the future [
3]. However, nickel contamination due to the increased use of batteries causes aquatic environment pollution; therefore, it is necessary to collect the baseline data of nickel that exists in the environment.
Graphite-furnace atomic-absorption spectrometry (GF-AAS) has high sensitivity and requires only a small sample volume of 10–20 µL. Although the sensitivity of GF-AAS is high enough to allow the detection of trace elements at low concentrations, thus allowing adherence to WHO guidelines, it is still low for detecting the presence of trace elements in environmental water such as river water and seawater. Similar to other atomic spectroscopic methods, GF-AAS is affected by coexisting components; thus, pretreatment, such as extraction, enrichment, and separation, is necessary.
Enrichment and separation techniques that require low sample volume include liquid-liquid extraction/back-microextractions [
4], cloud point extractions [
5,
6,
7], dispersive liquid–liquid extractions [
7,
8,
9], thermoresponsive polymer-mediated extractions [
10,
11], and homogeneous liquid–liquid extractions [
12,
13,
14]. The efficiency of these techniques depends on application of heat [
5,
6,
11,
13], use of chlorobenzene, chloroform, or xylene as an extractant [
4,
7,
8,
9]; and use of a fluorinated surfactant [
12] or dry ice [
14]. Herein, we propose an ion-associate phase (IAP) extraction technique [
15,
16] that improves sensitivity and removes interfering components, and use it as a pretreatment technique for atomic spectroscopy [
17,
18,
19,
20]. Through the proposed method, the analyte in environmental water can be converted to moderate hydrophobicity as needed, and an organic cation (Q
+) and an organic anion (OA
−) can be added to form an ion associate. Centrifugation separates the IAP and removes the aqueous phase. The IAP is dissolved in a water-miscible solvent (2-methoxyethanol) [
17] or a mixture of methanol and nitric acid [
18], or is back-microextracted with nitric acid [
19,
20], and the analyte is characterized through atomic spectroscopy.
As a chelating reagent of heavy metals for IAP extraction, a 2-(5-bromo-2-pyridylazo)-5-(
N-propyl-
N-sulfopropylamino)phenol (5-Br-PAPS) is typically used [
17,
18,
19], which is expensive. As chelating reagent of lithium for IAP extraction, a dipivaloylmethane (HDPM) is used [
20]. HDPM not only reacts selectively with lithium to form a chelating complex, but also acts as an OA
− for the proposed IAP extraction/back-microextraction [
20]; it is also inexpensive. Moreover, β-diketones are chosen as chelating reagents of heavy metals for IAP extraction, because they can also act as OA
−, which are the component of the IAP. Thenoyltrifluoroacetone (HTTA), a hydrophobic chelating agent, a type of β-diketones, serves as the OA
− in the developed IAP microextraction. The combined use of IAP microextraction and back-microextraction facilitates the beneficial integration of preconcentration and GF-AAS for nickel detection. To verify the applicability of the proposed method, we used it to measure the nickel concentration in river and seawater samples through GF-AAS.
2. Experimental
2.1. Reagents and Chemicals
The standard solution of nickel, 1000 mg L−1, was prepared by dissolving nickel(II) nitrate hexahydrate (99.9%, Wako Pure Chemical, Osaka, Japan) in 0.1 M nitric acid. HTTA solution was prepared by dissolving HTTA (99%, Aldrich, St. Louis, MO, USA) in acetone to make 0.1 M. Benzoylacetone (BzA) solution was prepared by dissolving BzA (1-phenyl-1,3-butanedione, Wako Pure Chemical, Osaka, Japan) in acetone to make 0.1 M. A phenolsulfonate (PS−) solution was prepared by dissolving sodium 4-hydroxybenzenesulfonate (Tokyo Chemical Industry, Tokyo, Japan) in water to make 0.5 M, and then purified. For purification, a 0.1% 1-(2-pyridylazo) 2-naphthol (PAN) ethanol solution was added to the PS− solution and the pH was adjusted to 9 to form a PAN complex, which was then filtered off. This was repeated again and the PS− solution was used. A tetramethylammonium hyhdroxide (TMAH) was diluted a TMAH solution (25%, AA-grade, Tama Chemicals, Kawasaki, Japan) to 12.5% with water. Q+ solutions were prepared as follows. The bromide of benzyldodecyldimethylammonium (C12BzDMA+), chlorides of benzyldimethyltetradecylammonium (C14BzDMA+), and benzethonium (Ben+), were dissolved in water to make 0.1 M. A nitric acid solution for back-microextraction, was prepared by dissolving 0.5 g of ammonium dihydrogen phosphate in 100 mL of 2 M nitric acid.
