Probing the Dynamics of Solvation and Structure of the OH- Ion in Aqueous Solution from Picosecond Transient Absorption Measurements

Abstract
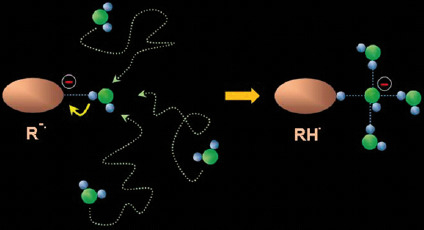
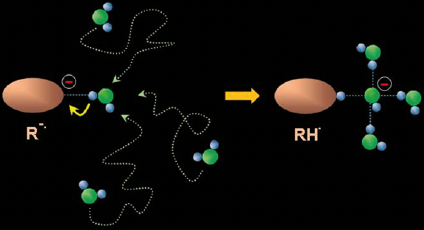
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
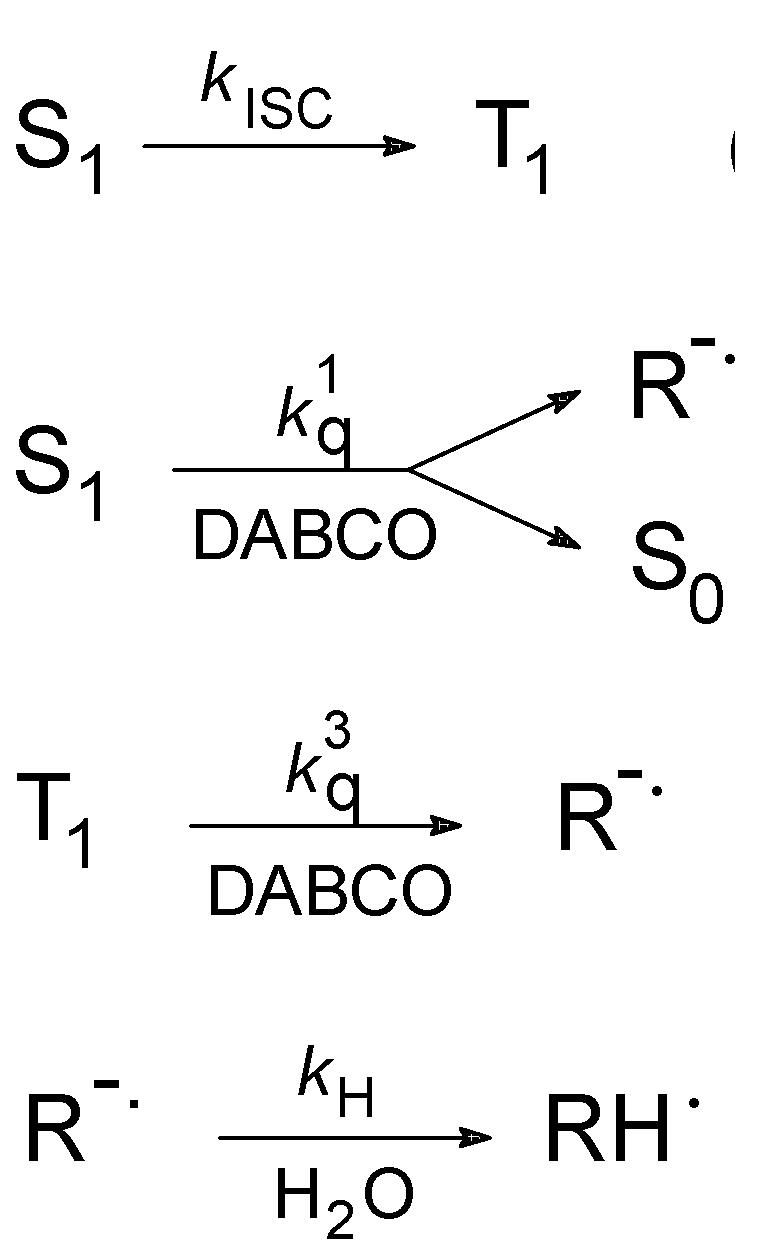{kind=link}
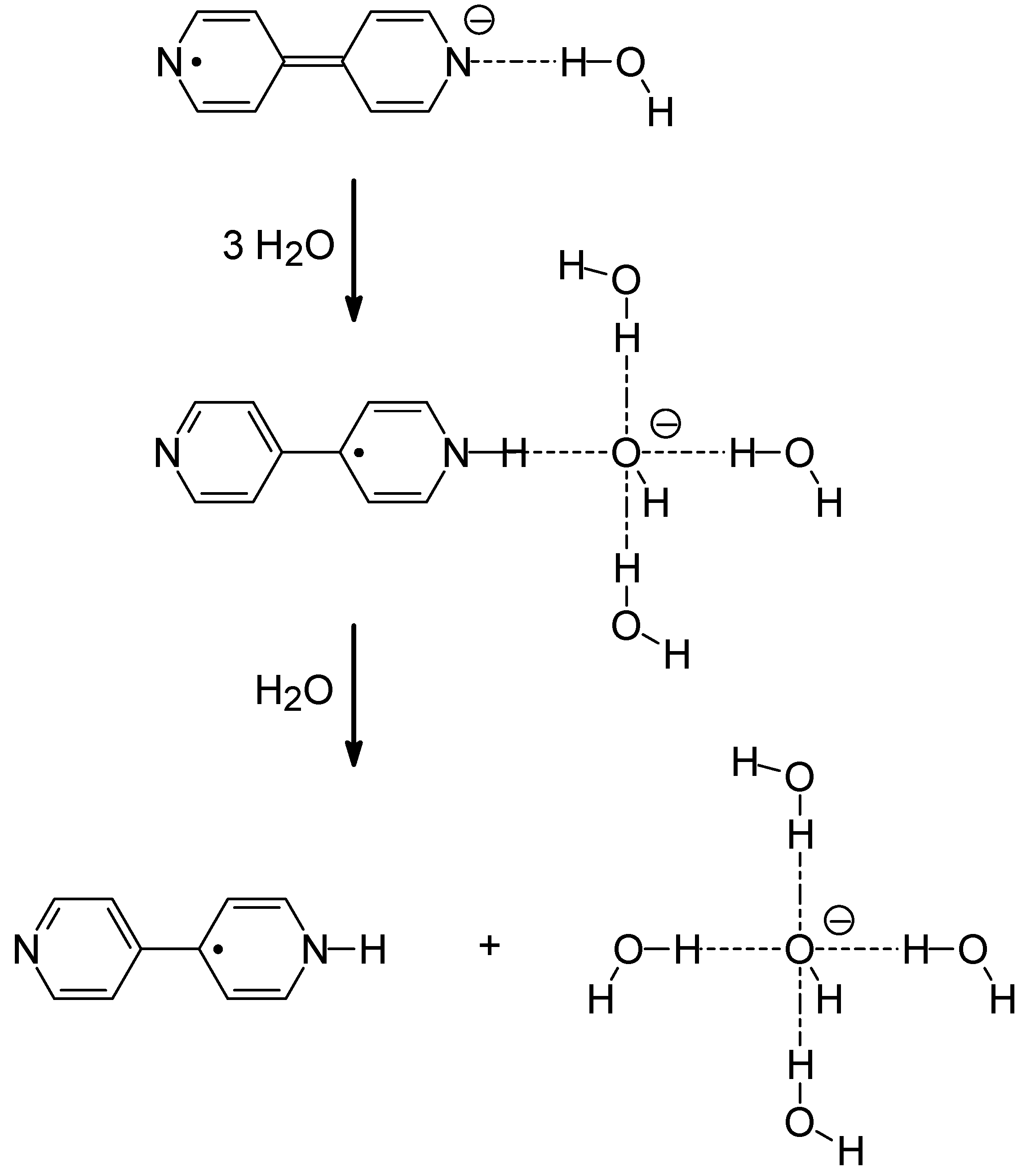{kind=link}
1. Introduction

2. Results and Discussion






3. Experimental
3.1. General
3.2. Transient absorption measurements
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- Eisenberg, D.; Kauzmann, W. The Structure and Properties of Water; Oxford University Press: Oxford, UK, 1969; p. 225. [Google Scholar]
- de Grotthuss, C.J.T. Sur la décomposition de l'eau et des corps qu'elle tient en dissolution à l'aide de l'électricité galvanique. Ann. Chim. 1806, 58, 54–74. [Google Scholar]
- Atkins, P.; de Paula, J. Atkins’s Physical Chemistry, 7th ed; Oxford University Press: Oxford, UK, 2002; p. 837, Chapter 24. [Google Scholar]
- Halle, B.; Karlström, G. Prototropic charge migration in water. Part 1.—Rate constants in light and heavy water and in salt solution from oxygen-17 spin relaxation. J. Chem. Soc., Faraday Trans. 2 1983, 79, 1031–1046. [Google Scholar] [CrossRef]
- Eigen, M. Proton transfer, acid-base catalysis, and enzymatic hydrolysis. Part I: elementary processes. Angew. Chem. Int. Ed. 1964, 3, 1–19. [Google Scholar] [CrossRef]
- Schiöberg, D.; Zundel, G. Very polarisable hydrogen bonds in solutions of bases having infra-red absorption continua. J. Chem. Soc., Faraday Trans. 2 1973, 69, 771–781. [Google Scholar] [CrossRef]
- Zundel, G. Hydrogen bonds with large proton polarizability and proton transfer processes in electrochemistry and biology. Adv. Chem. Phys. 2000, 111, 1–217. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Laasonen, K.; Sprik, M.; Parrinello, M. Ab initio simulations of water and water ions. J. Phys.: Condens. Matter 1994, 6, A93–A100. