Entropic Stabilization of Cas4 Protein SSO0001 Predicted with Popcoen



Abstract
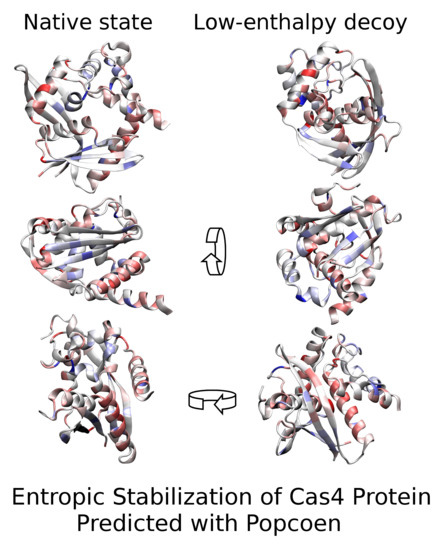
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Decoy Structures Acquisition
4.2. Free-Energy Calculation
4.3. Popcoen
4.4. Structure Characterization
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nobel Media, AB. The Nobel Prize in Chemistry 2013. Awarded Jointly to Martin Karplus, Michael Levitt, and Arieh Warshel. Available online: http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2013/ (accessed on 28 June 2018).
- Adcock, S.A.; McCammon, J.A. Molecular Dynamics: Survey of Methods for Simulating the Activity of Proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, T.; Karplus, M. Effective Energy Function for Proteins in Solution. Proteins Struct. Funct. Bioinform. 1999, 35, 133–152. [Google Scholar] [CrossRef]
- Goethe, M.; Fita, I.; Rubi, J.M. Thermal motion in proteins: Large effects on the time-averaged interaction energies. AIP Adv. 2016, 6, 035020. [Google Scholar] [CrossRef] [Green Version]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Pokala, N.; Handel, T.M. Energy Functions for Protein Design: Adjustment with Protein-Protein Complex Affinities, Models for the Unfolded State, and Negative Design of Solubility and Specificity. J. Mol. Biol. 2005, 347, 203–227. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.A.; Strauss, C.E.; Misura, K.M.; Baker, D. Protein Structure Prediction using Rosetta. Methods Enzymol. 2004, 383, 66–93. [Google Scholar] [PubMed]
- Schwieters, C.D.; Kuszewski, J.J.; Tjandra, N.; Clore, G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003, 160, 65–73. [Google Scholar] [CrossRef]
- Cheng, T.M.; Blundell, T.L.; Fernández-Recio, J. pyDock: Electrostatics and desolvation for effective scoring of rigid-body protein–protein docking. Proteins Struct. Funct. Bioinf. 2007, 68, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Suárez, M.; Tortosa, P.; Jaramillo, A. PROTDES: CHARMM toolbox for computational protein design. Syst. Synth. Biol. 2008, 2, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, B.; Weng, Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins Struct. Funct. Bioinform. 2007, 67, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Thévenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tufféry, P. PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–W293. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, H.; Smith, L.J.; Mark, A.E.; van Gunsteren, W.F. Entropy Calculations on the Molten Globule State of a Protein: Side-Chain Entropies of α-Lactalbumin. Proteins Struct. Funct. Bioinform. 2002, 46, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Berezovsky, I.N.; Chen, W.W.; Choi, P.J.; Shakhnovich, E.I. Entropic Stabilization of Proteins and Its Proteomic Consequences. PLoS Comput. Biol. 2005, 1, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, J.S. On Side-Chain Conformational Entropy of Proteins. PLoS Comput. Biol. 2006, 2, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Goethe, M.; Fita, I.; Rubi, J.M. Vibrational Entropy of a Protein: Large Differences between Distinct Conformations. J. Chem. Theory Comput. 2015, 11, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Goethe, M.; Gleixner, J.; Fita, I.; Rubi, J.M. Prediction of Protein Configurational Entropy (Popcoen). J. Chem. Theory Comput. 2018, 14, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Andricioaei, I.; Karplus, M. On the calculation of entropy from covariance matrices of the atomic fluctuations. J. Chem. Phys. 2001, 115, 6289–6292. [Google Scholar] [CrossRef]