All other reagents (e.g., nitric acid, acetone, ethanol, methanol, 2-methoxyethanol) were of analytical grade and were used as received. An ultrapure water with a specific resistance of 18.2 MΩ cm (Direct-Q3 UV system, Millipore, Tokyo, Japan) was used throughout.
2.2. Apparatus
Trace nickel in the aqueous solutions was measured using a Hitachi (Tokyo, Japan) Z-8000 GF-AAS. The instrumental conditions for GF-AAS were an analytical wavelength of 232.0 nm, a sample volume of 10 μL, and a slit of 0.4 nm.
For centrifugation, Kubota (Type 5420, Tokyo, Japan) and Kokusan (Type H-80R, Saitama, Japan) centrifuges were used.
The measurement of the UV spectra was performed with a Hitachi (Tokyo, Japan) Model U-2000A, and a Shimadzu (Kyoto, Japan) Model UV-2450 spectrophotometers (1 cm quartz cell).
Mixing and shaking of samples for back-microextraction was performed using Taitec (Koshigaya, Japan) test tube mixer (Present Mixer).
A 50 mL plastic centrifuge tube (Violamo, AS ONE, Osaka, Japan) with a centrifugal strength of 20,000 G was used during the entire sample preparation process. A glass centrifuge tube was used to measure the UV absorption spectrum.
3. Results and Discussion
3.1. Chelate Complex Formation
As chelating reagents for the extraction of nickel from water, dithiocarbamates [
4,
9], 2-(5-bromo-2-pyridylazo)-5-dimethylaminoaniline [
6], PAN [
7,
8,
21], 8-quinolinol [
10], and 5-Br-PAPS [
19] have been used apart from β-diketones, such as HTTA [
22,
23,
24,
25].
HTTA and benzoylacetone (BzA) were used as chelating reagents because they make stable complexes with nickel and are β-diketones [
26], which are a constituent of the IAP and an OA
−. The acid dissociation constant, p
Ka, of HTTA is 6.2 [
27], and of BzA is 8.88 [
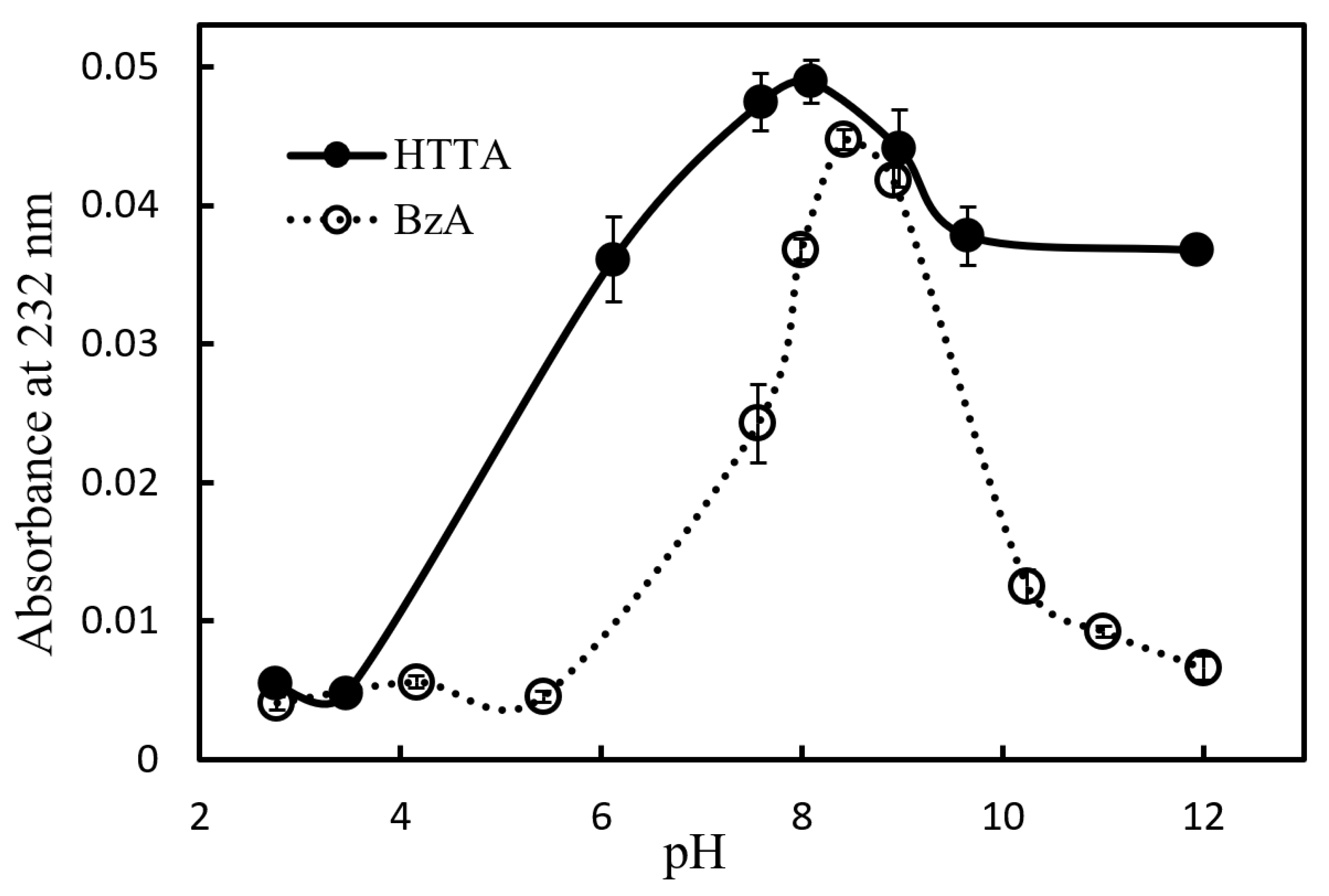
27]. The effect of pH on the complexation of the β-diketones chelating reagent HA (HTTA or BzA) with nickel was investigated. Here, pH was adjusted using phenol sulphonate and TMAH solutions. The effect of pH on the formation of nickel chelate complexes when HTTA and BzA are used as chelating reagents is shown in
Figure 1. The recoveries of both HTTA and BzA decrease with increasing acidity or basicity. β-Diketones cannot be coordinated in metals unless they are deprotonated to the enol form [
