
A Statistical Journey through the Topological Determinants of the β2 Adrenergic Receptor Dynamics

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics Simulations
2.2. Protein Contact Networks
2.3. Statistical Analysis of Molecular Dynamics Simulations
3. Results
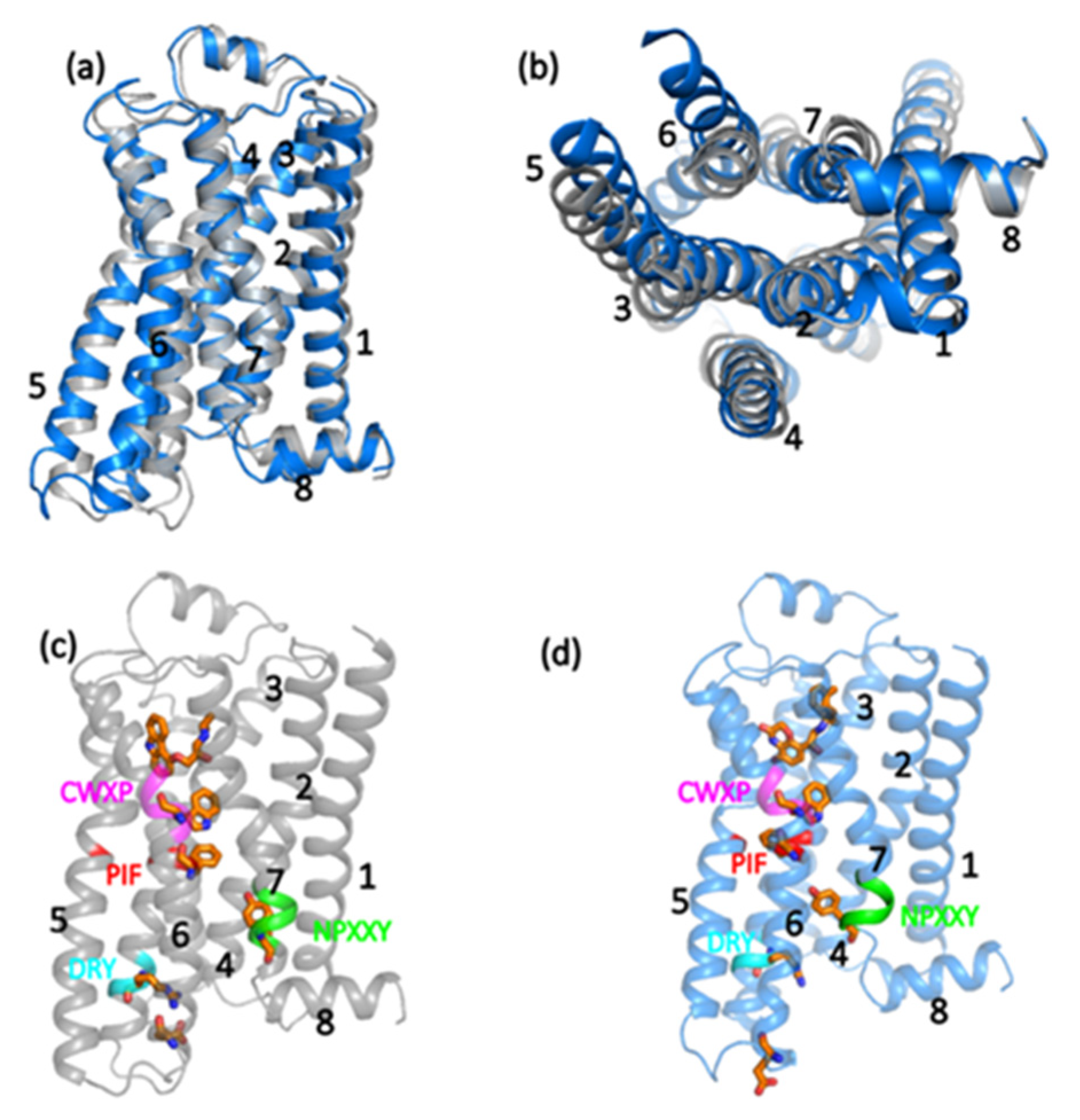
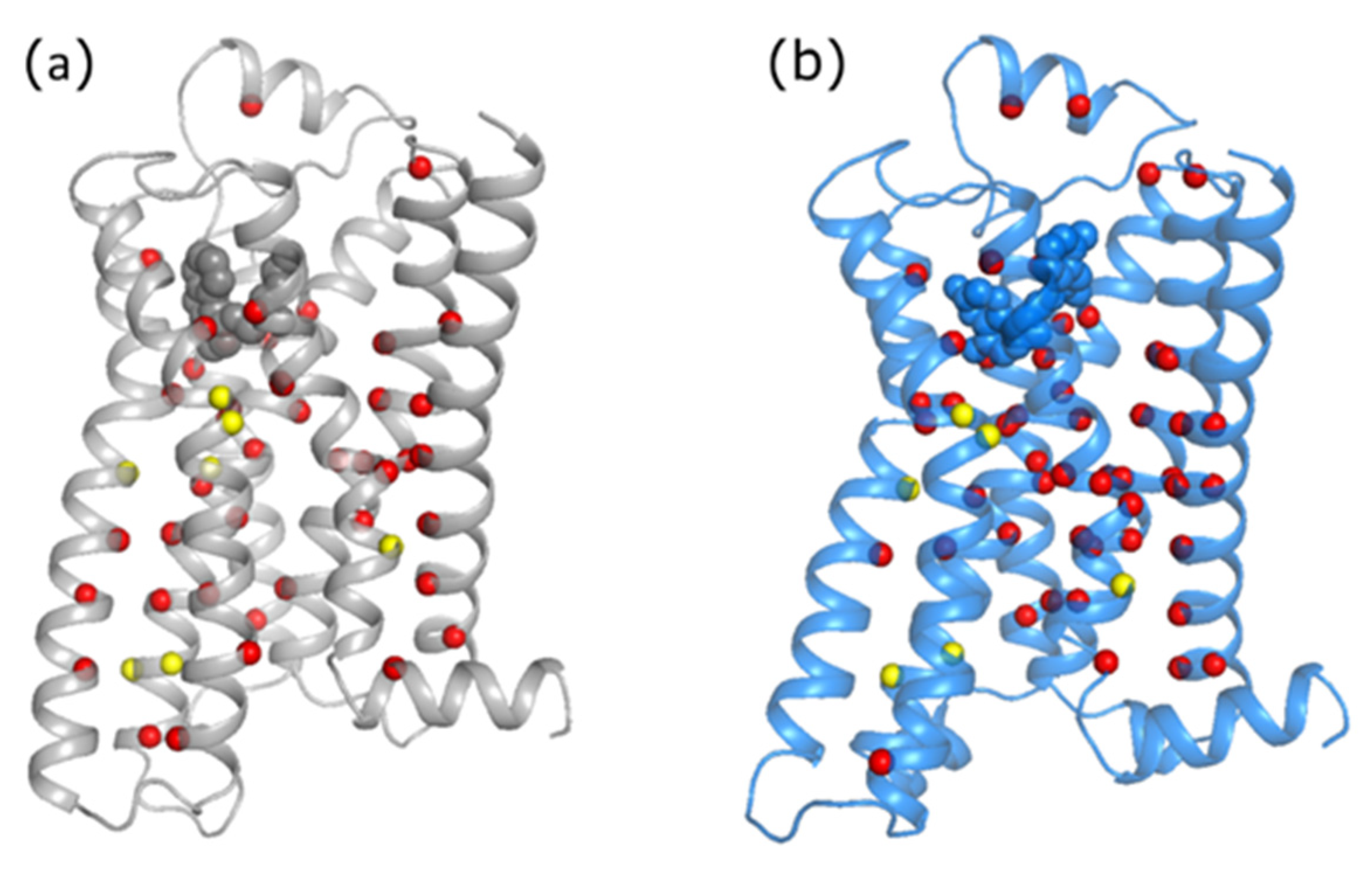
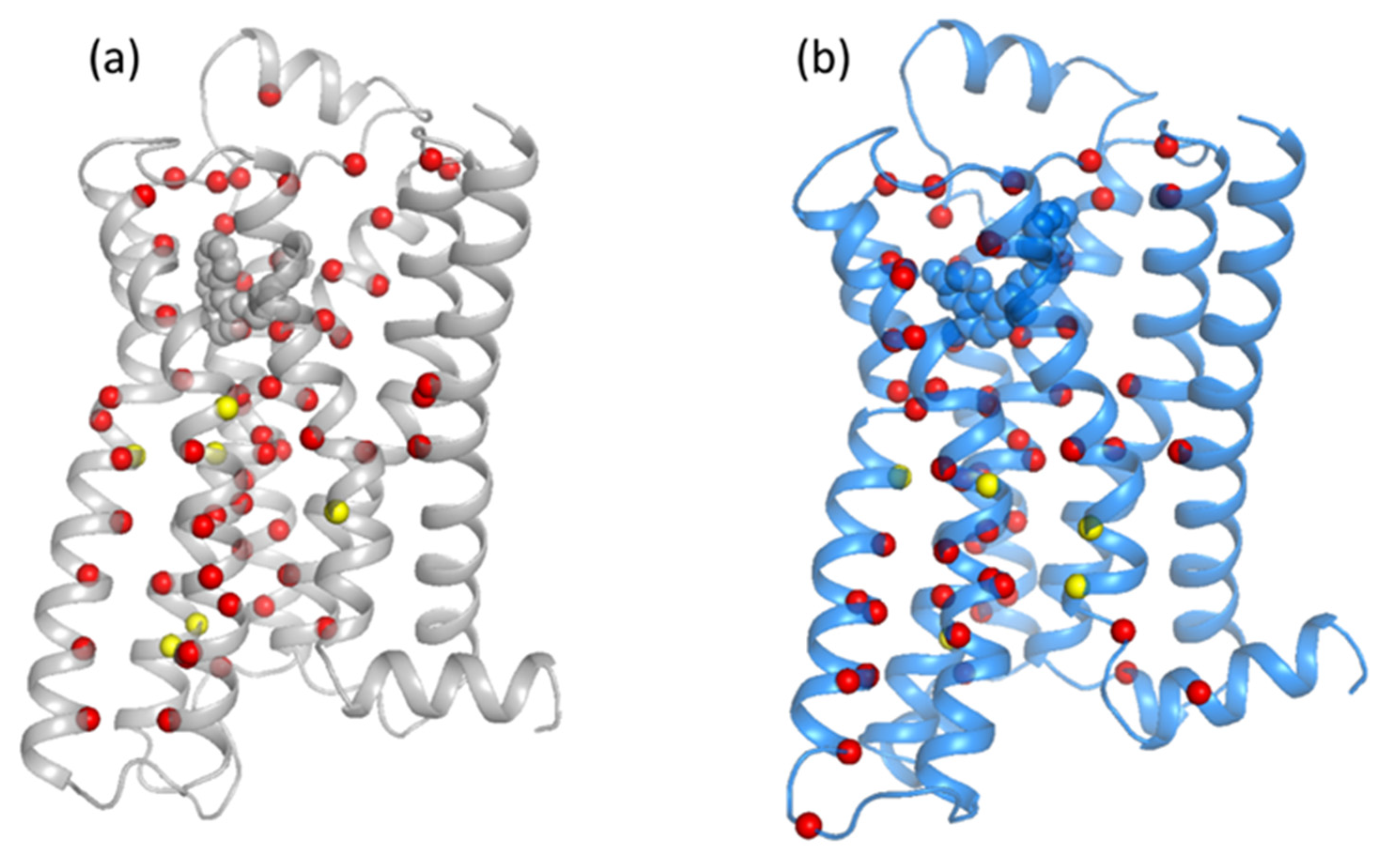
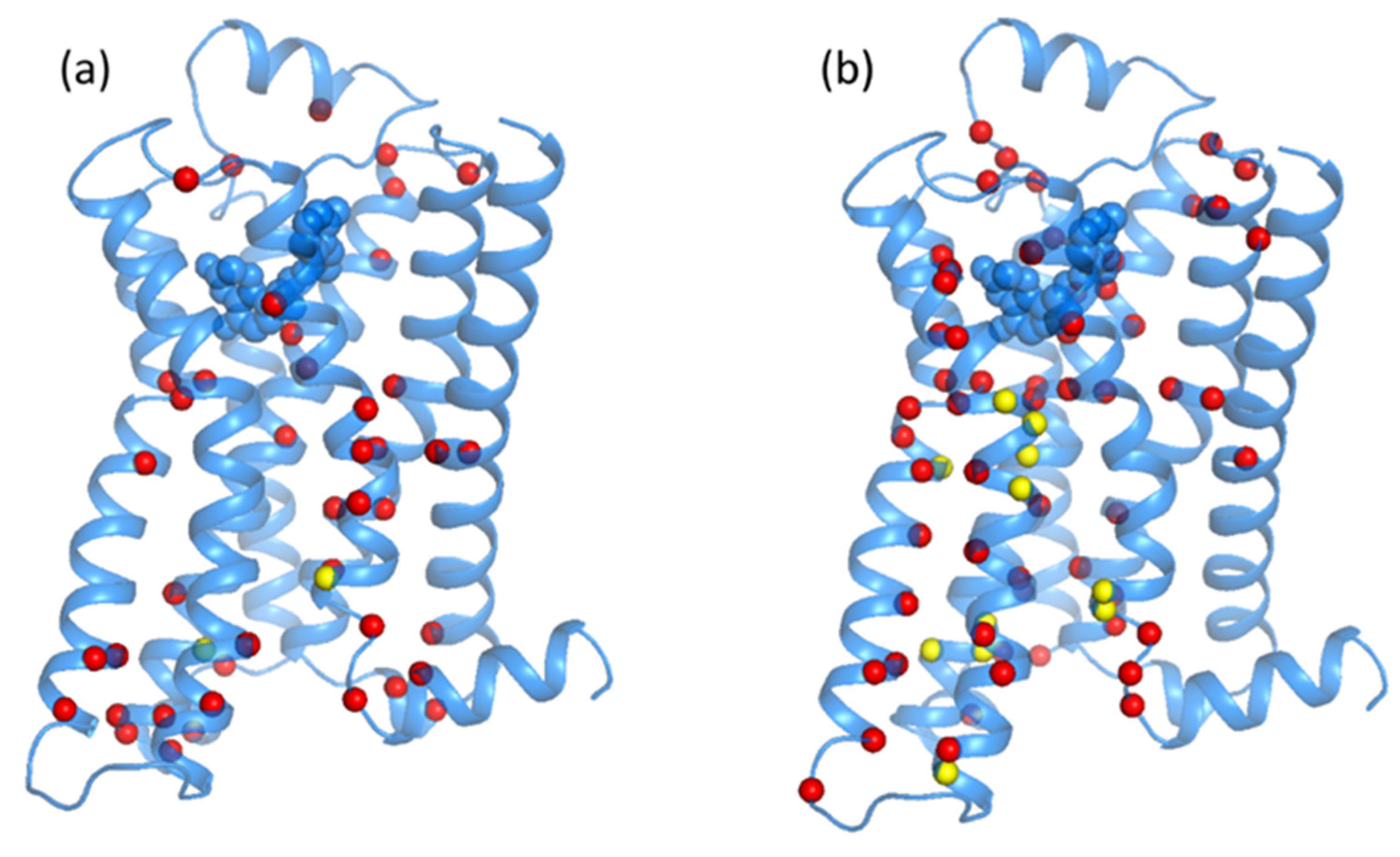
