
Energy Metabolism as a Therapeutic Target in Cancer: The Role of Coenzyme Q10

 and
and 
Abstract
:1. Introduction
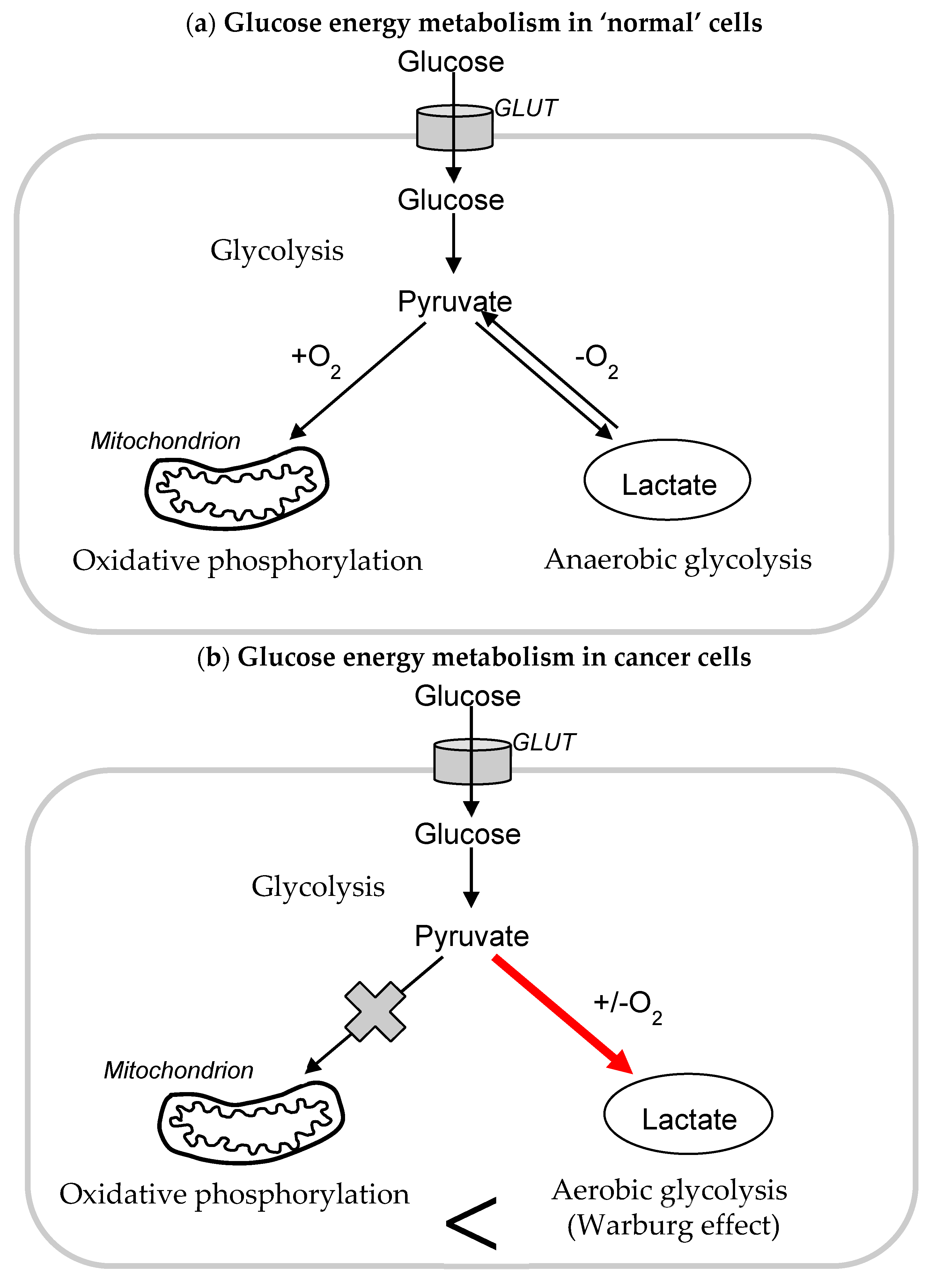
1.1. Glycolysis and Cancer
1.2. Mitochondria and Cancer
1.3. Mitochondria as a Target for Cancer Therapy
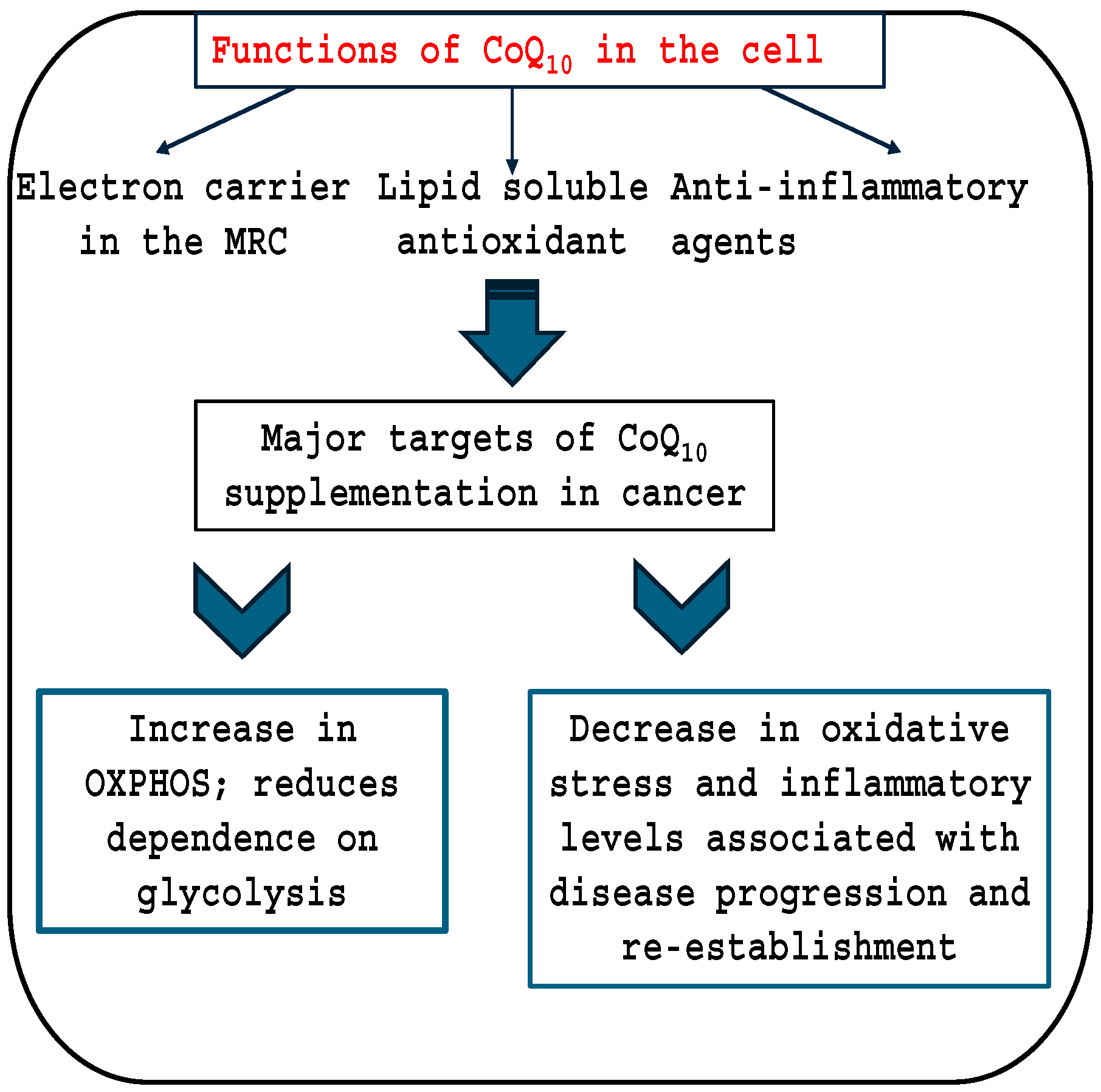
1.4. Coenzyme Q10 and Cancer
2. OXPHOS; Oxidative Phosphorylation
2.1. Clinical Studies
2.2. Studies in Animal Models
2.3. Cell Culture Studies
2.4. CoQ10 and the Prevention of Chemotherapy Side-Effects
2.5. Conclusions
Funding
Conflicts of Interest
References
- Orang, A.V.; Petersen, J.; McKinnon, R.A.; Michael, M.Z. Micromanaging aerobic respiration and glycolysis in cancer cells. Mol. Metab. 2019, 23, 98–126. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Duan, Z.; Li, Z.; Ge, F.; Wei, R.; Kong, L. The significance of glycolysis in tumor progression and its relationship with the tumor microenvironment. Front. Pharmacol. 2022, 13, 1091779. [Google Scholar] [CrossRef] [PubMed]
- Desideri, E.; Ciccarone, F.; Ciriolo, M.R. Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy. Nutrients 2019, 11, 1926. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Lanaspa, M.A.; Millan, I.S.; Fini, M.; Rivard, C.J.; Sanchez-Lozada, L.G.; Andres-Hernando, A.; Tolan, D.R.; Johnson, R.J. Fructose contributes to the Warburg effect for cancer growth. Cancer Metab. 2020, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Chelakkot, V.S.; Shin, Y.; Song, K. Modulating glycolysis to improve cancer therapy. Int. J. Mol. Sci. 2023, 24, 2606. [Google Scholar] [CrossRef]
- Sun, X.; Fan, T.; Sun, G.; Zhou, Y.; Huang, Y.; Zhang, N.; Zhao, L.; Zhong, R.; Peng, Y. 2-Deoxy-D-glucose increases the sensitivity of glioblastoma cells to BCNU through the regulation of glycolysis, ROS and ERS pathways: In vitro and in vivo validation. Biochem. Pharmacol. 2022, 199, 115029. [Google Scholar] [CrossRef]
- Zhang, Z.; Rohweder, P.J.; Ongpipattanakul, C.; Basu, K.; Bohn, M.-F.; Dugan, E.J.; Steri, V.; Hann, B.; Shokat, K.M.; Craik, C.S. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell 2022, 40, 1060–1069.e7. [Google Scholar] [CrossRef]
- Harrington, J.S.; Ryter, S.W.; Plataki, M.; Price, D.R.; Choi, A.M.K. Mitochondria in health, disease, and aging. Physiol. Rev. 2023, 103, 2349–2422. [Google Scholar] [CrossRef]
- Patel, P.S.; Castelow, C.; Patel, D.S.; Bhattacharya, S.K.; Kuscu, C.; Kuscu, C.; Makowski, L.; Eason, J.D.; Bajwa, A. Mitochondrial role in oncogenesis and potential chemotherapeutic strategy of mitochondrial infusion in breast cancer. Int. J. Mol. Sci. 2022, 23, 12993. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Bai, R.; Cui, J. Mitochondrial immune regulation and anti-tumor immunotherapy strategies targeting mitochondria. Cancer Lett. 2023, 564, 216223. [Google Scholar] [CrossRef] [PubMed]
- Bazhin, A.V. Mitochondria and cancer. Cancers 2020, 12, 2641. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sunkel, B.; Wang, M.; Kang, S.; Wang, T.; Gnanaprakasam, J.N.R.; Liu, L.; Cassel, T.A.; Scott, D.A.; Muñoz-Cabello, A.M.; et al. Succinate dehydrogenase/complex II is critical for metabolic and epigenetic regulation of T cell proliferation and inflammation. Sci. Immunol. 2022, 7, eabm8161. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 2008, 129, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and mitochondrial ROS in cancer: Novel targets for anticancer therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Wilson, M.H.; Wright, M.H. Redox regulation of cell proliferation: Bioinformatics and redox proteomics approaches to identify redox-sensitive cell cycle regulators. Free Radic. Biol. Med. 2018, 122, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Dadali, T.; Diers, A.R.; Kazerounian, S.; Muthuswamy, S.K.; Awate, P.; Ng, R.; Mogre, S.; Spencer, C.; Krumova, K.; Rockwell, H.E.; et al. Elevated levels of mitochondrial CoQ10 induce ROS-mediated apoptosis in pancreatic cancer. Sci. Rep. 2021, 11, 5749. [Google Scholar] [CrossRef] [PubMed]