26]. However, when the pH is too high, hydroxide complexes are formed, and the formation of β-diketonate complexes is considered to be inhibited. In this study, HTTA was selected because the recovery for HTTA is higher than that for BzA at the optimum pH. pH 8 was chosen as the optimal pH because at this value, the highest recovery was achieved. At pH 8, calculations at p
Ka = 6.2 [
27] show that this chelating reagent exists mostly as an anion, TTA
−.
HTTA is soluble in water only at 0.02 g per 100 mL [
26]. The organic solvents used to dissolve and prepare HTTA, namely, acetone, methanol, ethanol, and 2-methoxyethanol, were investigated for their effect on the determination of nickel. As a solvent for preparing HTTA, acetone recovered nickel almost quantitatively, while methanol and ethanol recovered about 80% and 2-methoxyethanol about 50% Ni. Thus, in this study, acetone was selected.
The recovery was determined using double extraction. The recovery (R) is:
where
C1 is the concentration of nickel or absorbance at 232 nm during the first extraction into 100 µL of nitric acid solution and
C2 is the concentration of nickel or absorbance at 232 nm during the second extraction into 100 µL of nitric acid solution.
When water was added to the acetone solution of HTTA to reduce the volatilisation of acetone, the nickel recovery was decreased. Thus, in this study, the HTTA acetone solution was stored and used as is without diluting it with water.
The Ni-TTA complex was formed and extracted quantitatively in to the IAP at pH 8. For increasing the pH value from 0.01 M nitric acid solution (about pH 2) to pH 8 acidity, four different pH buffers and pH adjusters, (1) a mixture of tris(hydroxymethyl)aminomethane (Tris) (p
Ka2 = 8.20) [
27], hydrochloric acid and disodium carbonate, (2) potassium hydroxide, (3) disodium carbonate, (4) a mixture of phenolsulfonate (p
Ka2 = 8.56) [
27] and tetramethylammonium hydroxide (TMAH) were used for complex formation and their effect on the preconcentration and determination of nickel was investigated. For pH adjustment, a mixture of phenolsulfonate and tetramethylammonium hydroxide (TMAH) was recommended because it afforded a quantitative recovery of nickel. The phenolsulfonate ion (PS
−) is not only a component of pH buffers, but also one of the OA
− that constitute IAP.
The reaction of nickel with TTA
− in a solution of pH 8 follows Equations (2)–(4).