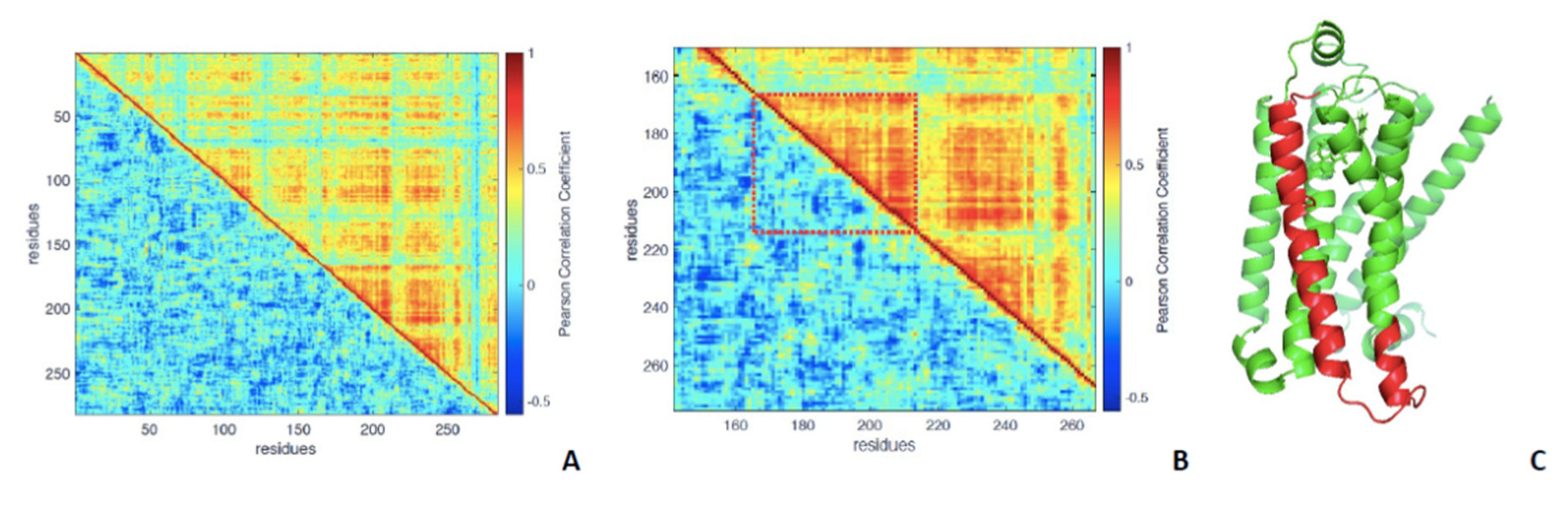
3.1. Analysis of Equilibrated Forms (Active and Inactive) of -AR
3.2. Statistical Analysis of Molecular Dynamics Simulations of -AR Forms (Active/Inactive)