- Eniafe, J.; Jiang, S. The functional roles of TCA cycle metabolites in cancer. Oncogene 2021, 40, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, e02935. [Google Scholar] [CrossRef] [PubMed]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Dyason, J.C.; Freeman, R.; Dong, L.-F.; Prochazka, L.; Wang, X.-F.; Scheffler, I.; Ralph, S.J. Mitocans as anti-cancer agents targeting mitochondria: Lessons from studies with vitamin E analogues, inhibitors of complex II. J. Bioenerg. Biomembr. 2007, 39, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Dong, L.F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef]
- Villa-Ruano, N.; Anaya-Ruiz, M.; Villafaña-Diaz, L.; Barron-Villaverde, D.; Perez-Santos, M. Drug repurposing of Mito-Atovaquone for cancer treatment. Pharm. Pat. Anal. 2023, 12, 143–149. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Xu, Z.; Huang, Z.; Tang, Y.; Yang, D.; Huang, J.; He, L.; Liu, M.; Chen, Z.; Teng, Y. CPI-613 rewires lipid metabolism to enhance pancreatic cancer apoptosis via the AMPK-ACC signaling. Exp. Clin. Cancer Res. 2020, 39, 73. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Zhou, C.; Lu, L.; Liu, B.; Ding, Y. Elesclomol: A copper ionophore targeting mitochondrial metabolism for cancer therapy. J. Exp. Clin. Cancer Res. 2022, 41, 271. [Google Scholar] [CrossRef]
- Belzacq, A.S.; El Hamel, C.; Vieira, H.L.A.; Cohen, I.; Haouzi, D.; Métivier, D.; Marchetti, P.; Brenner, C.; Kroemer, G. Adenine nucleotide translocator mediates the mitochondrial membrane permeabilization induced by lonidamine, arsenite and CD437. Oncogene 2001, 20, 7579–7587. [Google Scholar] [CrossRef]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.-C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys. Acta 2016, 1866, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Madak, J.T.; Bankhead, A.; Cuthbertson, C.R.; Showalter, H.D.; Neamati, N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol. Ther. 2019, 195, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, V.G.; Yuvaraj, S.; Vijayasarathy, K.; Gangadaran, S.G.D.; Sachdanandam, P. Effect of coenzyme Q10, riboflavin and niacin on serum CEA and CA 15-3 levels in breast cancer patients undergoing tamoxifen therapy. Biol. Pharm. Bull. 2007, 30, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.T.; Huang, Y.C.; Cheng, S.-B.; Huang, Y.-T.; Lin, P.-T. Effects of coenzyme Q10 supplementation on antioxidant capacity and inflammation in hepatocellular carcinoma patients after surgery: A randomized, placebo-controlled trial. Nutr. J. 2016, 15, 85. [Google Scholar] [CrossRef]
- Takaki, A.; Yamamoto, K. Control of oxidative stress in hepatocellular carcinoma: Helpful or harmful? World J. Hepatol. 2015, 7, 968–979. [Google Scholar] [CrossRef]
- Zahrooni, N.; Hosseini, S.A.; Ahmadzadeh, A.; Angali, K.A.; Assarehzadegan, M.A. The Effect of Coenzyme Q10 Supplementation on Vascular Endothelial Growth Factor and Serum Levels of Interleukin 6 and 8 in Women with Breast Cancer: A Double-Blind, Placebo-Controlled, Randomized Clinical Trial. Ther. Clin. Risk Manag. 2019, 15, 1403–1410. [Google Scholar] [CrossRef]
- Iarussi, D.; Auricchio, U.; Agretto, A.; Murano, A.; Giuliano, M.; Casale, F.; Indolfi, P.; Iacono, A. Protective effect of coenzyme Q10 on anthracyclines cardiotoxicity: Control study in children with acute lymphoblastic leukemia and non-Hodgkin lymphoma. Mol. Asp. Med. 1994, 15 (Suppl. S1), s207–s212. [Google Scholar] [CrossRef]
- Alimohammadi, M.; Rahimi, A.; Faramarzi, F.; Golpour, M.; Jafari-Shakib, R.; Alizadeh-Navaei, R.; Rafiei, A. Effects of coenzyme Q10 supplementation on inflammation, angiogenesis, and oxidative stress in breast cancer patients: A systematic review and meta-analysis of randomized controlled- trials. Inflammopharmacology 2021, 29, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Lesser, G.J.; Case, D.; Stark, N.; Williford, S.; Giguere, J.; Garino, L.A.; Naughton, M.J.; Vitolins, M.Z.; Lively, M.O.; Shaw, E.G. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J. Support. Oncol. 2013, 11, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.; Kawaguchi, T.; Yotsumoto, D.; Doi, T.; Miyara, K.; Odagiri, H.; Kitamura, K.; Ariyoshi, K.; Miyaji, T.; Ishiki, H.; et al. Efficacy and safety of an amino acid jelly containing coenzyme Q10 and L-carnitine in controlling fatigue in breast cancer patients receiving chemotherapy: A multi-institutional, randomized, exploratory trial (JORTC-CAM01). Support. Care Cancer 2016, 24, 637–646. [Google Scholar] [CrossRef]