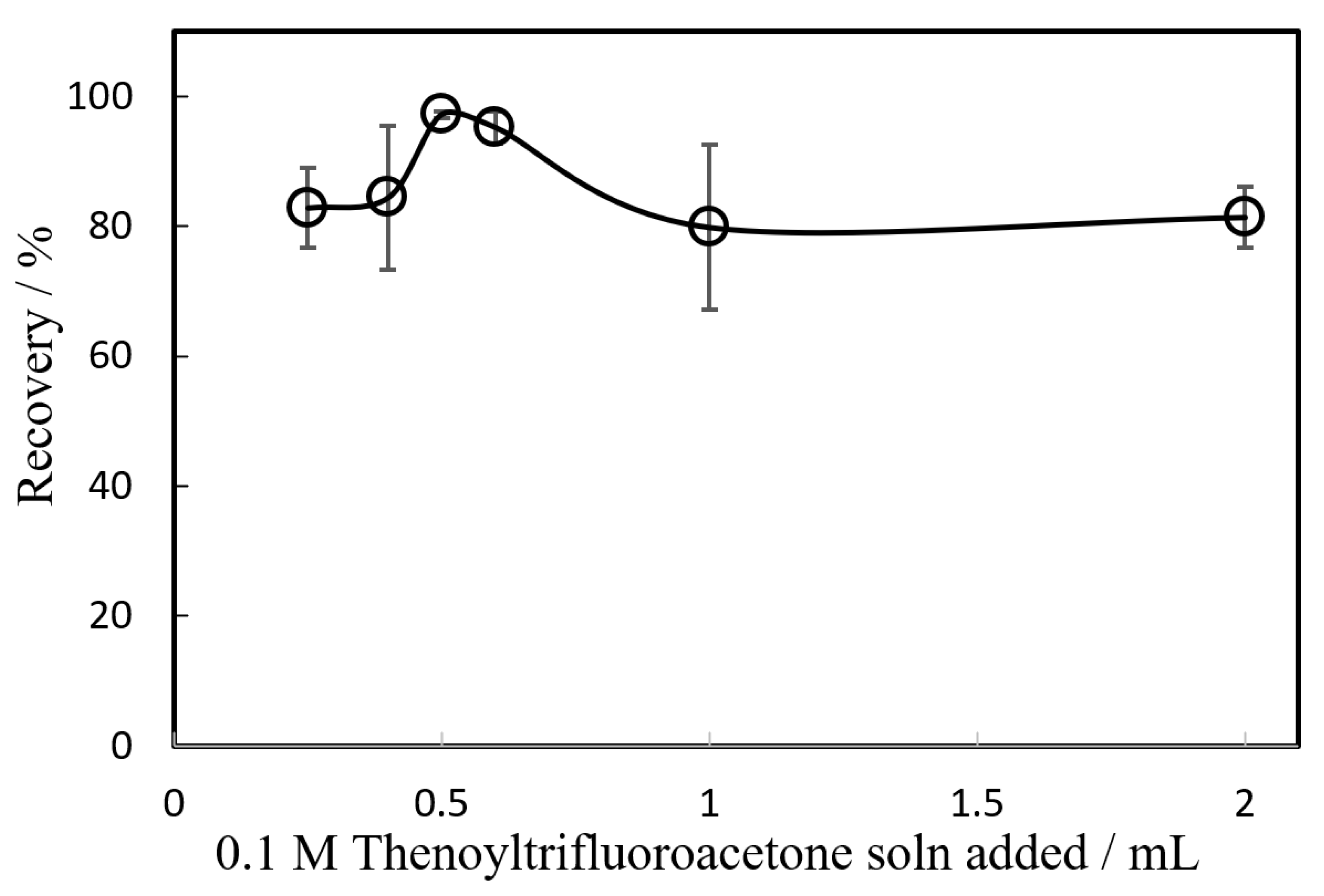
The optimal amount of the chelating reagent HTTA for nickel was investigated (
Figure 2). The amount of the HTTA acetone solution added was changed from 0.25 to 2.0 mL. The highest recovery was observed at 0.5 mL, and under these experimental conditions, TTA was present tens of thousands of times more moles than nickel. The decrease in recovery with increasing amounts of the HTTA acetone may be due to the slight solubilisation of the IAP in the presence of acetone. Thus, in this study, the addition of 0.5 mL HTTA acetone solution was recommended.
3.2. Ion-Associate Phase (IAP) Microextraction
At pH 8, where Ni is extracted into IAP, from the p
Ka value, thenoyltrifluoroacetone is mostly (98%) present as TTA
−, pH buffer component is about 80% as PS
− and the remaining 20% as PS
2−. In the proposed IAP microextraction method, TTA
− (the chelating reagent) and PS
− (the component of the pH buffer) act as the constituent ions (OA
−) of the IAP. When Q
+ is added, the IAP is formed. Therefore, the number of different reagents to be added can be reduced. The C
12BzDMA
+, C
14BzDMA
+, and Ben
+ were investigated as possible Q
+ constituents of the IAP (
Figure 3). C
12BzDMA
+ was chosen as the Q
+ because of the recovery of nickel was highest for C
12BzDMA
+.
