
Drugs Drug Candidates 2024, 3(3), 615-637; https://doi.org/10.3390/ddc3030035 - 13 Sep 2024
Viewed by 632
Abstract
►
Show Figures
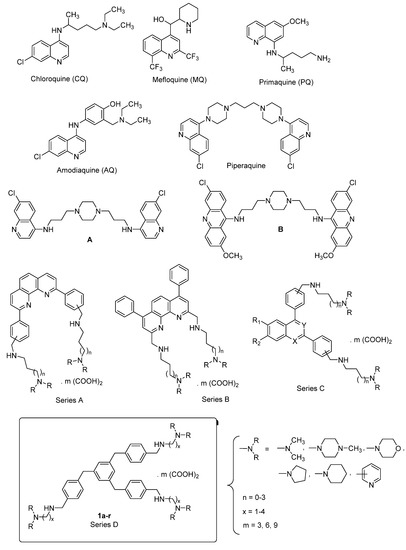
By taking into account our previously described series of 1,3,5-tris[(4-(substituted-aminomethyl)phenyl)methyl]benzene compounds, we have now designed, prepared, and evaluated in vitro against Plasmodium falciparum a novel series of structural analogues of these molecules, i.e., the 1,3,5-tris[(4-(substituted-aminomethyl)phenoxy)methyl]benzene derivatives. The pharmacological data
[...] Read more.
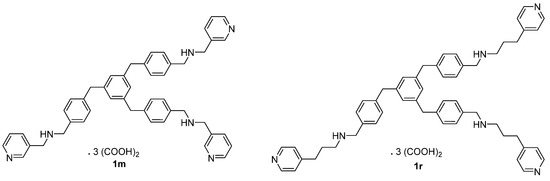
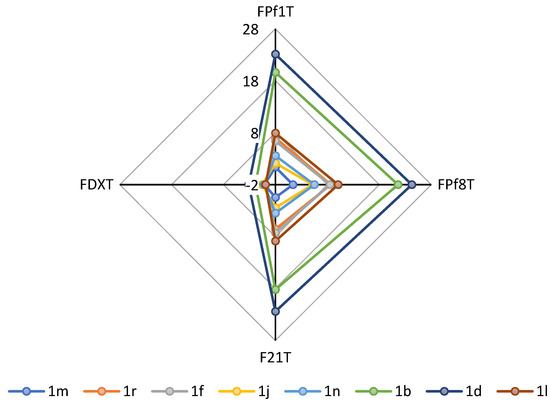
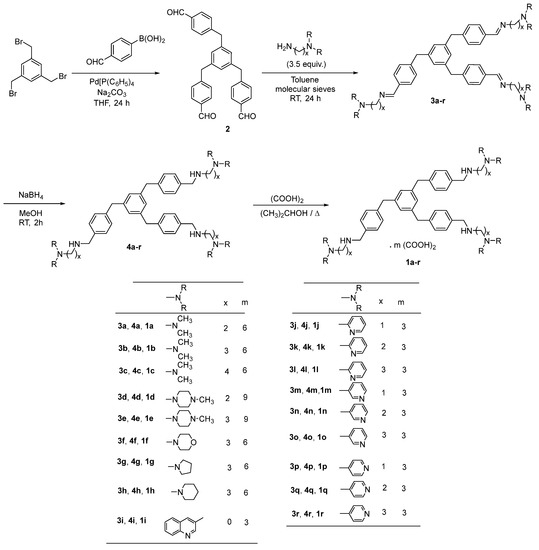
By taking into account our previously described series of 1,3,5-tris[(4-(substituted-aminomethyl)phenyl)methyl]benzene compounds, we have now designed, prepared, and evaluated in vitro against Plasmodium falciparum a novel series of structural analogues of these molecules, i.e., the 1,3,5-tris[(4-(substituted-aminomethyl)phenoxy)methyl]benzene derivatives. The pharmacological data showed antimalarial activity with IC50 values in the sub and μM range. The in vitro cytotoxicity of these new nitrogen polyphenoxymethylbenzene compounds was also evaluated on human HepG2 cells. The 1,3,5-tris[(4-(substituted-aminomethyl)phenoxy)methyl]benzene derivative 1m was found as one of the most potent and promising antimalarial candidates with favorable cytotoxic to antiprotozoal properties in the P. falciparum strains W2 and 3D7. In conclusion, this 1,3,5-tris[(4-(pyridin-3-ylmethylaminomethyl)phenoxyl)methyl]benzene 1m (IC50 = 0.07 μM on W2, 0.06 μM on 3D7, and 62.11 μM on HepG2) was identified as the most promising antimalarial derivative with selectivity indexes (SI) of 887.29 on the W2 P. falciparum chloroquine-resistant strain, and of 1035.17 on the chloroquine-sensitive and mefloquine decreased sensitivity strain 3D7. It has been previously described that the telomeres of P. falciparum could represent potential targets for these types of polyaromatic compounds; therefore, the capacity of our novel derivatives to stabilize the parasitic telomeric G-quadruplexes was assessed using a FRET melting assay. However, with regard to the stabilization of the protozoal G-quadruplex, we observed that the best substituted derivatives 1, which exhibited some interesting stabilization profiles, were not the most active antimalarial compounds against the two Plasmodium strains. Thus, there were no correlations between their antimalarial activities and selectivities of their respective binding to G-quadruplexes.
Full article

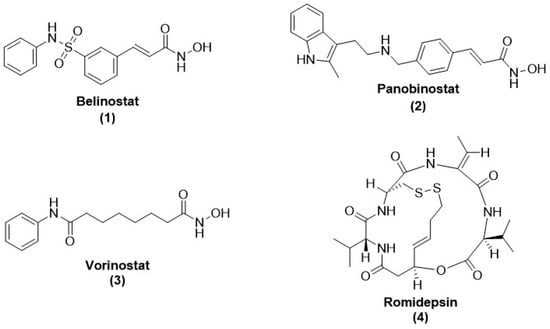
Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}