- Hoenjet, K.M.; Dagnelie, P.C.; Delaere, K.; Wijckmans, N.; Zambon, J.; Oosterhof, G. Effect of a nutritional supplement containing vitamin E, selenium, vitamin c and coenzyme Q10 on serum PSA in patients with hormonally untreated carcinoma of the prostate: A randomised placebo-controlled study. Eur. Urol. 2005, 47, 433–439. [Google Scholar] [CrossRef]
- Lockwood, K.; Moesgaard, S.; Hanioka, T.; Folkers, K. Apparent partial remission of breast cancer in ‘high risk’ patients supplemented with nutritional antioxidants, essential fatty acids and coenzyme Q10. Mol. Asp. Med. 1994, 15 (Suppl. S1), s231–s240. [Google Scholar] [CrossRef] [PubMed]
- Hertz, N.; Lister, R.E. Improved survival in patients with end-stage cancer treated with coenzyme Q10 and other antioxidants: A pilot study. J. Int. Med. Res. 2009, 37, 1961–1971. [Google Scholar] [CrossRef]
- Akagi, J.; Baba, H. Hydrogen gas activates coenzyme Q10 to restore exhausted CD8+ T cells, especially PD-1+Tim3+terminal CD8+ T cells, leading to better nivolumab outcomes in patients with lung cancer. Oncol. Lett. 2020, 20, 258. [Google Scholar] [CrossRef]
- Rusciani, L.; Proietti, I.; Paradisi, A.; Rusciani, A.; Guerriero, G.; Mammone, A.; De Gaetano, A.; Lippa, S. Recombinant interferon alpha-2b and coenzyme Q10 as a postsurgical adjuvant therapy for melanoma: A 3-year trial with recombinant interferon-alpha and 5-year follow-up. Melanoma Res. 2007, 17, 177–183. [Google Scholar] [CrossRef]
- Folkers, K. Relevance of the biosynthesis of coenzyme Q10 and of the four bases of DNA as a rationale for the molecular causes of cancer and a therapy. Biochem. Biophys. Res. Commun. 1996, 224, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Folkers, K.; Osterborg, A.; Nylander, M.; Morita, M.; Mellstedt, H. Activities of vitamin Q10 in animal models and a serious deficiency in patients with cancer. Biochem. Biophys. Res. Commun. 1997, 234, 296–299. [Google Scholar] [CrossRef]
- Palan, P.R.; Mikhail, M.S.; Shaban, D.W.; Romney, S.L. Plasma concentrations of coenzyme Q10 and tocopherols in cervical intraepithelial neoplasia and cervical cancer. Eur. J. Cancer Prev. 2003, 12, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Cobanoglu, U.; Demir, H. Lipid peroxidation, DNA damage and coenzyme Q10 in lung cancer patients--markers for risk assessment? Asian Pac. J. Cancer Prev. 2011, 12, 1399–1403. [Google Scholar] [PubMed]
- Chan, I.S.; Knútsdóttir, H.; Ramakrishnan, G.; Padmanaban, V.; Warrier, M.; Ramirez, J.C.; Dunworth, M.; Zhang, H.; Jaffee, E.M.; Bader, J.S.; et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J. Cell Biol. 2020, 219, e202001134. [Google Scholar] [CrossRef] [PubMed]
- Shidal, C.; Yoon, H.S.; Zheng, W.; Wu, J.; Franke, A.A.; Blot, W.J.; Shu, X.; Cai, Q. Prospective study of plasma levels of coenzyme Q10 and lung cancer risk in a low-income population in the Southeastern United States. Cancer Med. 2021, 10, 1439–1447. [Google Scholar] [CrossRef]
- Cooney, R.V.; Dai, Q.; Gao, Y.-T.; Chow, W.-H.; Franke, A.A.; Shu, X.-O.; Li, H.; Ji, B.; Cai, Q.; Chai, W.; et al. Low plasma coenzyme Q10 levels and breast cancer risk in Chinese women. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1124–1130. [Google Scholar] [CrossRef]
- Portakal, O.; Özkaya, Ö.; Erden Inal, M.; Bozan, B.; Koşan, M.; Sayek, I. Coenzyme Q10 concentrations and antioxidant status in tissues of breast cancer patients. Clin. Biochem. 2000, 33, 279–284. [Google Scholar] [CrossRef]