By adding C
12BzDMA
+, the IAP was formed by Equations (5)–(7),
where subscript iap refers to the IAP.
The quantity of 0.1 M C
12BzDMA
+ solution added as a Q
+, a constituent of the IAP, was determined (
Figure 4). When the quantity of 0.1 M C
12BzDMA
+ solution added was 0.3–0.5 mL, the recovery of nickel was the highest. Thus, the optimal quantity of added Q
+ was 0.4 mL.
TTA
− forms a valence-saturated chelate, Ni(TTA)
2·2H
2O, with nickel(I1) but this chelate cannot be extracted into inert solvents such as 1,2-dichloroethane unless there is a synergistic effect [
22,
26]. However, Noriki reported that in the presence of quaternary ammonium ions (Q
+) such as C
14BzDMA
+, the nickel(II)-TTA chelate can be extracted into 1,2-dichloroethane as [Ni(TTA)
3 ]
−·Q
+ [
22,
26]. Sekine et al. reported that tetrabutylammonium ion (tba
+) enhanced the extraction of Ni-TTA complex into chloroform by forming ternary complex, [Ni(TTA)
3]
−·tba
+ [
22]. These suggest that nickel is extracted in the form of C
12BzDMA
+·[Ni(TTA)
3]
− rather than Ni(TTA)
2 in this IAP microextraction system and that C
12BzDMA
+ is not only a constituent of IAP, but also facilitates the extraction of Ni-TTA complex. The Ni-TTA chelate is extracted into IAP as follows.
Nickel chelates with TTA−; the addition of C12BzDMA+ causes the formation of an ion associate with TTA− and the extraction of the Ni- TTA− complex into the IAP. When nitric acid is added, nickel is extracted into the nitric acid phase by reverse microextraction.
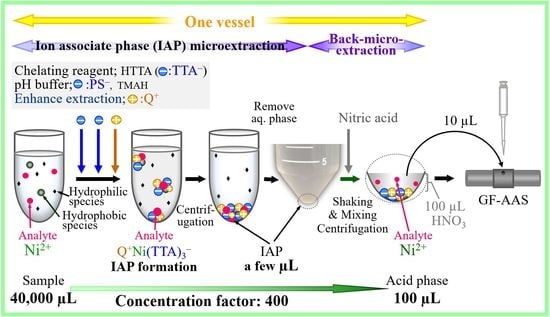
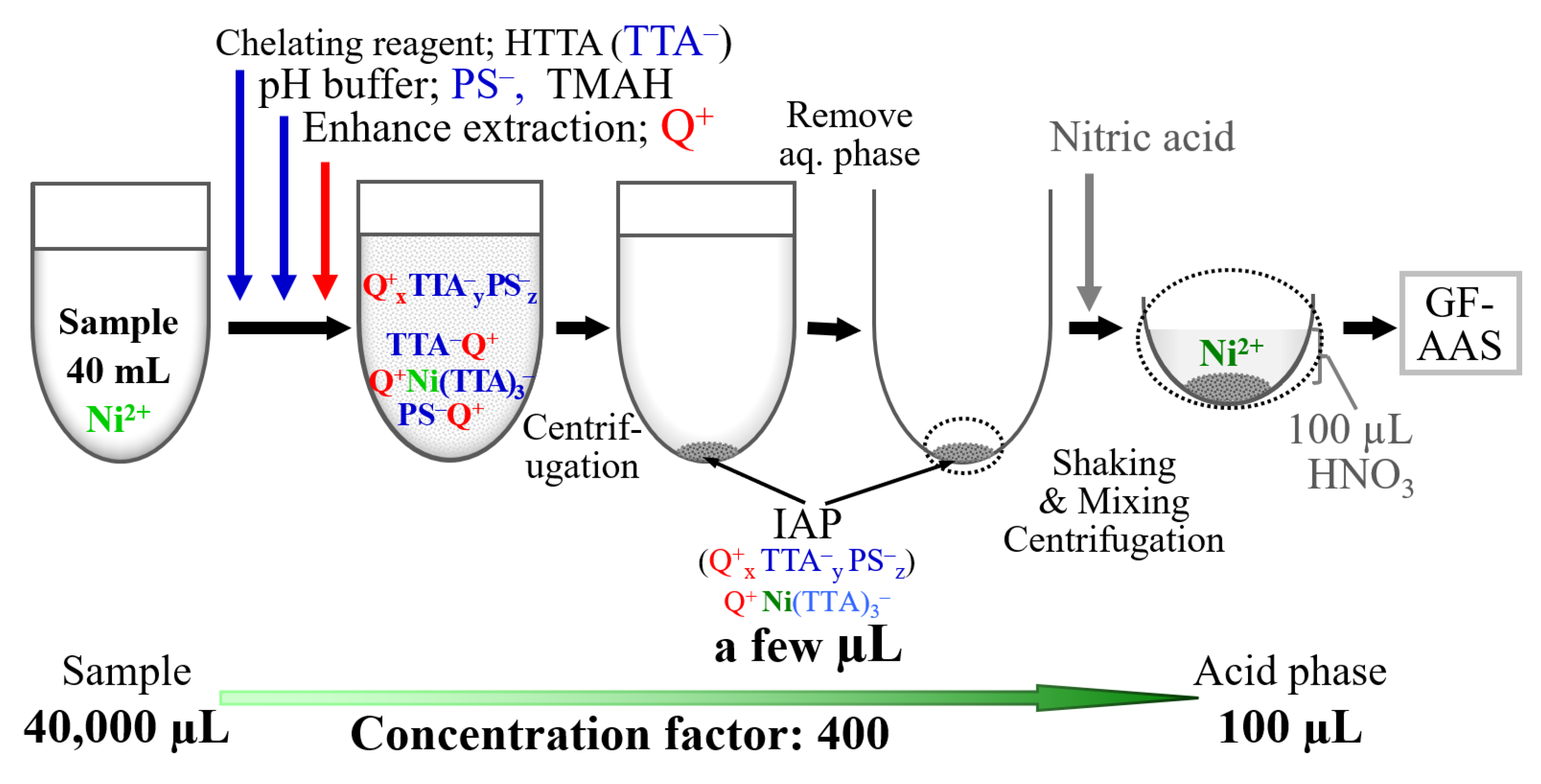
The illustration of IAP microextraction/back-microextraction system for nickel in the proposed system were shown in
Figure 5. When TTA
− and PS
− are added, the Ni-TTA complex is formed, then when C
12BzDMA
