1. Introduction
Access to clean and safe water is crucial for health and improved life quality for people all over the world. Unfortunately, contaminated drinking water and poor sanitation and hygiene are still major sources of diseases and cause approximately six percent of deaths worldwide [
1,
2]. Waterborne diseases are caused by polluted drinking water and contaminated water used in the production process of industrial sectors, such as the food or pharmaceutical industry. Therefore, to improve water safety, fast, cheap, and on-site identification methods are required. The current procedures for the identification and quantification of bacteria are either cultivation or molecular methods. Cultivation-based approaches are time-consuming and need up to 72 h.
In contrast, molecular methods such as polymerase chain reactions (PCR) are faster but more expensive [
3]. Several biosensors were developed in the last decades to detect pathogens in water) [
4,
5]. However, the complexity, high cost, and the requirement of high sensitivity standards are still limiting factors for the deployment of biosensors in the analysis of environmental samples.
We investigated the use of horseradish peroxidase-conjugated to streptavidin (HRP-Strep) for the direct detection of pathogens. The metalloenzyme horseradish peroxidase (HRP) is one of the most sensitive reporter enzymes known [
6]. HRP is an important commercial enzyme extensively studied as a model for oxidation reactions and is frequently used in various applications such as biosensors, immunoassays, and medical diagnostics [
7,
8,
9,
10,
11,
12,
13]. The enzyme binds noncovalently to a heme cofactor that contains four structurally essential disulfide bonds, nine N-linked glycosylation sites, and two Ca
2+ ions [
7,
14]. It catalyzes the H
2O
2-dependent oxidation of a wide variety of substrates. Conversion of a chromogenic substrate, such as 2,2-azinobis-(3-ethylbenzothiazoline)-6-sulfonic acid (ABTS) or 3,3′,5,5′-tetramethylbenzidine (TMB) results in color changes visible with the naked eye [
15].
HRP naturally occurs in the horseradish roots, which exist in at least 15 different isoforms. The most abundant isoenzyme is C1A [
16]. For industrial use, HRP is still extracted from the plant, which results in a diverse composition of isoenzymes in the final product. The plant-derived isoenzyme C1A is glycosylated with (Xyl)Man
3GlcNAc
2(Fuc) at eight of the nine available asparagine sites. To apply HRP for biotechnological applications such as diagnostics, its recombinant production is required. One option is the isolation from the yeast species
Pichia pastoris or
Saccharomyces cerevisiae, which results in correct folding, disulfide bond formation, and heterogenous hypermannosylation (Man
xGlcNAc
2) [
17,
18]. Another possibility is the expression in
E. coli. However, this requires refolding, resulting in unglycosylated HRP with reduced stability [
19].
For use as a reporter enzyme, HRP is commonly conjugated to antibodies, proteins, and other chemical molecules, most commonly to streptavidin. Streptavidin is a protein derived from the bacterium
Streptomyces avidinii and resembles a homotetramer. It can bind four biotin molecules. The streptavidin-biotin interaction is one of the strongest known non-covalent interactions (Kd 10–14 M) [
20,
21] and therefore widely used in different biotechnological applications [
21]. Bacteria naturally contain biotinylated proteins in their cell membrane [
22]. Therefore, we hypothesized that streptavidin can be used to recognize bacteria directly as it can bind to pathogenic bacteria via biotin. We exploited this feature and used HRP-Strep for the direct detection of bacteria in water. In this context, different detection platforms were evaluated (
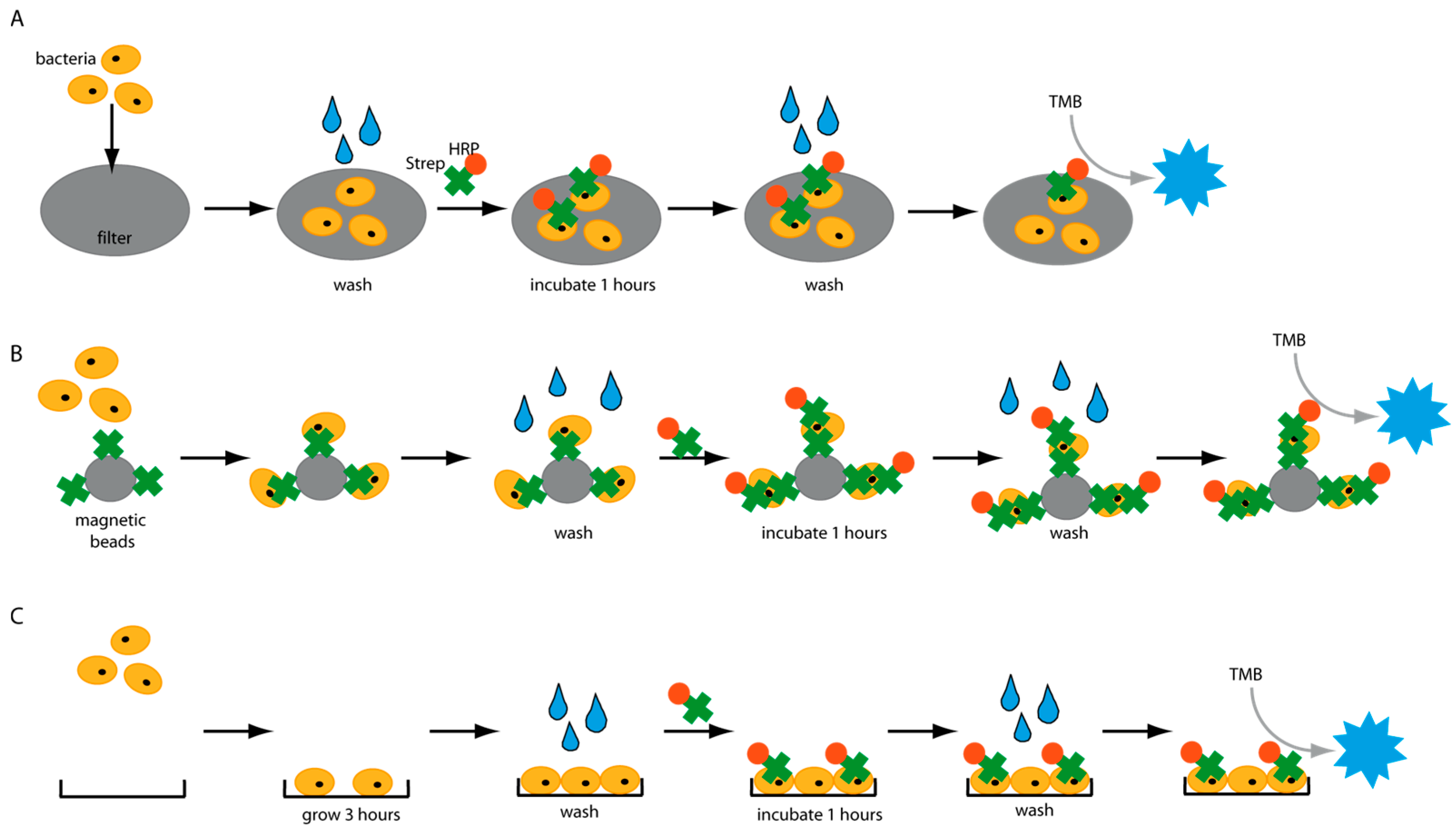