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Laasonen, K.; Sprik, M.; Parrinello, M. Ab initio molecular dynamics simulation of the solvation and transport of H3O+ and OH- ions in water. J. Phys. Chem. 1995, 99, 5749–5752. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Laasonen, K.; Sprik, M.; Parrinello, M. Ab initio molecular dynamics simulation of the solvation and transport of hydronium and hydroxyl ions in water. J. Chem. Phys. 1995, 103, 150–161. [Google Scholar] [CrossRef]
- Agmon, N. The Grotthuss mechanism. Chem. Phys. Lett. 1995, 244, 456–462. [Google Scholar] [CrossRef]
- Ando, K.; Hynes, J.T. HCl acid ionization in water: A theoretical molecular modelling. J. Molec. Liquids. 1995, 64, 25–34. [Google Scholar]
- Ando, K.; Hynes, J.T. Molecular mechanism of HCl acid ionization in water. Ab initio potential energy surfaces and Monte Carlo simulations. J. Phys. Chem. B 1997, 101, 10464–10478. [Google Scholar] [CrossRef]
- Lobaugh, J.; Voth, G.A. The quantum dynamics of an excess proton in water. J. Chem. Phys. 1996, 104, 2056–2069. [Google Scholar] [CrossRef]
- Vuilleummier, R.; Borgis, D. Transport and spectroscopy of the hydrated proton: A molecular dynamics study. J. Chem. Phys. 1999, 111, 4251–4266. [Google Scholar] [CrossRef]
- Schmitt, U.; Voth, G.A. The computer simulation of proton transport in water. J. Chem. Phys. 1999, 111, 9361–9381. [Google Scholar] [CrossRef]
- Marx, D.; Tuckerman, M.E.; Hutter, J.; Parrinello, M. The nature of the hydrated excess proton in water. Nature (London) 1999, 397, 601. [Google Scholar] [CrossRef]
- Hynes, J.T. The protean proton in water. Nature (London) 1999, 397, 565–567. [Google Scholar] [CrossRef]
- Marx, D.; Tuckerman, M.E.; Parrinello, M. Solvated excess protons in water: quantum effects on the hydration structure. J. Phys.: Condens. Matter 2000, 12, A153–A159. [Google Scholar] [CrossRef]
- Lapid, H.; Agmon, N.; Petersen, M.K.; Voth, G.A. a bond-order analysis of the mechanism for hydrated proton mobility in liquid water. J. Chem. Phys. 2005, 122, 014506/1–014506/11. [Google Scholar]
- Asthagiri, D.; Pratt, L.R.; Kress, J.D. Ab initio molecular dynamics and quasichemical study of H+(aq). Proc. Natl. Acad. Sci. USA 2005, 102, 6704–6708. [Google Scholar]