- Jolliet, P.; Simon, N.; Barré, J.; Pons, J.Y.; Boukef, M.; Paniel, B.J.; Tillement, J.P. Plasma coenzyme Q10 concentrations in breast cancer: Prognosis and therapeutic consequences. Int. J. Clin. Pharmacol. Ther. 1998, 36, 506–509. [Google Scholar]
- Yen, H.C.; Chen, B.S.; Yang, S.-L.; Wu, S.-Y.; Chang, C.-W.; Wei, K.-C.; Hsu, J.-C.; Hsu, Y.-H.; Yen, T.-H.; Lin, C.-L. Levels of Coenzyme Q10 and Several COQ Proteins in Human Astrocytoma Tissues Are Inversely Correlated with Malignancy. Biomolecules 2022, 12, 336. [Google Scholar] [CrossRef]
- Kanda, M.; Nomoto, S.; Oya, H.; Hashimoto, R.; Takami, H.; Shimizu, D.; Sonohara, F.; Kobayashi, D.; Tanaka, C.; Yamada, S.; et al. Decreased expression of prenyl diphosphate synthase subunit 2 correlates with reduced survival of patients with gastric cancer. J. Exp. Clin. Cancer Res. 2014, 33, 88. [Google Scholar] [CrossRef]
- Chai, W.; Cooney, R.V.; Franke, A.A.; Shvetsov, Y.B.; Caberto, C.P.; Wilkens, L.R.; Le Marchand, L.; Henderson, B.E.; Kolonel, L.N.; Goodman, M.T. Plasma coenzyme Q10 levels and postmenopausal breast cancer risk: The multiethnic cohort study. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2351–2356. [Google Scholar] [CrossRef]
- El-Attar, E.; Kamel, A.; Karmouty, A.; Wehida, N.; Nassra, R.; El Nemr, M.; Kandil, N.S. Assessment of Serum CoQ10 Levels and other Antioxidant Markers in Breast Cancer. Asian Pac. J. Cancer Prev. 2020, 21, 465–471. [Google Scholar] [CrossRef]
- Ambrosone, C.B.; Zirpoli, G.R.; Hutson, A.D.; McCann, W.E.; McCann, S.E.; Barlow, W.E.; Kelly, K.M.; Cannioto, R.; Sucheston-Campbell, L.E.; Hershman, D.L.; et al. Dietary Supplement Use During Chemotherapy and Survival Outcomes of Patients with Breast Cancer Enrolled in a Cooperative Group Clinical Trial (SWOG S0221). J. Clin. Oncol. 2020, 38, 804–814. [Google Scholar] [CrossRef]
- Mohamed, H.A.; Said, R.S. Coenzyme Q10 attenuates inflammation and fibrosis implicated in radiation enteropathy through suppression of NF-kB/TGF-β/MMP-9 pathways. Int. Immunopharmacol. 2021, 92, 107347. [Google Scholar] [CrossRef]
- Ki, Y.; Kim, W.; Kim, Y.H.; Kim, D.; Bae, J.S.; Park, D.; Jeon, H.; Lee, J.H.; Lee, J.; Nam, J. Effect of Coenzyme Q10 on Radiation Nephropathy in Rats. J. Korean Med. Sci. 2017, 32, 757–763. [Google Scholar] [CrossRef]
- Abdel-Latif, M.; Saidan, S.; Morsy, B.M. Coenzyme Q10 attenuates rat hepatocarcinogenesis via the reduction of CD59 expression and phospholipase D activity. Cell Biochem. Funct. 2020, 38, 490–499. [Google Scholar] [CrossRef]
- Kokawa, T.; Shiota, K.; Oda, K.; Okubo, S.; Okamoto, Y.; Okubo, H. Coenzyme Q10 in cancer chemotherapy--experimental studies on augmentation of the effects of masked compounds, especially in the combined chemotherapy with immunopotentiators. Gan Kagaku Ryoho 1983, 10, 768–774. [Google Scholar]
- Perumal, S.S.; Shanthi, P.; Sachdanandam, P. Combined efficacy of tamoxifen and coenzyme Q10 on the status of lipid peroxidation and antioxidants in DMBA induced breast cancer. Mol. Cell. Biochem. 2005, 273, 151–160. [Google Scholar] [CrossRef]
- Burlaka, A.P.; Ganusevich, I.; Golotiuk, V.V.; Vovk, A.V.; Lukin, S.M. Superoxide- and NO-dependent mechanisms of antitumor and antimetastatic effect of L-arginine hydrochloride and coenzyme Q10. Exp. Oncol. 2016, 38, 31–35. [Google Scholar] [CrossRef]
- Sakano, K.; Takahashi, M.; Kitano, M.; Sugimura, T.; Wakabayashi, K. Suppression of azoxymethane-induced colonic premalignant lesion formation by coenzyme Q10 in rats. Asian Pac. J. Cancer Prev. 2006, 7, 599–603. [Google Scholar] [PubMed]
- Frontiñán-Rubio, J.; Llanos-González, E.; García-Carpintero, S.; Peinado, J.R.; Ballesteros-Yáñez, I.; Rayo, M.V.; de la Fuente, J.; Pérez-García, V.M.; Perez-Romasanta, L.A.; Malumbres, M.; et al. CoQ10 reduces glioblastoma growth and infiltration through proteome remodeling and inhibition of angiogenesis and inflammation. Cell. Oncol. 2023, 46, 65–77. [Google Scholar] [CrossRef]