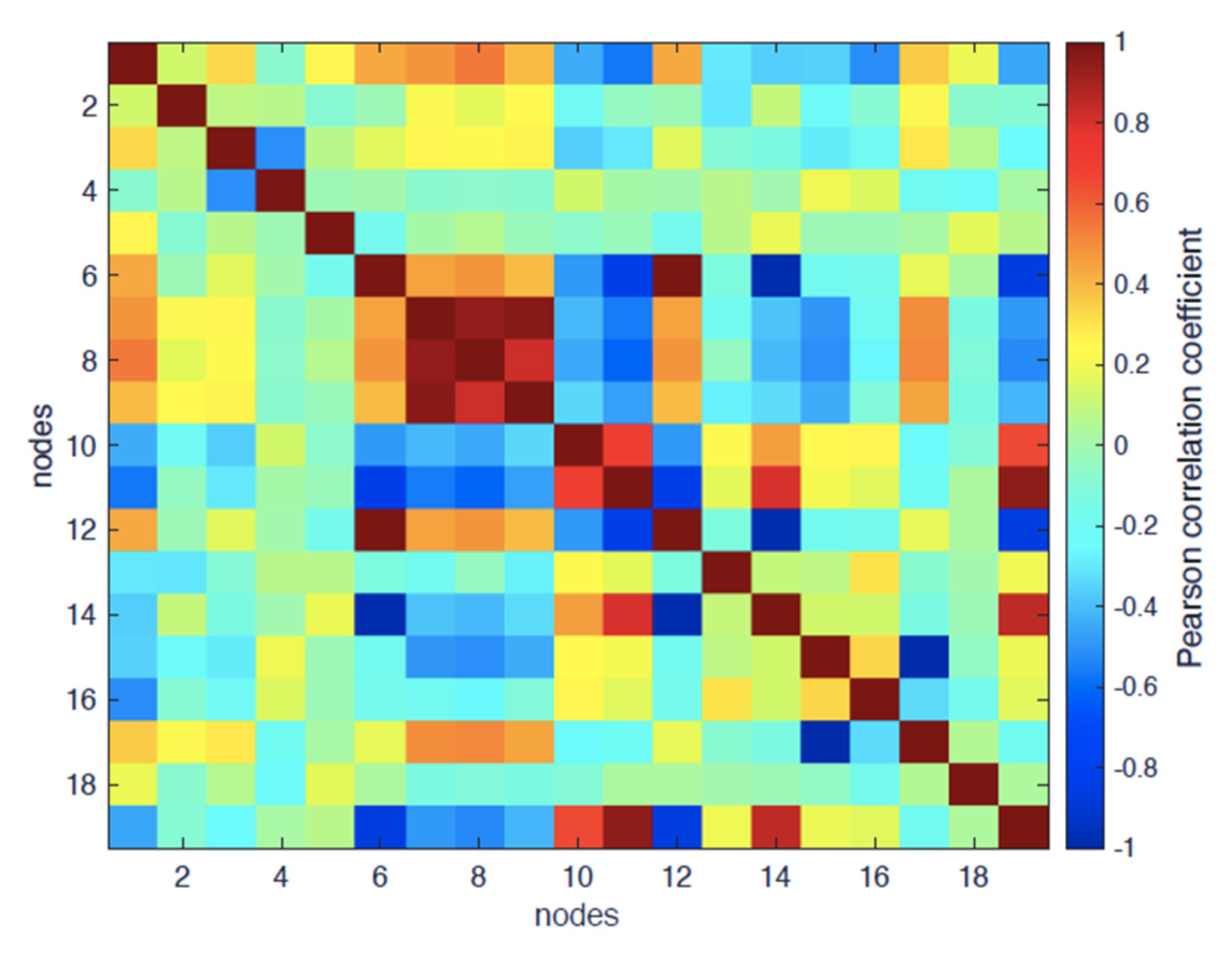
- Degree-based: adeg and E (considering that corr(E, adeg) = 0.95, meaning that E is practically overlapping with adeg);
- Shortest-path-based: abtw, aclose and asp.