- Rini, M.; Magnes, B.Z.; Pines, E.; Nibbering, E.T.J. Real-time observation of bimodal proton transfer in acid-base pairs in water. Science 2003, 301, 349–352. [Google Scholar] [CrossRef]
- Mohammed, O.F.; Pines, D.; Dreyer, J.; Pines, E.; Nibbering, E.T.J. Sequential proton transfer through water bridges in acid-base reactions. Science 2005, 310, 83–86. [Google Scholar] [CrossRef]
- Mohammed, O.F.; Pines, D.; Nibbering, E.T.J.; Pines, E. Based-induced solvent switches in acid-base reactions. Angew. Chem. Int. Ed. 2007, 46, 1458–1461. [Google Scholar] [CrossRef]
- Smiechowski, M.; Stangret, J. Hydroxide ion hydration in aqueous solutions. J. Phys. Chem. A 2007, 111, 2889–2897. [Google Scholar] [CrossRef]
- Stillinger, F.H. Theoretical Chemistry: Advances and Perspectives; Eyring, H., Henderson, D., Eds.; Academic Press: New York, NY, USA, 1978. [Google Scholar]
- Tunon, I.; Rinaldi, D.; Ruiz-Lopez, M.F.; Rivail, J.L. Hydroxide ion in liquid water: structure, energetics, and proton transfer using a mixed discrete-continuum ab initio model. J. Phys. Chem. 1995, 99, 3798–3805. [Google Scholar]
- Meot-Ner, M.; Speller, C.V. Filling of solvent shells about ions. 1. Thermochemical criteria and the effects of isomeric clusters. J. Phys. Chem. 1986, 90, 6616–6624. [Google Scholar] [CrossRef]
- Meot-Ner, M. Multicomponent cluster ions. 2. Comparative stabilities of cationic and anionic hydrogen-bonded networks. Mixed clusters of water-methanol. J. Am. Chem. Soc. 1986, 108, 6189–6197. [Google Scholar] [CrossRef]
- Robertson, W.H.; Diken, E.G.; Price, E.A.; Shin, J.-W.; Johnson, M.A. Snapshots of water at work. Science 2003, 299, 1367–1372. [Google Scholar] [CrossRef]
- Tuckermann, M.E.; Marx, D.; Parrinello, M. The nature and transport mechanism of hydrated hydroxide ions in aqueous solution. Nature 2002, 417, 925–929. [Google Scholar] [CrossRef]
- Asthagiri, D.; Pratt, L.R.; Kress, J.D.; Gomez, M.A. The hydration state of HO-(aq). Chem. Phys. Lett. 2003, 380, 530–535. [Google Scholar] [CrossRef]