+ is added, IAP is formed and the Ni complex is extracted into the IAP.
Assuming no interaction between each compound, the composition of the IAP was determined from the absorption spectrum. The results showed that TTA
− and PS
− were present in almost equal concentrations in the IAP before back-microextraction. In addition, thenoyltrifluoroacetone in IAP was mostly present as TTA
− before back-microextraction and mostly present as HTTA after back-microextraction. To balance the charge. tetramethylammonium ion (TMA
+) may also constitute the IAP. In other words, the formation of IAP is presumed to be in Equation (9), not Equations (5)–(7),
where m and
n were approximately equal, if we assume that the interactions between compounds in the UV spectrum are negligible. The volume of the IAP was visually measured and found to be less than 10 µL. If the volume is less than 10 µL, the concentration of TTA and PS in the IAP would be more than 1 M each. Ni was extracted into the nitric acid phase.
3.3. Back-Microextraction
Nickel was back-microextracted from the IAP that extracted the nickel complex into 2 M nitric acid solution containing phosphate (100 µL), and then analysed by GF-AAS. Nickel extracted into the IAP is back-microextracted into the acid solution by adding acid (HX), as shown in the following equation.
In previous studies [
19,
20], nitric acid has been chosen as the acid of choice for the back-extraction of metal ions from IAP. As the nitrate ion is a moderately large ion, it can extract metals quantitatively and do not form insoluble compounds with the organic cations that consist of the IAP. A nitric acid solution is the preferred solution for GF-AAS measurements. Therefore, nitric acid was chosen qua the optimal acid for back-microextraction of nickel from the IAP.