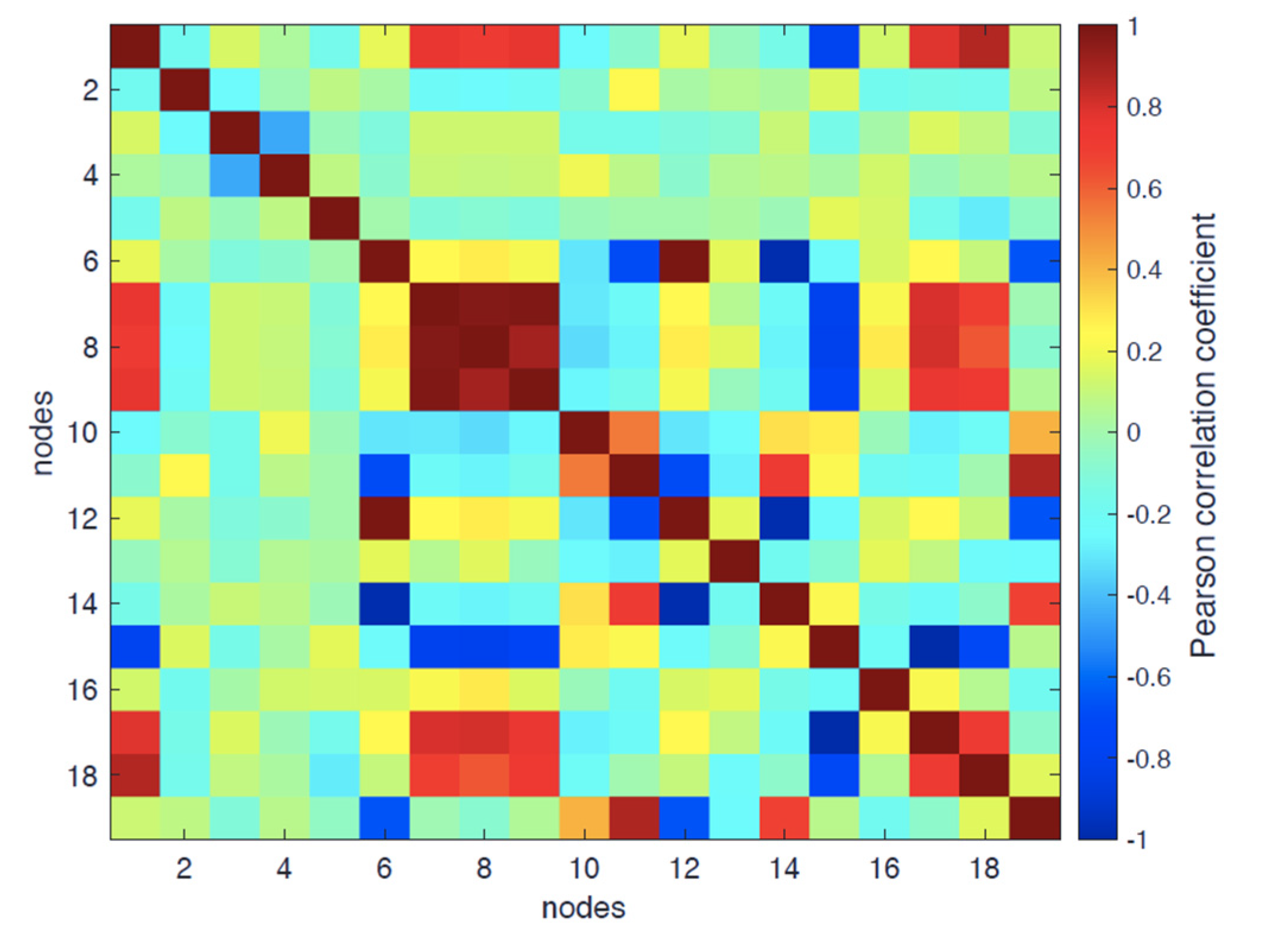
- Degree-based: adeg and E (corr(E, adeg) = 0.95);
- Shortest-path-based: abtw, aclose, and asp.