- Asthagiri, D.; Pratt, L.R.; Kress, J.D.; Gomez, M.A. Hydration and mobility of HO-(aq). Proc. Natl. Acad. Sci. 2004, 101, 7229–7233. [Google Scholar]
- Tuckermann, M.E.; Chandra, A.; Marx, D. Structure and dynamics of OH-(aq). Acc. Chem. Res. 2006, 39, 151–158. [Google Scholar] [CrossRef]
- Ludwig, R. New insight into the transport mechanism of hydrated hydroxide ions in water. Angew. Chem., Int. Ed. 2003, 42, 258–260. [Google Scholar] [CrossRef]
- Chandra A.; Tuckerman, M.E.; Marx, D. Connecting solvation shell structure to proton transport kinetics in hydrogen-bonded networks via population correlation functions. Phys. Rev. Lett. 2007, 99, 145901.1–145901.4. [Google Scholar]
- Marx, D.; Chandra, A.; Tuckerman, M.E. Aqueous basic solutions: hydroxide solvation, structural diffusion, and comparison to the hydrated proton. Chem. Rev. 2010, 110, 2174–2216. [Google Scholar] [CrossRef]
- Botti, A.; Bruni, F.; Imberti, S.; Ricci, M.A.; Soper, A.K. Solvation of hydroxyl ions in water. J. Chem. Phys. 2003, 119, 5001–5004. [Google Scholar] [CrossRef]
- Imberti, S.; Botti, A.; Bruni, F.; Cappa, G.; Ricci, M.A.; Soper, A.K. Ions in water: The microscopic structure of concentrated hydroxide solutions. J. Chem. Phys. 2005, 122, 194509:1–194509:9. [Google Scholar]
- Megyes, T.; Bálint, S.; Grósz, T.; Radnai, T.; Bakó I. The structure of aqueous sodium hydroxide solutions: A combined solution x-ray diffraction and simulation study. J. Chem. Phys. 2008, 128, 044501:1–044501:12. [Google Scholar]
- Cappa, C.D.; Smith, J.D.; Messer, B.M.; Cohen, R.C.; Saykally, R.J. Nature of the aqueous hydroxide ion probed by X-ray absorption spectroscopy. J. Phys. Chem. A 2007, 111, 4776–4785. [Google Scholar]
- Aziz, E.F.; Ottosson, N.; Fubel, M.; Hertel, I.V.; Winter, B. Interaction between liquid water and hydroxide revealed by core-hole de-excitation. Nature 2008, 455, 89–91. [Google Scholar]
- Petersen, C.; Thogersen, J.; Jensen, S.K.; Keiding, S.R. Electron detachment and relaxation of OH-(aq). J. Phys. Chem. A 2007, 111, 11410–11420. [Google Scholar] [CrossRef]