Figure 1), and several factors were identified which have a great impact on the detection assays. An important factor negatively influencing the sensitivity of diagnostic tests is unintended background signals. Thus, we analyzed the molecular interactions of HRP with various surfaces in different platforms and identified a wide range of settings in which HRP causes false-positive signals. Our findings could have a significant effect on the development of HRP-based diagnostic assays. Finally, an enzyme-linked immunosorbent-like assay (ELISA-like) was developed and could detect different pathogens in water in less than 5 h.
2. Materials and Methods
2.1. Materials
Unless otherwise specified, all chemicals were purchased from Merck KGaA (Darmstadt, Germany). DNA was purchased from Integrated DNA Technologies (IDT) (Leuven, Belgium). Enzymes were acquired from Thermo Fisher Scientific (Waltham, MA, USA).
2.2. Filter Experiments
Bacterial cells were grown overnight in a 2xYT medium. The next day, the cells were adjusted to OD600 of 1 with PBS. This corresponds to a concentration of 109 cells/mL. 10-fold serial dilutions were prepared in PBS, and 1 mL of each concentration was applied to different syringe filters made with PVDF-, PES- or polypropylene-membrane. The filter was washed three times by passing 1 mL PBS through the filter. HRP-conjugated streptavidin (HRP-Strep, N100/Thermo Fisher Scientific, Waltham, MA, USA) was prepared in a 1:104 dilution in PBS, and 1 mL was used to apply on the filter. Then, the filter is incubated for 1 h at room temperature (RT). Subsequently, the filters were washed five times with 10 mL PBS. Finally, 1 mL TMB substrate solution (1-step ultra TMB-ELISA substrate solution, Thermo Fisher Scientific, Waltham, MA, USA) was passed through each filter. The filters were incubated for 10 min at RT before the development of a blue color was observed and a picture was taken.
HRP from horseradish was purchased from Merck KGaA (P8375, Darmstadt, Germany). Unglycosylated HRP was expressed and purified from
E. coli as described in Humer et al. [
19]. HRP was cloned in-frame with the α-factor into pPICZαB, linearized by digestion with SacI and transformed into competent
P. pastoris X-33 (Invitrogen, Waltham, MA, USA) or
P. pastoris SuperMan5 (Biogrammatics, Carlsbad, CA, USA) by heat shock. Yeast cultures were grown in BMMY medium supplemented with 10 µM hemin for 80 h, and expression was induced by the addition of 1% methanol every 24 h. The cells were harvested, and the HRP-containing supernatant was applied to the filter.
2.3. Detection of Bacteria on Lateral Flow Device (LFD)
Briefly, bacterial cells were first grown overnight in TSB media. The next day, the cells were adjusted to OD 600 of 1 using 0.1 M sodium phosphate buffer, pH 7.4 (PBS), corresponding to a concentration of ~109 cell/mL. 10-fold bacterial serial dilution was prepared in PBS to a final volume of 1mL in PBS. Lateral flow dipsticks already prefabricated at AIT made of Nitrocellulose were used. Briefly, a drop of a suspension of bacteria in PBS at different concentrations was pipetted on different nitrocellulose dipsticks. The dipsticks were incubated at room temperature for 1 h to let the PBS evaporate, and the bacteria stuck to the nitrocellulose dipstick. 1 mL of PBS was running through the dipstick from left to right (direction of the flow). Subsequentially, HRP-conjugated streptavidin (N100/ThermoFisher Scientific, Waltham, MA, USA) was prepared in a dilution range of 1:104 in PBS, and 1 mL of it was running through the dipstick in the direction of the flow. 3 to 5 washing steps were then made by running 1 mL of PBS in the direction of the flow. TMB substrate solution (1 mL; 1-step ultra TMB-ELISA substrate solution, ThermoFisher Scientific, 34028, Waltham, MA, USA) was running through the dipstick in the direction of the flow. After 10 min of incubation at room temperature, the filters were inspected, and the development of blue color was observed. If the bacterial cells were detected after 10 min of incubation at room temperature, a formation of blue color was observed where the cells were pipetted. Negative control was prepared by following all the previous steps but without the addition of bacterial cells.