- (a)
- PC1 (accounting for 37.2% of total variance): t = −0.67; abtw = −0.83, RG = −0.78, RGh = −0.80, RGp = −0.7; acc = −0.68, adeg = −0.88; aclose = 0.78; = 0.5; E = 0.86; this component accounts for the relaxation dynamics (negative correlation with t), driving all listed topological and structural variables.
- (b)
- PC2 (accounting for 14.4% of total variance): aclose = 0.59, ρ = −0.66; this component variance is not addressed by the linear trend toward the equilibrium state and points to time-invariant features of the structure.
- (a)
- PC1 (accounting for 34.8% of total variance): t = −0.80; abtw = 0.56, RG = −0.89, RGh = 0.89, RGp = 0.85 adeg = −0.51; asp = 0.56, aclose = −0.54; = −0.87; = 0.87, AS = 0.71; this component accounts mainly for the linear trend, driving all listed topological and structural variables;
- (b)
- PC2 (scoring 11% of total variance): abtw = −0.74; adeg = 0.72, aclose = 0.76, E = 0.81; again, this variance is not addressed by relaxation dynamics.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patrick, J.W.; Boone, C.D.; Liu, W.; Conover, G.M.; Liu, Y.; Cong, X.; Laganowsky, A. Allostery revealed within lipid binding events to membrane proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 2976–2981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cournia, Z.; Chatzigoulas, A. Allostery in membrane proteins. Curr. Opin. Struct. Biol. 2020, 62, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Choi, S.; Hyeon, C. Mapping the intramolecular signal transduction of G-protein coupled receptors. Proteins Struct. Funct. Bioinform. 2014, 82, 727–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusach, A.; Maslov, I.; Luginina, A.; Borshchevskiy, V.; Mishin, A.; Cherezov, V. Beyond structure: Emerging approaches to study GPCR dynamics. Curr. Opin. Struct. Biol. 2020, 63, 18–25. [Google Scholar] [CrossRef]
- O’Brien, E.S.; Fuglestad, B.; Lessen, H.J.; Stetz, M.A.; Lin, D.W.; Marques, B.S.; Gupta, K.; Fleming, K.G.; Wand, A.J. Membrane Proteins Have Distinct Fast Internal Motion and Residual Conformational Entropy. Angew. Chem. -Int. Ed. 2020, 59, 11108–11114. [Google Scholar] [CrossRef]
- Basith, S.; Lee, Y.; Choi, S. Understanding G protein-coupled receptor allostery via molecular dynamics simulations: Implications for drug discovery. In Methods in Molecular Biology; Humana Press: Totova, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Rasmussen, S.G.F.; Devree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Dror, R.O.; Arlow, D.H.; Borhani, D.W.; Jensen, M.; Piana, S.; Shaw, D.E. Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA 2009, 106, 4689–4694. [Google Scholar] [CrossRef] [Green Version]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Tee, W.-V.; Berezovsky, I.N. Learning about Allosteric Drugs and Ways to Design Them. J. Mol. Biol. 2022. in press. Available online: https://www.sciencedirect.com/science/article/pii/S0022283622002844?casa_token=GTChrX3mp_8AAAAA:vpqfy2zgFmYXx8z24lu3C3INRLwkzY2TU4k-NZgBqzDNyXaLpeomzULCmp9c7isXM1-2ufrO7lg#f0005 (accessed on 5 July 2022).
- Poudel, H.; Leitner, D.M. Activation-Induced Reorganization of Energy Transport Networks in the β2Adrenergic Receptor. J. Phys. Chem. B 2021, 125, 6522–6531. [Google Scholar] [CrossRef]
- di Paola, L.; de Ruvo, M.; Paci, P.; Santoni, D.; Giuliani, A. Protein contact networks: An emerging paradigm in chemistry. Chem. Rev. 2013, 113, 1598–1613. [Google Scholar] [CrossRef]
- Cimini, S.; di Paola, L.; Giuliani, A.; Ridolfi, A.; de Gara, L. GH32 family activity: A topological approach through protein contact networks. Plant Mol. Biol. 2016, 92, 401–410. [Google Scholar] [CrossRef]
- Hadi-Alijanvand, H.; di Paola, L.; Hu, G.; Leitner, D.; Verkhivker, G.; Sun, P.; Poudel, H.; Giuliani, A. Biophysical Insight into the SARS-CoV2 Spike–ACE2 Interaction and Its Modulation by Hepcidin through a Multifaceted Computational Approach. ACS Omega 2022, 7, 17024–17042. [Google Scholar] [CrossRef] [PubMed]
- di Paola, L.; Hadi-Alijanvand, H.; Song, X.; Hu, G.; Giuliani, A. The Discovery of a Putative Allosteric Site in the SARS-CoV-2 Spike Protein Using an Integrated Structural/Dynamic Approach. J. Proteome Res. 2020, 19, 4576–4586. [Google Scholar] [CrossRef] [PubMed]
- Minicozzi, V.; di Venere, A.; Nicolai, E.; Giuliani, A.; Caccuri, A.M.; di Paola, L.; Mei, G. Non-symmetrical structural behavior of a symmetric protein: The case of homo-trimeric TRAF2 (tumor necrosis factor-receptor associated factor 2). J. Biomol. Struct. Dyn. 2021, 39, 319–329. [Google Scholar] [CrossRef] [PubMed]