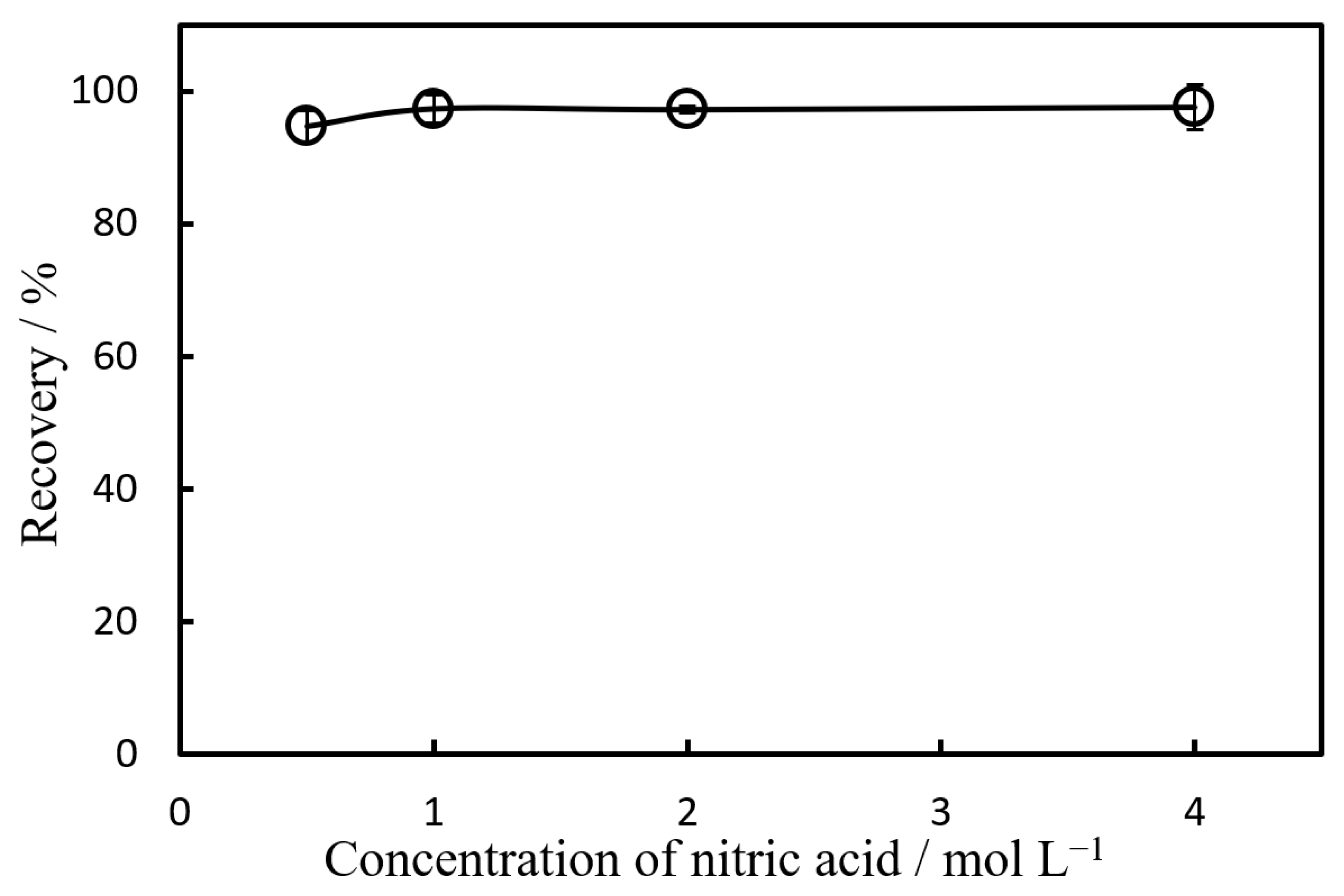
The effect of the concentration of nitric acid on the quantitative back-microextraction of nickel was investigated. The results are shown in
Figure 6; with nitric acid concentrations of 1.0 M or higher, nickel was quantitatively back-microextracted into the nitric acid phase. Therefore, we used a nitric acid concentration of 2.0 M qua the optimal solvent for back-microextraction.
In the IAP microextraction, the extraction of the analyte was instantaneous because of the formation of an ion associate from the aqueous phase; thus, shaking was not necessary. In contrast, back-microextraction often required shaking to facilitate sufficient distribution quickly because of the involvement of two phases, the IAP and the nitric acid phase. In the proposed method, shaking and mixing were considered for quantitative back-extraction. The shaking time for back-extraction into the nitric acid solution was investigated (
Figure 7). The maximum absorbance was obtained at a shaking time of 2–4 min in the test tube mixer. Shaking and mixing were chosen for 2–4 min using a test tube mixer.
The nickel-extracted IAP was treated by two methods: one was back-microextraction with acid (the proposed method), and the other was dissolution in an organic solvent (see Ref. [
17]), and the results measured by GF-AAS were compared. When nickel was back-microextracted from the IAP with the nitric acid solution, the sensitivity was about 1.5 times higher than when IAP was dissolved in an organic solvent (100 μL of 2-methoxyethanol). The detection limits for the dissolution and back- microextraction methods were 0.03 µg L
−1 and 0.02 µg L
−1, respectively. These results indicate that the acid back-microextraction method (this method) is effective than the dissolution method by 2-methoxyethanol for GF-AAS-based determination of nickel.