2.4. Magnetic Bead Experiments
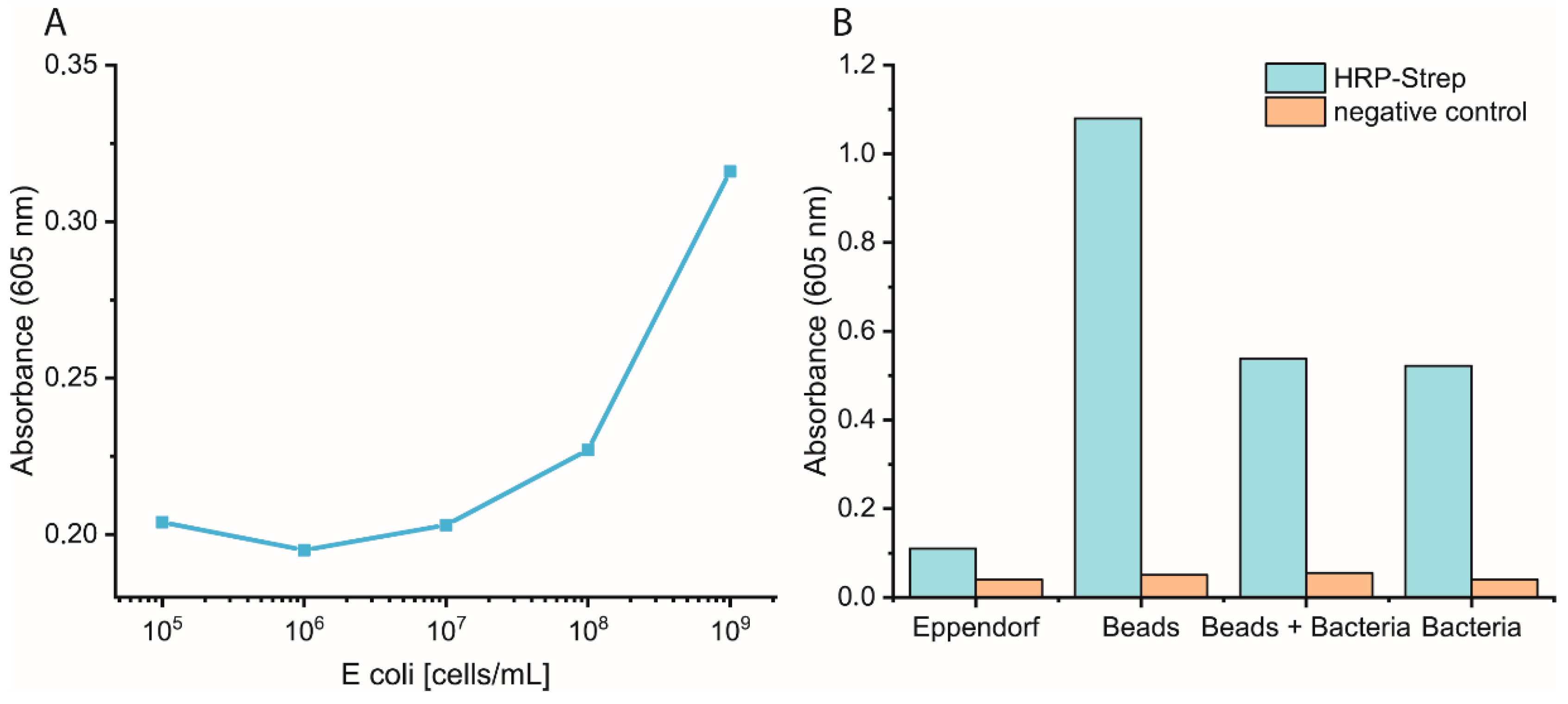
Bacterial cells were prepared as described previously. On the day of the experiment, a 10-fold dilution of bacteria in PBS was prepared, and 1 mL was distributed in either normal Eppendorf tubes (EP0030120086) or low-binding Eppendorf tubes (EP0030108116). Streptavidin magnetic beads (SMB, 11641778001, Merck KGaA, Darmstadt, Germany) were washed three times with PBS, and then a 1:10 dilution of the beads was prepared. Each bacterial dilution was mixed with 100 µL diluted SMB and shaken at 700 rpm for 1 h at RT. Then, the supernatant was removed, and the beads were washed three times with 1 mL PBS. Afterwards, 1 mL of 0.1 µM HRP-Strep was added and shaken at 700 rpm for 1 h at RT. The beads were washed thrice with 1 mL PBS, then 200 µL TMB was added. Finally, 100 µL supernatant was transferred into a 96-well plate, and absorbance was measured at 605 nm using a plate reader (EPOCH, Agilent Biotek, Santa Clara, CA, USA).