- Hnizdo, V.; Darian, E.; Fedorowicz, A.; Demchuk, E.; Li, S.; Singh, H. Nearest-neighbor nonparametric method for estimating the configurational entropy of complex molecules. J. Chem. Theory 2007, 28, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Killian, B.J.; Kravitz, J.Y.; Gilson, M.K. Extraction of configurational entropy from molecular simulations via an expansion approximation. J. Chem. Phys. 2007, 127, 024107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnizdo, V.; Tan, J.; Killian, B.J.; Gilson, M.K. Efficient calculation of configurational entropy from molecular simulations by combining the mutual-information expansion and nearest-neighbor methods. J. Comput. Chem. 2008, 29, 1605–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, B.M.; Tidor, B. MIST: Maximum Information Spanning Trees for dimension reduction of biological data sets. Bioinformatics 2009, 25, 1165–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggins, D.J. Estimating Translational and Orientational Entropies using the k-Nearest Neighbors Algorithm. J. Chem. Theory Comput. 2014, 10, 3617–3625. [Google Scholar] [CrossRef] [PubMed]
- Fogolari, F.; Corazza, A.; Fortuna, S.; Soler, M.A.; VanSchouwen, B.; Brancolini, G.; Corni, S.; Melacini, G.; Esposito, G. Distance-Based Configurational Entropy of Proteins from Molecular Dynamics Simulations. PLoS ONE 2015, 10, e0132356. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.H.; Ham, S. New Computational Approach for External Entropy in Protein–Protein Binding. J. Chem. Theory Comput. 2016, 12, 2509–2516. [Google Scholar] [CrossRef] [PubMed]
- Gyimesi, G.; Závodszky, P.; Szilágyi, A. Calculation of configurational entropy differences from conformational ensembles using Gaussian mixtures. J. Chem. Theory Comput. 2017, 13, 29–41. [Google Scholar] [CrossRef] [PubMed]
- van der Oost, J.; Westra, E.R.; Jackson, R.N.; Wiedenheft, B. Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nat. Rev. Microbiol. 2014, 12, 479. [Google Scholar] [CrossRef] [PubMed]
- Rath, D.; Amlinger, L.; Rath, A.; Lundgren, M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015, 117, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhou, Y.; Taylor, D.W.; Sashital, D.G. Cas4-Dependent Prespacer Processing Ensures High-Fidelity Programming of CRISPR Arrays. Mol. Cell 2018, 70, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Lemak, S.; Beloglazova, N.; Nocek, B.; Skarina, T.; Flick, R.; Brown, G.; Popovic, A.; Joachimiak, A.; Savchenko, A.; Yakunin, A.F. Toroidal structure and DNA cleavage by the CRISPR-associated [4Fe-4S] cluster containing Cas4 nuclease SSO0001 from Sulfolobus solfataricus. J. Am. Chem. Soc. 2013, 135, 17476–17487. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.J.; Tai, C.H.; Huang, Y.J.; Block, J.; Bai, H.; Kryshtafovych, A.; Montelione, G.T.; Lee, B. Definition and classification of evaluation units for CASP10. Proteins Struct. Funct. Bioinf. 2014, 82, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kasciukovic, T.; White, M.F. The CRISPR associated protein Cas4 Is a 5’ to 3’ DNA exonuclease with an iron-sulfur cluster. PLoS ONE 2012, 7, e47232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.; (FoldX consortium, Barcelona, Spain). Private communication, 2016.