3.4. Optimized Procedure
We placed 40 mL of the sample solution (acidified with HNO3 to 0.01 M) into a 50 mL plastic centrifuge tube. Next, 0.5 mL of 0.1 M HTTA acetone solution was added as a chelating reagent and an OA−. Then, 2 mL of a p-phenolsulfonate solution (0.5 M) was added and pH 8 was adjusted with 12.5% TMAH solution to form a nickel complex. Subsequently, 0.4 mL of the C12BzDMA+ solution (0.1 M) was added to form the IAP and the chelating complex was extracted. The aqueous phase was then discarded after centrifugation at 4500 rpm for 15 min. When the amount of remaining water was reduced, the aqueous phase was aspirated with a syringe.
Next, 100 µL of 2 M nitric acid solution containing ammonium dihydrogen phosphate was added to the IAP after the extraction of the nickel chelate to back-microextract the nickel ion into the nitric acid phase (upper phase). The solution was shaken with a test tube mixer for 2–4 min and then centrifuged at 2500 rpm for 10 min. The concentration of nickel in the acid phase was then measured via GF-AAS.
3.5. Analytical Figure of Merit
The calibration graph of Ni by proposed method was linear with R2 values 0.9986 over the range of 0.1–2.0 µg L−1.
The sample solution was concentrated from 40 mL to 100 µL of nitric acid solution, so the enrichment factor was 400 times. The calibration curve obtained by 400-fold enrichment using the proposed method was compared with the calibration curve without enrichment. Comparison of the slopes of the calibration curves showed that 97% of the nickel was recovered by this IAP microextraction/back-microextraction. Based on this recovery, the actual enrichment factor was 390 times.
The limits of detection (3σb) and the limits of quantification (10σb) on two different days (5 replicates each) were 0.017 µg L−1 and 0.056 µg L−1, and 0.031 µg L−1 and 0.10 µg L−1, respectively.
The relative standard deviation (RSD) was 4.5% at 0.1 µg L−1, 7.1% at 0.5 µg L−1, and 3.3% at 1.0 µg L−1 (3 replicates each).
3.6. Application to River Water and Seawater Samples
The proposed method was applied to real environmental water samples. River water was collected from the bridges closest to the mouths of the Jinzu and Joganji rivers, which flow into Toyama Bay in the Japan Sea, and seawater was collected from Toyama Bay in the Japan Sea. The acidity of all three real water samples was adjusted to 0.01 M nitric acidity by adding 0.6 mL of concentrated nitric acid to 1 L of the sample. IAP microextraction/back-microextraction was performed according to the optimized procedure. The nickel concentrations in the water samples were 0.24 µg L
−1 in the Jinzu River, 0.05 µg L
−1 in the Joganji River, and 0.13 µg L
−1 in the seawater, respectively. The standard addition method was performed for the river water samples and compared with the calibration curve for the standard solution without river water (
Figure 8). The calibration curves obtained by adding the standard solution to the Jinzu River and the Joganji River were almost identical to the slope of the calibration curve of the standard solution.
Spike recovery tests were performed at concentrations of 0.13 and 0.25 µg L
−1 using the seawater sample (
Table 1). Nickel at the submicrogram per liter level could be recovered quantitatively from the seawater sample. The quantitative recovery of nickel was achieved from the Toyama Bay seawater. Moreover, nickel at the submicrogram per liter level could be recovered quantitatively from the seawater sample. Based on these results, we confirmed that nickel can be extracted effectively without being affected by the components contained in seawater.
The proposed method was also applied for the detection of certified reference materials (NASS 3) in seawater. The nickel concentration in the reference sample was 0.257 ± 0.027 µg L−1 and that detected through the proposed method was 0.244 ± 0.014 µg L−1 (mean ± standard deviation, number of replicates: 3). The nickel concentration detected by the proposed method was in good agreement with the certified value.
4. Conclusions
A novel system for the enrichment and separation of nickel in environmental water based on IAP microextraction/back-microextraction was developed and applied to measurements via GF-AAS. Nickel at submicrogram per liter levels was successfully detected in river water and seawater samples.
As with dipivaloylmethane for lithium [
20], β-diketones chosen for this study, 2-thenoyltrifluoroacetone (HTTA), were found to act not only as a chelating reagent but also as one of the organic anion in the IAP. In this study, TTA
− and phenolsulphonate ions played a double role: pH buffer and one of the organic anion in the IAP. In addition, the benzyldodecyldimethylammonium ion (C
12BzDMA
+) was involved in the extraction of nickel complexes (C
12BzDMA
+·[Ni(TTA)
3]
−) as well as served as a component of the IAP, i.e., the organic cation.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}