2.5. Eppendorf Experiments
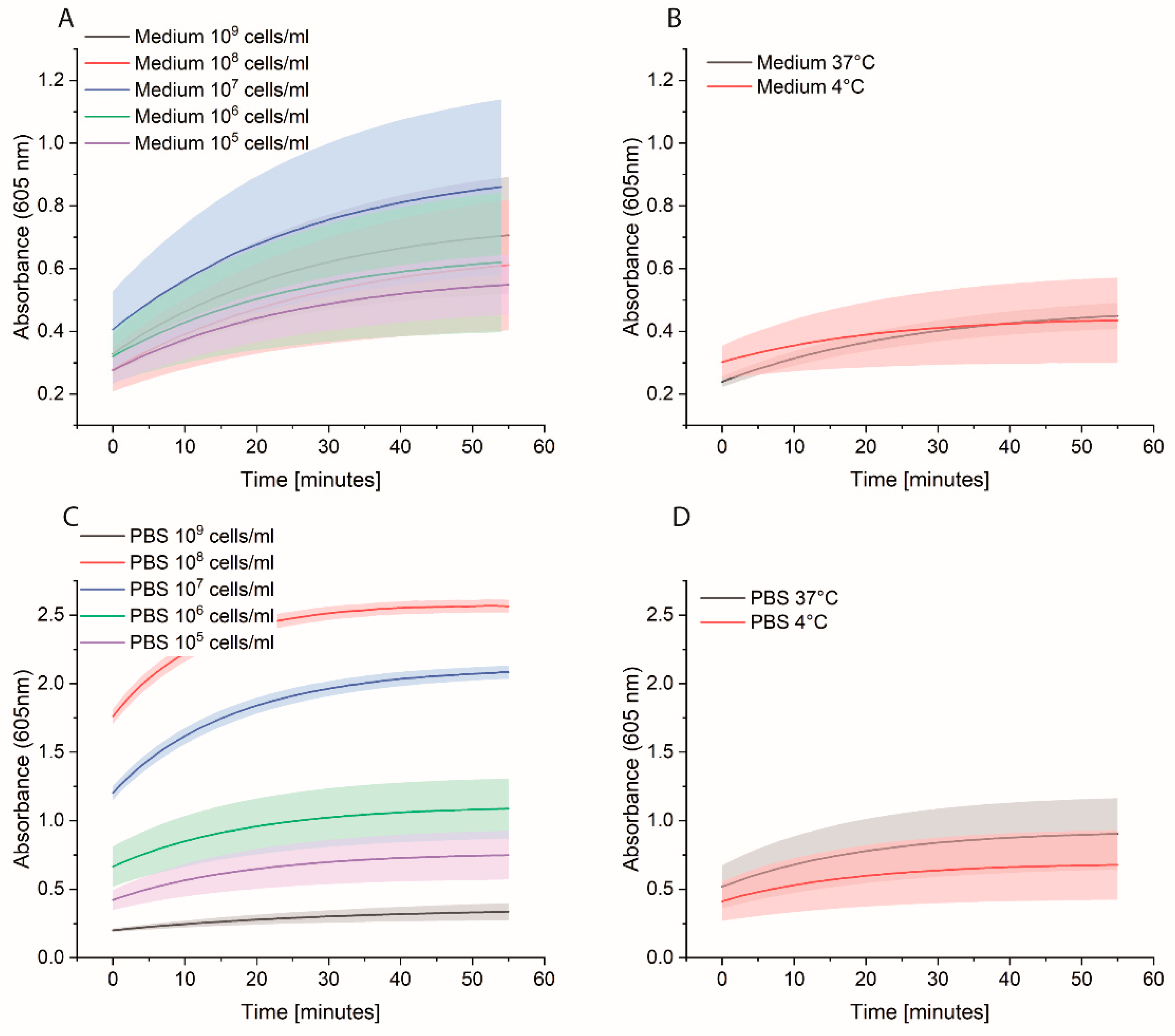
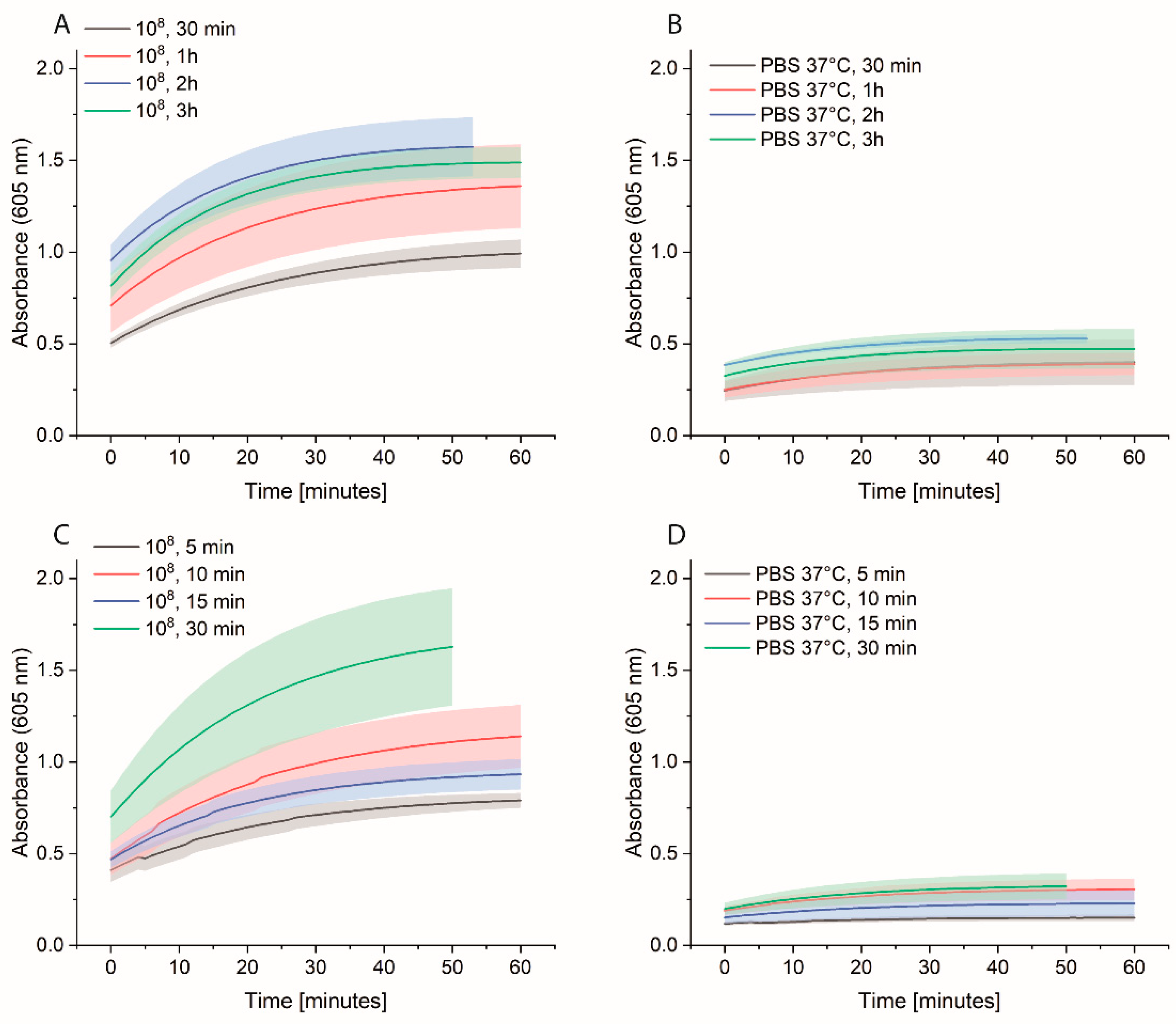
Bacterial cells were first grown overnight in 2xYT medium. The next day, the cells were washed with PBS and adjusted to OD600 of 1 (correlate to 109 cells/mL) in medium or PBS. 10-fold serial dilutions were prepared, and 1 mL was distributed in each Eppendorf tube (EP0030120086). The plate was incubated at 37 °C for 3 h at 600 rpm. The cells were washed once with 1 mL PBS, and then 1 mL of 0.1 nM HRP-Strep was added and incubated for 1 h at RT at 600 rpm. The cells were washed with 2 × 1 mL of PBS and 200 µL TMB substrate solution. After 10 min incubation, 100 µL were transferred in a standard 96-well plate added, and absorbance was measured at 605 nm using a plate reader (EPOCH, Agilent Biotek, Santa Clara, CA, USA).
2.6. Spiked Water Experiments
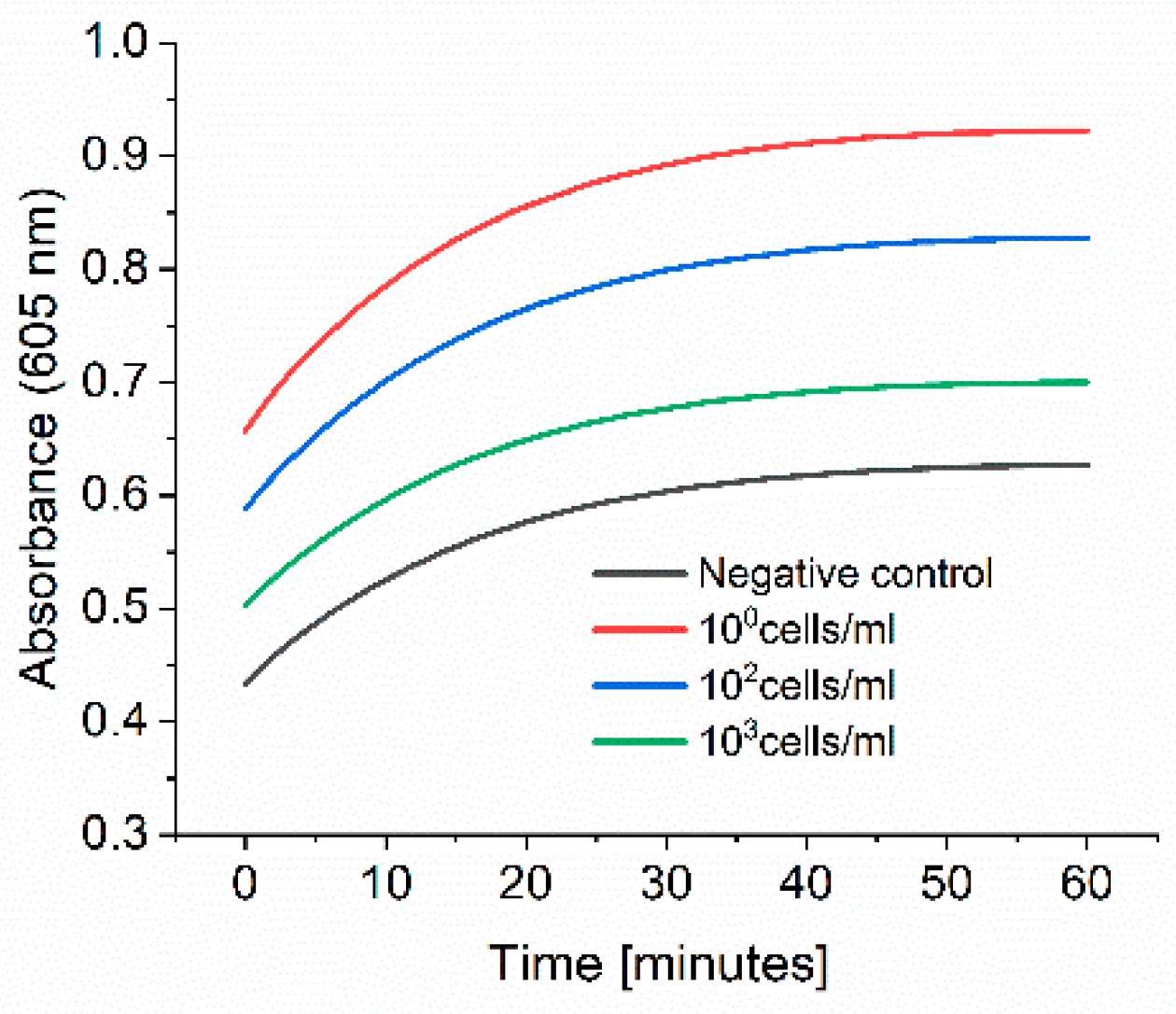