- Meyer, T.; D’Abramo, M.; Hospital, A.; Rueda, M.; Ferrer-Costa, C.; Pérez, A.; Carrillo, O.; Camps, J.; Fenollosa, C.; Repchevsky, D.; et al. MoDEL (Molecular Dynamics Extended Library): A Database of Atomistic Molecular Dynamics Trajectories. Structure 2010, 18, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.A.; Harvey, S.C. Dynamics of Proteins and Nucleic Acids; Cambridge University Press: Cambridge, UK, 1987. [Google Scholar]
- Li, D.W.; Meng, D.; Brüschweiler, R. Short-Range Coherence of Internal Protein Dynamics Revealed by High-Precision in Silico Study. J. Am. Chem. Soc. 2009, 131, 14610–14611. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Shrake, A.; Rupley, J.A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef]
- Sokal, R.; Michener, C. A statistical method for evaluating systematic relationships. Univ. Kansas Sci. Bull. 1958, 38, 1409–1438. [Google Scholar]



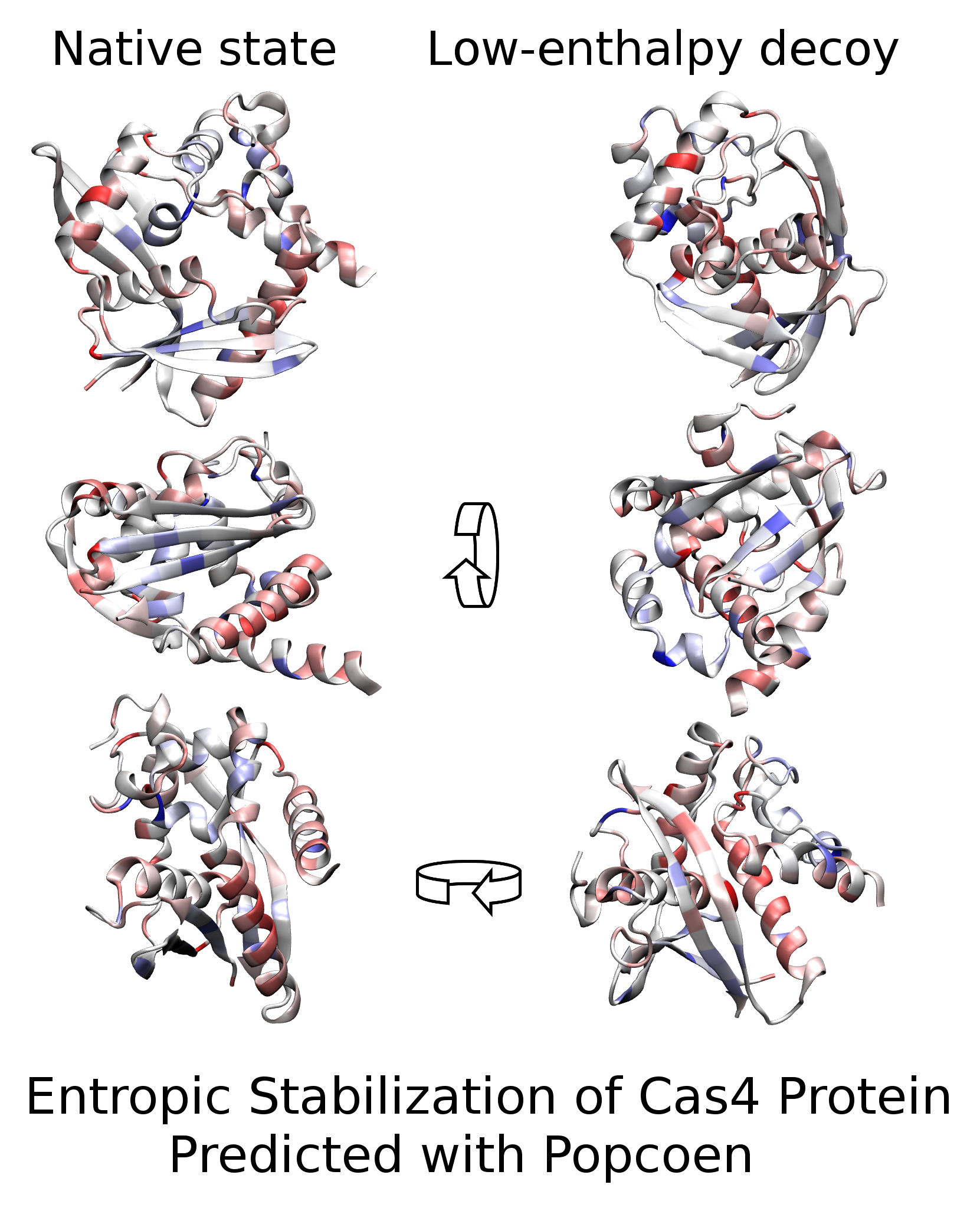{kind=link}
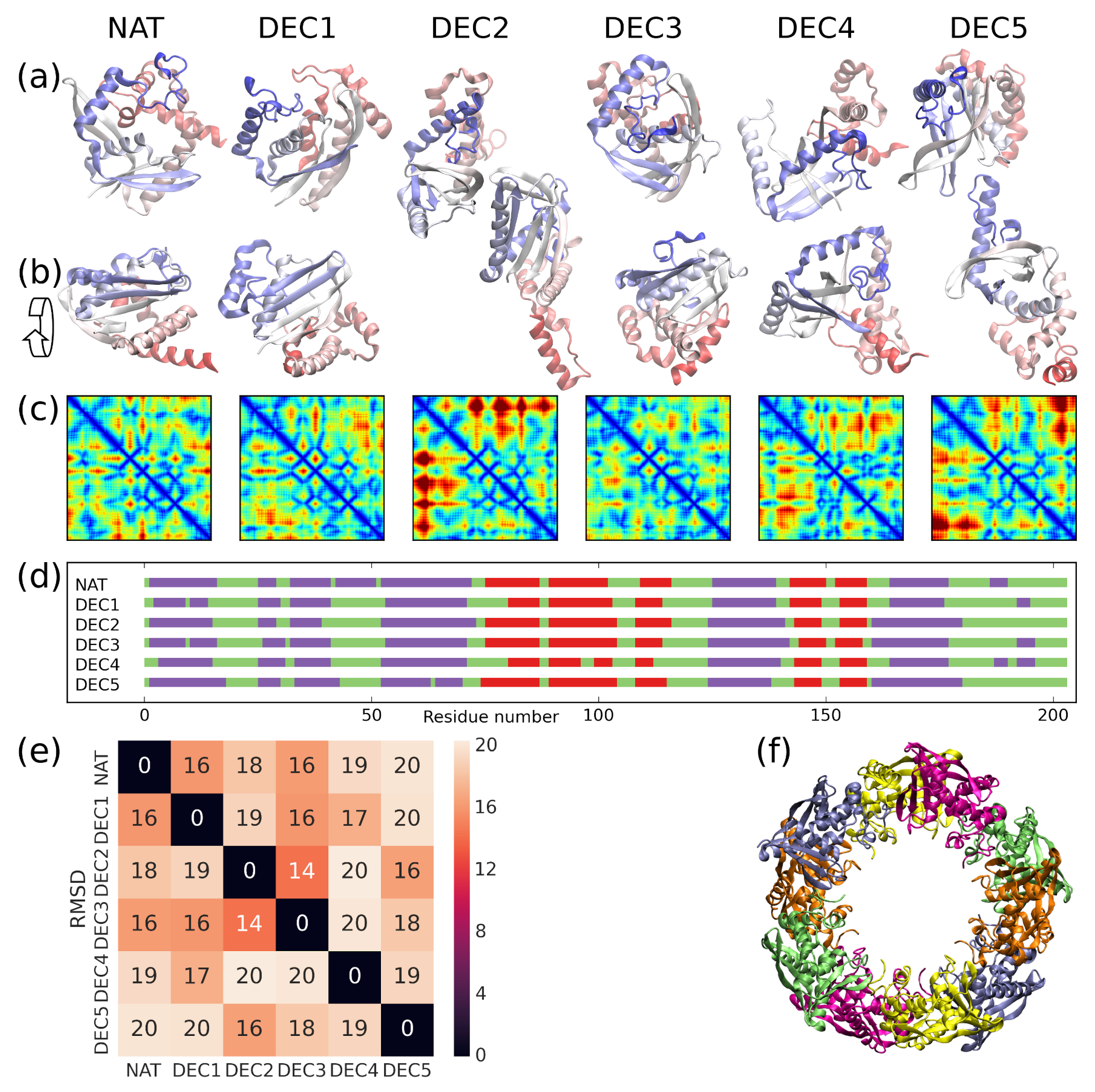{kind=link}
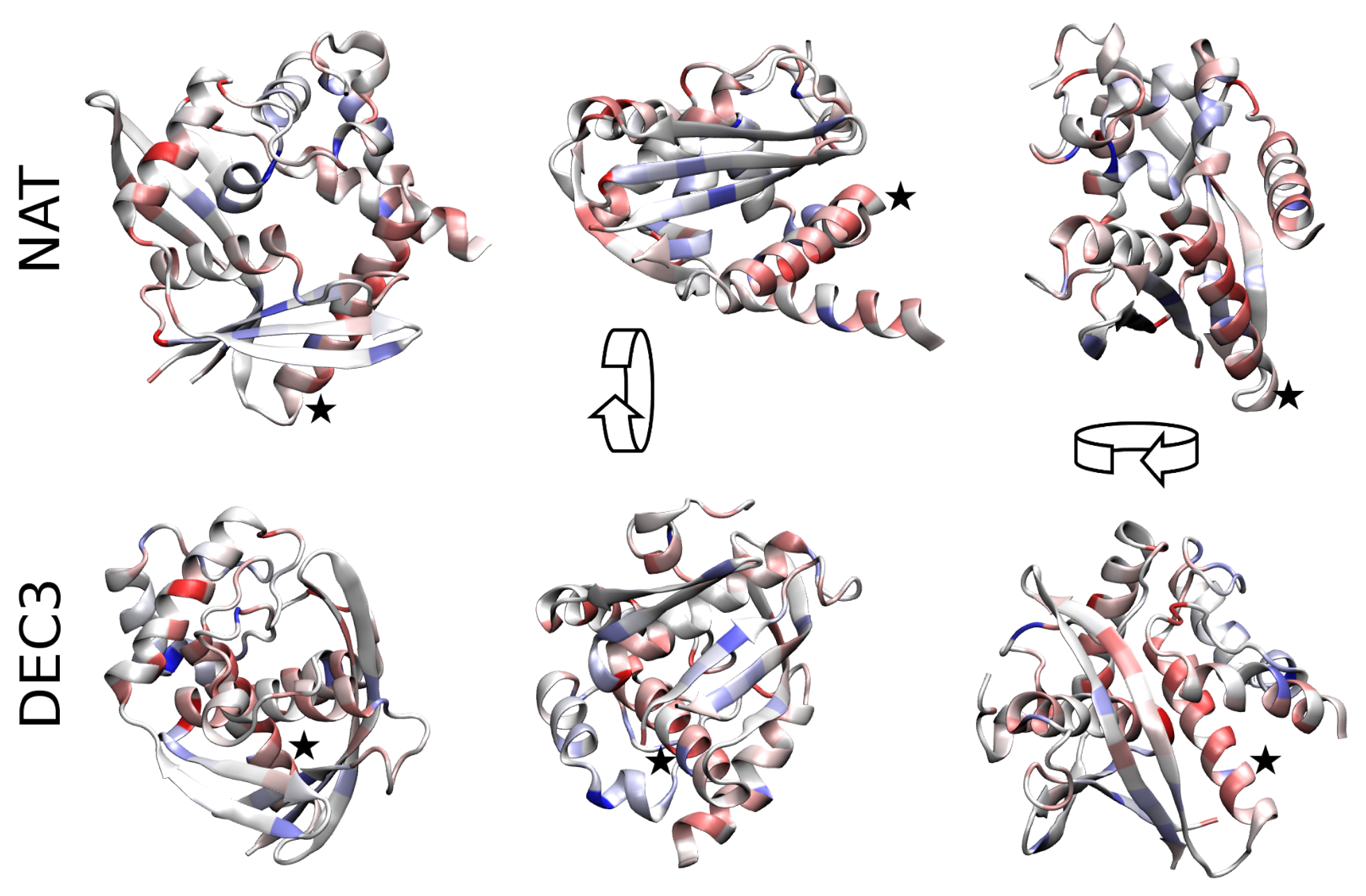{kind=link}
| Conformation | |||||
|---|---|---|---|---|---|
| NAT | 6.6 [5] | 1.3 [2] | 0.0 [1] | 0.4 [2] | 0.0 [1] |
| DEC1 | 0.0 [1] | 17.0 [6] | 7.3 [5] | 9.5 [5] | 16.4 [6] |
| DEC2 | 0.8 [2] | 9.5 [4] | 6.6 [3] | 2.8 [4] | 9.0 [4] |
| DEC3 | 0.9 [3] | 6.6 [3] | 8.7 [6] | 0.0 [1] | 8.3 [3] |
| DEC4 | 5.6 [4] | 14.0 [5] | 0.5 [2] | 12.1 [6] | 12.2 [5] |
| DEC5 | 8.5 [6] | 0.0 [1] | 7.0 [4] | 1.0 [3] | 7.6 [2] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goethe, M.; Fita, I.; Rubi, J.M. Entropic Stabilization of Cas4 Protein SSO0001 Predicted with Popcoen. Entropy 2018, 20, 580. https://doi.org/10.3390/e20080580
Goethe M, Fita I, Rubi JM. Entropic Stabilization of Cas4 Protein SSO0001 Predicted with Popcoen. Entropy. 2018; 20(8):580. https://doi.org/10.3390/e20080580
Chicago/Turabian StyleGoethe, Martin, Ignacio Fita, and J. Miguel Rubi. 2018. "Entropic Stabilization of Cas4 Protein SSO0001 Predicted with Popcoen" Entropy 20, no. 8: 580. https://doi.org/10.3390/e20080580
APA StyleGoethe, M., Fita, I., & Rubi, J. M. (2018). Entropic Stabilization of Cas4 Protein SSO0001 Predicted with Popcoen. Entropy, 20(8), 580. https://doi.org/10.3390/e20080580