- Sun, J.; Patel, C.B.; Jang, T.; Merchant, M.; Chen, C.; Kazerounian, S.; Diers, A.R.; Kiebish, M.A.; Vishnudas, V.K.; Gesta, S.; et al. High levels of ubidecarenone (oxidized CoQ10) delivered using a drug-lipid conjugate nanodispersion (BPM31510) differentially affect redox status and growth in malignant glioma versus non-tumor cells. Sci. Rep. 2020, 10, 13899. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Patra, D.; Ghosh, P.; Banerjee, S.; Mishra, S.; Chakraborty, P.; Chowdhury, K.D.; Basu, A.; Sadhukhan, G.C. Protective role of Decylubiquinone against secondary melanoma at lung in B16F10 induced mice by reducing E-cadherin expression and ameliorating ROCKII-Limk1/2-Cofiliin mediated metastasis. Cell. Signal. 2023, 101, 110486. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.L.; Quistorff, B.; Spang-Thomsen, M.; Kristjansen, P.E.G. Effect of radiation therapy on small-cell lung cancer is reduced by ubiquinone intake. Folia Microbiol. 1998, 43, 505–506. [Google Scholar] [CrossRef]
- Said, R.S.; Mohamed, H.A.; Kamal, M.M. Coenzyme Q10 mitigates ionizing radiation-induced testicular damage in rats through inhibition of oxidative stress and mitochondria-mediated apoptotic cell death. Toxicol. Appl. Pharmacol. 2019, 383, 114780. [Google Scholar] [CrossRef] [PubMed]
- Björkhem-Bergman, L.; Acimovic, J.; Torndal, U.-B.; Parini, P.; Eriksson, L.C. Lovastatin prevents carcinogenesis in a rat model for liver cancer. Effects of ubiquinone supplementation. Anticancer. Res. 2010, 30, 1105–1112. [Google Scholar] [PubMed]
- Quiles, J.L.; Farquharson, A.J.; Ramírez-Tortosa, M.C.; Grant, I.; Milne, L.; Huertas, J.R.; Battino, M.; Mataix, J.; Wahle, K.W.J. Coenzyme Q differentially modulates phospholipid hydroperoxide glutathione peroxidase gene expression and free radicals’ production in malignant and non-malignant prostate cells. Biofactors 2003, 18, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.N.; Wang, K.Y.; Zhang, X.-S.; Yang, C.; Li, X.-Y. 4-Hydroxybenzoic acid (4-HBA) enhances the sensitivity of human breast cancer cells to adriamycin as a specific HDAC6 inhibitor by promoting HIPK2/p53 pathway. Biochem. Biophys. Res. Commun. 2018, 504, 812–819. [Google Scholar] [CrossRef]
- Somers-Edgar, T.J.; Rosengren, R.J. Coenzyme Q0 induces apoptosis and modulates the cell cycle in estrogen receptor negative breast cancer cells. Anti-Cancer Drugs 2009, 20, 33–40. [Google Scholar] [CrossRef]
- Frontiñán-Rubio, J.; Santiago-Mora, R.M.; Nieva-Velasco, C.M.; Ferrín, G.; Martínez-González, A.; Gómez, M.V.; Moreno, M.; Ariza, J.; Lozano, E.; Arjona-Gutiérrez, J.; et al. Regulation of the oxidative balance with coenzyme Q10 sensitizes human glioblastoma cells to radiation and temozolomide. Radiother. Oncol. 2018, 128, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Gotorbe, C.; Durivault, J.; Meira, W.; Cassim, S.; Ždralević, M.; Pouysségur, J.; Vučetić, M. Metabolic Rewiring toward Oxidative Phosphorylation Disrupts Intrinsic Resistance to Ferroptosis of the Colon Adenocarcinoma Cells. Antioxidants 2022, 11, 2412. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Kawabata-Iwakawa, R.; Ida, S.; Mito, I.; Tada, H.; Chikamatsu, K. Upregulated glycolysis correlates with tumor progression and immune evasion in head and neck squamous cell carcinoma. Sci. Rep. 2021, 11, 17789. [Google Scholar] [CrossRef] [PubMed]
- Liparulo, I.; Bergamini, C.; Bortolus, M.; Calonghi, N.; Gasparre, G.; Kurelac, I.; Masin, L.; Rizzardi, N.; Rugolo, M.; Wang, W.; et al. Coenzyme Q biosynthesis inhibition induces HIF-1α stabilization and metabolic switch toward glycolysis. FEBS J. 2021, 288, 1956–1974. [Google Scholar] [CrossRef] [PubMed]
- Bahar, M.; Khaghani, S.; Pasalar, P.; Paknejad, M.; Khorramizadeh, M.R.; Mirmiranpour, H.; Nejad, S.G. Exogenous coenzyme Q10 modulates MMP-2 activity in MCF-7 cell line as a breast cancer cellular model. Nutr. J. 2010, 9, 62. [Google Scholar] [CrossRef]
- Hu, C.; Huang, Y.; Luo, P.; Yang, Y. Effect of antioxidants coenzyme Q10 and β-carotene on the cytotoxicity of vemurafenib against human malignant melanoma. Oncol. Lett. 2021, 21, 208. [Google Scholar] [CrossRef] [PubMed]