E. coli M/11407 was grown overnight at 37 °C in a 2xYT medium. Cells were then centrifuged and resuspended in tap water to an OD600 of 1. Different Eppendorf tubes were prepared to contain 100, 102 and 103 cell/mL in tap water. Each 1 mL of each cell concentration was added to 4 mL of 2xYT medium and incubated overnight at 37 °C. The day after, the cells were centrifuged and resuspended in 1 mL of PBS in Eppendorf tubes, which were then incubated at 37 °C for 3 h at 600 rpm. The cells were washed once with 1 mL PBS, and then 1 mL of 0.1 nM HRP-Strep was added and incubated for 1 h at RT at 600 rpm. The cells were washed with 2 × 1 mL of PBS and 200 µL TMB substrate solution was added. After 10 min incubation, 100 µL were transferred in a standard 96-well plate added, and absorbance was measured at 605 nm using a plate reader (EPOCH, Agilent Biotek, Santa Clara, CA, USA).
2.7. Plate Experiments
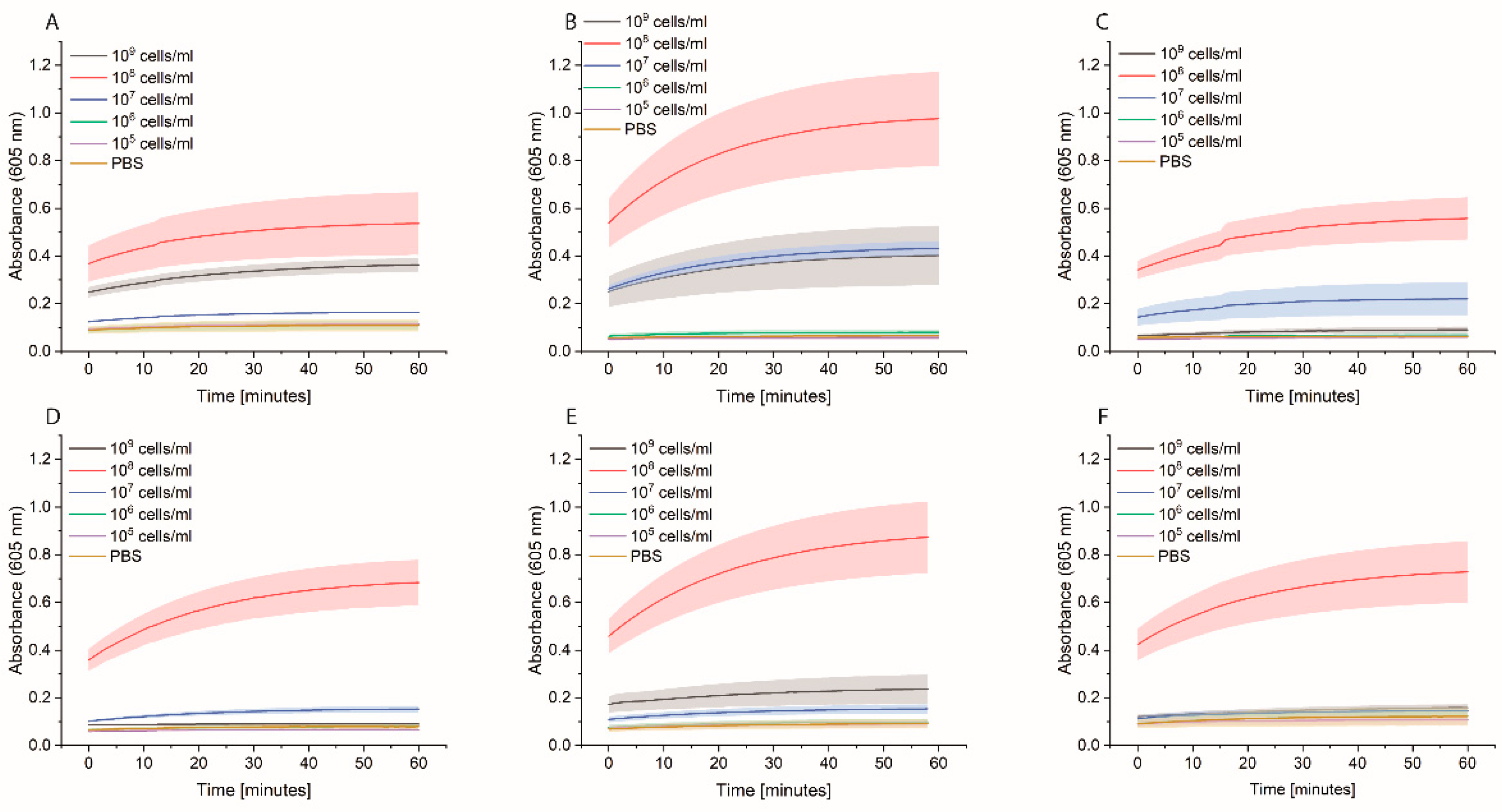