- di Venere, A.; Nicolai, E.; Minicozzi, V.; Caccuri, A.M.; di Paola, L.; Mei, G. The Odd Faces of Oligomers: The Case of TRAF2-C, A Trimeric C-Terminal Domain of TNF Receptor-Associated Factor. Int. J. Mol. Sci. 2021, 22, 5871. [Google Scholar] [CrossRef]
- Rasmussen, S.G.F.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.; Rasmussen, S.G.; Choi, H.-J.; Kuhn, P.; Weis, W.; Kobika, B.; Stevens, R.C. High-Resolution Crystal Structure of an Engineered Human β. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Sali, A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef]
- Allouche, A. Software News and Updates Gabedit—A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem. 2012, 32, 174–182. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- di Paola, L.; Mei, G.; di Venere, A.; Giuliani, A. Disclosing Allostery through Protein Contact Networks. In Methods in Molecular Biology; Humana Press Inc.: Totova, NJ, USA, 2021; pp. 7–20. [Google Scholar] [CrossRef]
- Kintali, S. Betweenness Centrality: Algorithms and Lower Bounds. arXiv 2008, arXiv:0809.1906. [Google Scholar]
- Guzzi, P.H.; di Paola, L.; Giuliani, A.; Veltri, P. PCN-Miner: An open-source extensible tool for the Analysis of Protein Contact Networks. Bioinformatics 2022, btac450. [Google Scholar] [CrossRef]
- Minicozzi, V.; di Venere, A.; Caccuri, A.M.; Mei, G.; di Paola, L. One for All, All for One: The Peculiar Dynamics of TNF-Receptor-Associated Factor (TRAF2) Subunits. Symmetry 2022, 14, 720. [Google Scholar] [CrossRef]
- Yeater, K.M.; Duke, S.E.; Riedell, W.E. Multivariate analysis: Greater insights into complex systems. Agron. J. 2015, 107, 799–810. [Google Scholar] [CrossRef] [Green Version]
- Gorban, A.N.; Tyukina, T.A.; Pokidysheva, L.I.; Smirnova, E.V. It is useful to analyze correlation graphs. Phys. Life Rev. 2022, 40, 15–23. [Google Scholar] [CrossRef]
- Broomhead, D.S.; King, G.P. Extracting qualitative dynamics from experimental data. Phys. D Nonlinear Phenom. 1986, 20, 217–236. [Google Scholar] [CrossRef]
- di Paola, L.; Paci, P.; Santoni, D.; de Ruvo, M.; Giuliani, A. Proteins as sponges: A statistical journey along protein structure organization principles. J. Chem. Inf. Model. 2012, 52, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, C.F. Relaxation Kinetics; Academic Press: Cambridge, MA, USA, 1976. [Google Scholar]
- Trulla, L.L.; Giuliani, A.; Zbilut, J.P.; Webber, C.L. Recurrence quantification analysis of the logistic equation with transients, Physics Letters. Sect. A Gen. At. Solid State Phys. 1996, 223, 255–260. [Google Scholar] [CrossRef]
- Giuliani, A.; Zbilut, J.P.; Conti, F.; Manetti, C.; Miccheli, A. Invariant features of metabolic networks: A data analysis application on scaling properties of biochemical pathways. Phys. A Stat. Mech. Its Appl. 2004, 337, 157–170. [Google Scholar] [CrossRef]
- Mojtahedi, M.; Skupin, A.; Zhou, J.; Castaño, I.G.; Leong-Quong, R.Y.Y.; Chang, H.; Trachana, K.; Giuliani, A.; Huang, S. Cell Fate Decision as High-Dimensional Critical State Transition. PLoS Biol. 2016, 14, e2000640. [Google Scholar] [CrossRef] [Green Version]
- Reid, K.M.; Yamato, T.; Leitner, D.M. Variation of Energy Transfer Rates across Protein-Water Contacts with Equilibrium Structural Fluctuations of a Homodimeric Hemoglobin. J. Phys. Chem. B 2020, 124, 1148–1159. [Google Scholar] [CrossRef] [Green Version]
- Ishikura, T.; Iwata, Y.; Hatano, T.; Yamato, T. Energy exchange network of inter-residue interactions within a thermally fluctuating protein molecule: A computational study. J. Comput. Chem. 2015, 36, 1709–1718. [Google Scholar] [CrossRef]
- Ota, K.; Yamato, T. Energy Exchange Network Model Demonstrates Protein Allosteric Transition: An Application to an Oxygen Sensor Protein. J. Phys. Chem. B 2019, 123, 768–775. [Google Scholar] [CrossRef]
- Leitner, D.M.; Yamato, T. Mapping Energy Transport Networks in Proteins; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; pp. 63–113. [Google Scholar] [CrossRef] [Green Version]
- Poudel, H.; Reid, K.M.; Yamato, T.; Leitner, D.M. Energy Transfer across Nonpolar and Polar Contacts in Proteins: Role of Contact Fluctuations. J. Phys. Chem. B 2020, 124, 9852–9861. [Google Scholar] [CrossRef]
- Enright, M.B.; Leitner, D.M. Mass fractal dimension and the compactness of proteins, Physical Review E-Statistical. Nonlinear Soft Matter Phys. 2005, 71, 011912. [Google Scholar] [CrossRef] [Green Version]