- Burić, S.S.; Podolski-Renić, A.; Dinić, J.; Stanković, T.; Jovanović, M.; Hadžić, S.; Ayuso, J.M.; Virumbrales-Muñoz, M.; Fernández, L.J.; Ochoa, I.; et al. Modulation of Antioxidant Potential with Coenzyme Q10 Suppressed Invasion of Temozolomide-Resistant Rat Glioma In Vitro and In Vivo. Oxid. Med. Cell Longev. 2019, 2019, 3061607. [Google Scholar] [CrossRef] [PubMed]
- Heidari-Kalvani, N.; Alizadeh-Fanalou, S.; Yarahmadi, S.; Fallah, S.; Alipourfard, I.; Farahmandian, N.; Barjesteh, F.; Bahreini, E. Investigation of the effects of catharanthine and Q10 on Nrf2 and its association with MMP-9, MRP1, and Bcl-2 and apoptosis in a model of hepatocellular carcinoma. Naunyn Schmiedebergs Arch. Pharmacol. 2023, 397, 2507–2522. [Google Scholar] [CrossRef]
- Jain, S.; Hu, C.; Kluza, J.; Ke, W.; Tian, G.; Giurgiu, M.; Bleilevens, A.; Campos, A.R.; Charbono, A.; Stickeler, E.; et al. Metabolic targeting of cancer by a ubiquinone uncompetitive inhibitor of mitochondrial complex I. Cell Chem. Biol. 2022, 29, 436–450.e15. [Google Scholar] [CrossRef]
- Brea-Calvo, G.; Rodríguez-Hernández, A.; Fernández-Ayala, D.J.; Navas, P.; Sánchez-Alcázar, J.A. Chemotherapy induces an increase in coenzyme Q10 levels in cancer cell lines. Free Radic. Biol. Med. 2006, 40, 1293–1302. [Google Scholar] [CrossRef]
- Zaal, E.A.; de Grooth, H.J.; Oudaert, I.; Langerhorst, P.; Levantovsky, S.; van Slobbe, G.J.J.; Jansen, J.W.A.; Menu, E.; Wu, W.; Berkers, C.R. Targeting coenzyme Q10 synthesis overcomes bortezomib resistance in multiple myeloma. Mol. Omics 2022, 18, 19–30. [Google Scholar] [CrossRef]
- McGregor, G.H.; Campbell, A.D.; Fey, S.K.; Tumanov, S.; Sumpton, D.; Blanco, G.R.; Mackay, G.; Nixon, C.; Vazquez, A.; Sansom, O.J.; et al. Targeting the Metabolic Response to Statin-Mediated Oxidative Stress Produces a Synergistic Antitumor Response. Cancer Res. 2020, 80, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Kaymak, I.; Maier, C.R.; Schmitz, W.; Campbell, A.D.; Dankworth, B.; Ade, C.P.; Walz, S.; Paauwe, M.; Kalogirou, C.; Marouf, H.; et al. Mevalonate Pathway Provides Ubiquinone to Maintain Pyrimidine Synthesis and Survival in p53-Deficient Cancer Cells Exposed to Metabolic Stress. Cancer Res. 2020, 80, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Li, W.; Liang, L.; Liu, S.; Yi, H.; Zhou, Y. FSP1: A key regulator of ferroptosis. Trends Mol. Med. 2023, 29, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Shin, D.; Roh, J.-L. Lipid metabolism alterations and ferroptosis in cancer: Paving the way for solving cancer resistance. Eur. J. Pharmacol. 2023, 941, 175497. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Huang, J.; Sun, W.; Wang, G.; Wu, Y.; Jiang, Z.; Lv, Y.; Wu, G.; Cao, J.; Liu, M.; et al. Zoledronic acid induces ferroptosis by reducing ubiquinone and promoting HMOX1 expression in osteosarcoma cells. Front. Pharmacol. 2023, 13, 1071946. [Google Scholar] [CrossRef] [PubMed]
- Cheu, J.W.; Lee, D.; Li, Q.; Goh, C.C.; Bao, M.H.-R.; Yuen, V.W.-H.; Zhang, M.S.; Yang, C.; Chan, C.Y.-K.; Tse, A.P.-W.; et al. Ferroptosis Suppressor Protein 1 Inhibition Promotes Tumor Ferroptosis and Anti-tumor Immune Responses in Liver Cancer. Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 133–159. [Google Scholar] [CrossRef]
- Arslanbaeva, L.; Tosi, G.; Ravazzolo, M.; Simonato, M.; Tucci, F.A.; Pece, S.; Cogo, P.; Santoro, M.M. UBIAD1 and CoQ10 protect melanoma cells from lipid peroxidation-mediated cell death. Redox Biol. 2022, 51, 102272. [Google Scholar] [CrossRef]
- Ortiz, T.; Villanueva-Paz, M.; Díaz-Parrado, E.; Illanes, M.; Fernández-Rodríguez, A.; Sánchez-Alcázar, J.A.; de Miguel, M. Amitriptyline down-regulates coenzyme Q10 biosynthesis in lung cancer cells. Eur. J. Pharmacol. 2017, 797, 75–82. [Google Scholar] [CrossRef]