Bacterial cells were first grown overnight in 2xYT medium. The next day, the cells were washed with PBS and adjusted to OD600 of 1 (correlate to 109 cells/mL) in medium or PBS, and 100 µL distributed in each well of a 96-well plate. Two types of plates were tested: standard 96-well plates (Nunc MicroWell 96-Well microplates, 269787, ThermoFisher Scientific, Waltham, MA, USA) or high-binding 96-well plates (Nunc Immuno MaxiSorp MicroWell 96-well solid plate, M9410, ThermoFisher Scientific, Waltham, MA, USA). The plates were incubated at 37 °C for 3 h at 600 rpm. The cells were washed once with 200 µL PBS, and then 200 µL of 0.1 nM HRP-Strep was added and incubated for 1 h at RT at 600 rpm. The cells were washed 2 × 200 µL of PBS and 100 µL TMB substrate solution. After 10 min incubation, absorbance was measured at 605 nm using a plate reader (SpectraMaxi iD3, Molecular Devices GmbH, Munich, Germany).
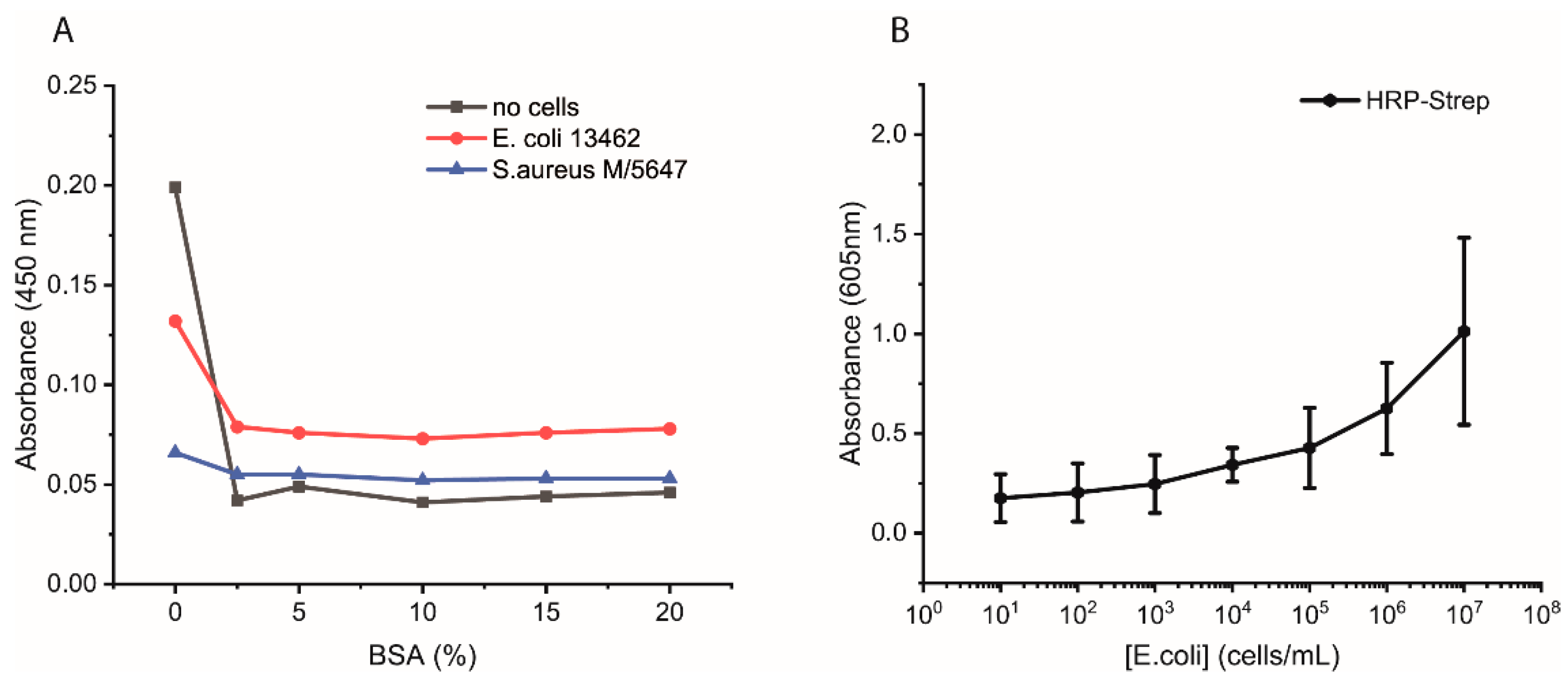
Additional experiments were performed as follows. Bacterial cells were first grown overnight in 2xYT medium. The next day, the cells were washed with PBS and adjusted to OD600 of 1 (correlate to 109 cells/mL) in PBS, and 100 µL distributed in each well of a high-binding 96-well plate. The plate was incubated overnight at 4 °C. On the day of the experiment, the wells were washed with 2 × 200 µL of PBS followed by blocking with 200 µL of 3% BSA or of 0, 5, 10, or 15% in PBS for 1.5 h at room temperature. Afterwards, the wells were washed with 3 × 200 µL of PBS, including 0.05% Tween-20. HRP-Strep or HRP-anti his tag antibody (A00612, GenScript, Rijswijk, The Netherlands) was prepared in a dilution range of 1:104 in PBS, and 100 µL was added to each well, followed by incubation at RT for 1 h. Subsequently, the wells were washed five times with 200 µL of PBS and then applied 100 µL of TMB substrate solution. After 15 min of incubation, the reaction was stopped by adding 50 µL/well of 1 M H2SO4. The optical density was measured at 450 nm using a plate reader.