- Leitner, D.M.; Pandey, H.D.; Reid, K.M. Energy Transport across Interfaces in Biomolecular Systems. J. Phys. Chem. B 2019, 123, 9507–9524. [Google Scholar] [CrossRef]
- Yu, X.; Leitner, D.M. Anomalous diffusion of vibrational energy in proteins. J. Chem. Phys. 2003, 119, 12673–12679. [Google Scholar] [CrossRef]
- Liu, X.; Li, D.; Ma, M.; Szymanski, B.K.; Stanley, H.E.; Gao, J. Network resilience. Phys. Rep. 2022, 971, 1–108. [Google Scholar] [CrossRef]
- di Paola, L.; Giuliani, A. Protein contact network topology: A natural language for allostery. Curr. Opin. Struct. Biol. 2015, 31, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, S.; Choi, S.; Hyeon, C. Ultraslow Water-Mediated Transmembrane Interactions Regulate the Activation of A2A Adenosine Receptor. Biophys. J. 2016, 111, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Kaindl, J.; Korczynska, M.; Stößel, A.; Dengler, D.; Stanek, M.; Hübner, H.; Clark, M.J.; Mahoney, J.; Matt, R.A.; et al. An allosteric modulator binds to a conformational hub in the β2 adrenergic receptor. Nat. Chem. Biol. 2020, 16, 749–755. [Google Scholar] [CrossRef]
- Liu, X.; Ahn, S.; Kahsai, A.W.; Meng, K.C.; Latorraca, N.R.; Pani, B.; Venkatakrishnan, A.J.; Masoudi, A.; Weis, W.I.; Dror, R.O.; et al. Mechanism of intracellular allosteric β2 AR antagonist revealed by X-ray crystal structure. Nature 2017, 548, 480–484. [Google Scholar] [CrossRef]
- Chen, K.Y.M.; Keri, D.; Barth, P. Computational design of G Protein-Coupled Receptor allosteric signal transductions. Nat. Chem. Biol. 2020, 16, 77–86. [Google Scholar] [CrossRef]
- Newman, M.E.J. Assortative Mixing in Networks. Phys. Rev. Lett. 2002, 89, 208701. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Arrigo, N.; Paci, P.; di Paola, L.; Santoni, D.; de Ruvo, M.; Giuliani, A.; Castiglione, F. Characterizing Protein Shape by a Volume Distribution Asymmetry Index. Open Bioinform. J. 2012, 6, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Perez, C.S.d.; Zaccaria, A.; di Matteo, T. Asymmetric Relatedness from Partial Correlation. Entropy 2022, 24, 365. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, A.; Bing, N.; Hoeschele, I.; Mendes, P. Discovery of meaningful associations in genomic data using partial correlation coefficients. Bioinformatics 2004, 20, 3565–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Short Description |
|---|---|
| Network Topology | |
| DBA | Degree-based assortativity |
| Dy | Diadicity |
| H | Heterophilicity |
| HBAKD | Hydrophobicity-based assortativity |
| abtw | Average node betweenness centrality |
| acc | Average node clustering coefficient |
| adeg | Average node degree |
| asp | Average shortest path |
| aclose | Average node closeness centrality |
| E | Graph energy |
| Molecular Structure | |
| Radius of gyration | |
| The radius of gyration of hydrophobic residues | |
| The radius of gyration of polar residues | |
| corrHBKD | Hydrophobic core probability |
| Mass density | |
| MFD | Mass fractal dimension |
| Porosity (void fraction) | |
| AS | Asymmetry index |
| Inactive | Active | |
|---|---|---|
| Structural properties | ||
| MFD | 2.70 | 2.52 |
| RG, Å | 10.07 | 10.20 |
| ε | 0.31 | 0.38 |
| AS | 0.53 | 0.46 |
| corrHb | −0.12 | −0.09 |
| Topological properties | ||
| adeg | 7.27 | 7.50 |
| abtw | 842 | 816 |
| asp | 5.28 | 5.24 |
| E | 587.2 | 591.9 |
| Jacc | 0.703 | |
| X1 | X2 | X3 | |
|---|---|---|---|
| X1 | 1 | 0.72 | 0.70 |
| X2 | 0.72 | 1 | 0.74 |
| X3 | 0.70 | 0.74 | 1 |
| X1 | X2 | X3 | |
|---|---|---|---|
| X1 | 1 | 0.54 | 0.91 |
| X2 | 0.54 | 1 | 0.55 |
| X3 | 0.91 | 0.55 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Paola, L.; Poudel, H.; Parise, M.; Giuliani, A.; Leitner, D.M. A Statistical Journey through the Topological Determinants of the β2 Adrenergic Receptor Dynamics. Entropy 2022, 24, 998. https://doi.org/10.3390/e24070998
Di Paola L, Poudel H, Parise M, Giuliani A, Leitner DM. A Statistical Journey through the Topological Determinants of the β2 Adrenergic Receptor Dynamics. Entropy. 2022; 24(7):998. https://doi.org/10.3390/e24070998
Chicago/Turabian StyleDi Paola, Luisa, Humanath Poudel, Mauro Parise, Alessandro Giuliani, and David M. Leitner. 2022. "A Statistical Journey through the Topological Determinants of the β2 Adrenergic Receptor Dynamics" Entropy 24, no. 7: 998. https://doi.org/10.3390/e24070998
APA StyleDi Paola, L., Poudel, H., Parise, M., Giuliani, A., & Leitner, D. M. (2022). A Statistical Journey through the Topological Determinants of the β2 Adrenergic Receptor Dynamics. Entropy, 24(7), 998. https://doi.org/10.3390/e24070998