- Yen, H.C.; Li, S.H.; Majima, H.J.; Huang, Y.-H.; Chen, C.-P.; Liu, C.-C.; Tu, Y.-C.; Chen, C.-W. Up-regulation of antioxidant enzymes and coenzyme Q(10) in a human oral cancer cell line with acquired bleomycin resistance. Free Radic. Res. 2011, 45, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Ma, L.; Li, Y.; Yang, J.; Yang, Q.; Yao, W.; Li, S. CoQ10 Improves Myocardial Damage in Doxorubicin-Induced Heart Failure in C57BL/6 Mice. Front. Biosci. 2022, 27, 244. [Google Scholar] [CrossRef] [PubMed]
- Folkers, K.; Liu, M.; Watanabe, T.; Porter, T.H. Inhibition by adriamycin of the mitochondrial biosynthesis of coenzyme Q10 and implication for the cardiotoxicity of adriamycin in cancer patients. Biochem. Biophys. Res. Commun. 1977, 77, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, Y.; Hansen, I.; Porter, T.H.; Folkers, K. Inhibition of coenzyme Q10-enzymes, succinoxidase and NADH-oxidase, by adriamycin and other quinones having antitumor activity. Biochem. Biophys. Res. Commun. 1974, 58, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Folkers, K. Prevention by coenzyme Q10 (NSC-140865) of the inhibition by adriamycin (NSC-123127) of coenzyme Q10 enzymes. Cancer Treat. Rep. 1976, 60, 223–224. [Google Scholar] [PubMed]
- Kishi, T.; Watanabe, T.; Folkers, K. Bioenergetics in clinical medicine: Prevention by forms of coenzyme Q of the inhibition by adriamycin of coenzyme Q10-enzymes in mitochondria of the myocardium. Proc. Natl. Acad. Sci. USA 1976, 73, 4653–4656. [Google Scholar] [CrossRef]
- HogenEsch, H.; Nikitin, A.Y. Challenges in pre-clinical testing of anti-cancer drugs in cell culture and in animal models. J. Control. Release 2012, 164, 183–186. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Reference | Cancer | Treatment | Outcome |
|---|---|---|---|
| Premkumar et al., 2007 [36] | Breast cancer (84 patients) | CoQ10 (100 mg/day) + riboflavin (10 mg/day) + niacin (50 mg/day) + tamoxifen (20 mg/day) for three months | Reduced the serum levels of tumour markers CEA and CA 15-3. |
| Liu et al., 2016 [37] | Hepatocellular cancer (41 patients) | CoQ10 (300 mg/day) for three months | Decreased the serum levels of oxidative stress and inflammation markers |
| Zahrooni et al., 2019 [39] | Breast cancer (30 patients and 29 controls) | CoQ10 (100 mg/day) for two months | Reduced the serum levels of inflammation markers IL-8 and IL-6. |
| Iarussi et al., 1994 [40] | Lymphoblastic leukemia or non-Hodgkin lymphoma (20 children) | CoQ10 (100 mg twice a day) for an unspecified period | Demonstrated the protective effects of CoQ on cardiac function during therapy with anthracyclines. |
| Alimohammadi et al., 2021 [41] | Breast cancer (pooled data from five eligible studies consisting of nine RCTs) | CoQ10 (100 mg/day) for 45–90 days | Reduced the levels of inflammatory markers and matrix metalloproteinases markers. |
| Lesser et al., 2013 [42] | Breast cancer (236 patients) | CoQ10 (300 mg/day) combined with vitamin E (300 IU/day), divided into three daily doses, for six months | Increased plasma CoQ10 levels but did not improve fatigue or QoL self-reports; there were no adverse effects on primary treatment. |
| Iwase et al., 2016 [43] | Breast cancer (59 patients undergoing chemotherapy) | Amino acid jelly Inner Power(®) containing CoQ10 and L-carnitine for three weeks | Significant differences between intervention and control groups in the worst level of fatigue, global fatigue scores, and current feelings of fatigue; there were no severe adverse effects. |
| Hoenjet et al., 2005 [44] | Prostate cancer (70 patients) | CoQ10 (100 mg twice a day), vitamin C (750 mg/day), vitamin E (350 mg/day), and selenium (200 mcg/day) for 21 weeks | No effect on serum levels of PSA or hormone levels in patients with hormonally untreated carcinoma of the prostate. |
| Lockwood et al., 1994 [45] | High-risk breast cancer (32 patients) | CoQ10 (90 mg/day), vitamin C (2850 mg/day), vitamin E (2500 IU/day), beta-carotene (32.5 IU/day), selenium (387 mcg/day), as well as secondary vitamins, minerals, and essential fatty acids as adjuvant treatment for 18 months | None of the patients died during the study period (the expected number was four); none of the patients showed signs of further distant metastases; quality of life was improved (no weight loss and reduced use of pain killers); six patients showed apparent partial remission. |