The efficiency of HRP-Strep to detect different number of bacterial cells were investigated by ELISA-like assay. Briefly, bacterial cells were first grown overnight in TSB media. On the next day, the cells were prepared in a 10-fold dilution (10–109 cells/mL) in PBS and immobilized on high-binding 96-well plates (Nunc Immuno MaxiSorp MicroWell 96-well solid plate, M9410, ThermoFisher Scientific, Waltham, MA, USA). The plate was incubated overnight at 4 °C. On the day of the experiment, the coated wells were washed with 2 × 200 µL of PBS, followed by blocking with 200 µL of 3% BSA for 1.5 h at room temperature. Afterwards, the wells were washed with 3 × 200 µL of PBS, including 0.02% Tween-20. HRP-conjugated streptavidin (N100/ThermoFisher Scientific, Waltham, MA, USA) was prepared in a dilution range of 1:104 in PBS, and 100 µL of it was applied to each well, followed by incubation at RT for 1 h. Subsequently, the wells were washed five times with 200 µL of PBS. TMB substrate solution of 100 µL was added to each well, and absorbance was measured at 605 nm using a plate (SpectraMaxi iD3, Molecular Devices GmbH, Munich, Germany).
4. Discussion
We showed that HRP conjugated to streptavidin can recognize bacterial cells and be directly used to detect different bacterial species. So far, HRP-Strep has been commonly used in ELISA assays [
28] to bind biotinylated antibodies, which recognize the target and generate a measurable signal with the HRP. Our results indicate that previous results where HRP-Strep or HRP were used could be misinterpreted as these proteins can directly bind to bacterial cells or surfaces and generate a signal. Independent of the target to detect, if HRP is applied in an assay, it is important to consider the used materials, as HRP could exhibit high unspecific binding affinities to various surfaces, which can greatly impact results. For example, we showed high HRP affinity to all the tested filter materials, even using filters with different chemical compositions and properties. It is only possible to filter HRP with a high loss, which is also relevant for protein purification, as several methods, such as column chromatography, require previous filtration. In general, purification of HRP is challenging due to factors such as glycosylation, the disulfide bonds, and the heme cofactor. Expression in
P. pastoris takes these factors into account as the resulting protein is correctly folded with disulfide bonds and also glycosylated, as mentioned before.
However, several common purification methods are not suitable to purify HRP (
Figure S1). For example, HRP with a His-tag is not binding to a Ni-NTA column, and even changing the tag length, the position and adjusting the linker length cannot improve the purification. Free heme can interact with the His-tag and inhibit binding to the resin. HRP is also found only in the flow-through in anion exchange chromatography, as the glycosylation shields the protein properties. Heterogenous hypermannosylated HRP expressed in
P. pastoris is eluting in various fractions from size exclusion chromatography columns. The nonuniform glycosylation also affects purification via ConA affinity chromatography and results in elution over several fractions. Also, concentrating HRP proteins by ultrafiltration will result in very low yields.
In summary, considering the high affinity of streptavidin to the cell membrane of bacteria, we explored various assays to detect bacteria. We faced different limitations with this approach as high background signals due to the unspecific binding of HRP to, e.g., magnetic beads.
In developing an assay to detect bacteria in water, several important factors were identified and optimized. An essential factor that requires cautious optimization is the number of washing steps. In our case, it was fundamental to wash at least two times after HRP-Strep incubation to remove unspecifically bound proteins and to reduce the background signal. Additionally, unspecific binding can be reduced by shortening the incubation time with HRP-Strep.
In conclusion, our assay detected various pathogenic bacterial strains within 5 h using HRP-Strep in an ELISA-like assay.
5. Conclusions
We developed a sensor based on the simple interaction of HRP-Strep with bacteria. The sensor showed many advantages and some limitations which have to be overcome in the future. One great advantage is the recognition of a broad spectrum of bacteria. As presented in
Figure 8, we were able to detect up to 6 different bacterial strains. On the other hand, we faced some limitations to detect low cell concentration. As reported in the experiment where we spiked tap water with
E. coli (
Figure 9), this limitation could be overcome with a pre-incubation step as we could detect low cell concentration in real samples. The time to result is relatively short as only 3 h of incubation of bacteria in PBS is needed. Moreover, there is no need for expensive materials and reagents as the assay is carried out in an Eppendorf tube. Anyway, a lab is still needed to carry out the protocol.
In conclusion, we developed a sensor able to detect bacteria by using only HRP-Strep. We faced some limitations due to the unspecific binding of HRP to different materials, but at the same time, we were able to develop a cheap, fast, and broad-spectrum biosensor.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