| Hertz and Lister, 2009 [46] | End-stage cancer, including breast, brain, lung, kidney, colon, pancreatic, skin, oesophagus, stomach, ovarian, and prostate (41 patients) | Daily doses of the following (divided into two administrations): 30 mg of CoQ10, 25,000 IU of vitamin A, 5.7 g of vitamin C, 1.625 g of vitamin E, 487 mcg of selenium, 5 mg of folic acid, and 76 mg of beta-carotene (not given to lung cancer patients) | Median predicted survival was 12 months; median actual survival was 17 months. Mean actual survival was 28.8 months versus 11.9 months for mean predicted survival. Treatments were very well tolerated with few adverse effects. |
| Akagi et al., 2020 [47] | Lung cancer (56 patients) | Lung cancer patients treated with nivolumab received hydrogen gas; hydrogen gas restored exhausted CD8+ T cells into active CD8+ T cells, possibly by activating mitochondria | Patients treated with hydrogen gas and nivolumab (n = 42) indicated a significantly longer overall survival compared with patients treated with nivolumab only (n = 14). |
| Rusciani et al., 2007 [48] | Stage I and II melanoma (small patient sample) | Low-dose recombinant interferon α-2b administered twice daily and CoQ10 (400 mg/day) for three years | Induced significantly decreased rates of recurrence and had negligible adverse effects. |
| Reference | Cancer | Results | Conclusions |
|---|---|---|---|
| Portakal et al., 2000 [56] | Breast cancer (21 patients with radical mastectomy) | Decreased CoQ10 concentrations in tumour tissues compared to the surrounding normal tissues; increased malondialdehyde levels in tumour tissues compared to non-cancerous tissues | Increased oxidative stress in malignant cells may cause CoQ10 consumption. The administration of oral CoQ10 may induce the protective effect of CoQ10 on breast tissue. |
| Jolliet et al., 1998 [57] | Breast cancer (80 patients) | CoQ10 deficiency noted both in carcinomas (n = 80) and non-malignant lesions (n = 120) | A correlation existed between the intensity of the deficiency and the bad prognosis of the breast disease. Ubiquinone supplementation in breast cancer could be relevant. |
| Yen et al., 2022 [58] | Brain cancer (40 patients) | CoQ10 levels were higher in nontumor controls than in all grades of astrocytoma tissues | Mitochondrial abnormalities are associated with impaired CoQ10 maintenance in human astrocytoma progression. |
| Kanda et al., 2014 [59] | Gastric cancer (238 patients) | Decreased expression in gastric carcinoma tissue of prenyl diphosphate synthase subunit 2, which is required for the biosynthesis of CoQ10 | Decreased precursor expression for CoQ10 biosynthesis is associated with the reduced survival of patients with gastric cancer. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantle, D.; Rowbottom, H.; Jones, J.; Potts, I.M.; Turton, N.; Dewsbury, M.; Lopez-Lluch, G.; Hargreaves, I.P. Energy Metabolism as a Therapeutic Target in Cancer: The Role of Coenzyme Q10. Oxygen 2024, 4, 122-138. https://doi.org/10.3390/oxygen4020008
Mantle D, Rowbottom H, Jones J, Potts IM, Turton N, Dewsbury M, Lopez-Lluch G, Hargreaves IP. Energy Metabolism as a Therapeutic Target in Cancer: The Role of Coenzyme Q10. Oxygen. 2024; 4(2):122-138. https://doi.org/10.3390/oxygen4020008
Chicago/Turabian StyleMantle, David, Hannah Rowbottom, Jacob Jones, Indiia Margarita Potts, Nadia Turton, Mollie Dewsbury, Guillermo Lopez-Lluch, and Iain P. Hargreaves. 2024. "Energy Metabolism as a Therapeutic Target in Cancer: The Role of Coenzyme Q10" Oxygen 4, no. 2: 122-138. https://doi.org/10.3390/oxygen4020008
APA StyleMantle, D., Rowbottom, H., Jones, J., Potts, I. M., Turton, N., Dewsbury, M., Lopez-Lluch, G., & Hargreaves, I. P. (2024). Energy Metabolism as a Therapeutic Target in Cancer: The Role of Coenzyme Q10. Oxygen, 4(2), 122-138. https://doi.org/10.3390/oxygen4020008