- Thogersen, J.; Jensen, S.K.; Petersen, C.; Keiding, S.R. Reorientation of hydroxide ions in water. Chem. Phys. Lett. 2008, 466, 1–5. [Google Scholar]
- Roberts, S.T.; Petersen, P.B.; Ramasesha, K.; Tokmakoff, A.; Ufimtsev, I.S.; Martinez, T.J. Observation of a Zundel-like transition state during proton transfer in aqueous hydroxide solutions. Proc. Natl. Acad. Sci. USA 2009, 106, 15154–15159. [Google Scholar]
- Boilet, L.; Buntinx, G.; Lefumeux, C.; Poizat, O. Picosecond dynamics of the photoreduction of 4,4’-bipyridine by 1,4-diazabicyclo[2.2.2]octane in water. J. Phys. Chem. A 2002, 106, 10222–10230. [Google Scholar]
- Poizat, O.; Buntinx, G.; Boilet, L. The photoreduction of 4,4’-bipyridine by amines in acetonitrile-water mixtures: influence of H-bonding on the ion-pair structure and dynamics. J. Phys. Chem. A 2005, 109, 10813–10823. [Google Scholar]
- Boilet, L.; Burdzinsky, G.; Buntinx, G.; Lefumeux, C.; Poizat, O. Picosecond absorption and resonance Raman investigation of the dynamics of the photoreduction of 4,4’-bipyridine by aliphatic amines in acetonitrile solution. J. Phys. Chem. A 2001, 105, 10271–10277. [Google Scholar] [CrossRef]
- Castella-Ventura, M.; Kassab, E. Vibrational analysis of some transient species implicated in the photoreduction of 4,4’-bipyridine based on ab initio and density functional calculations. J. Raman Spectrosc. 1998, 29, 511–536. [Google Scholar] [CrossRef]
- Lapouge, C.; Buntinx, G.; Poizat, O. Resonance Raman spectra simulation of the 4,4'-bipyridine anion radical and N-protonated radical. J. Phys. Chem. A 2002, 106, 4168–4175. [Google Scholar] [CrossRef]
- Buntinx, G.; Naskrecki, R.; Poizat, O. Sub-picosecond transient absorption analysis of the photophysics of 4,4’-bipyridine and 2,2’-bipyridine in solution. J. Phys. Chem. 1996, 100, 19380–19388. [Google Scholar] [CrossRef]
- Didierjean, C.; Dewaele, V.; Buntinx, G.; Poizat, O. The structure of the lowest excited singlet (S1) state of 4,4’-bipyridine: a picosecond time-resolved Raman analysis. Chem. Phys. 1998, 237, 169–181. [Google Scholar] [CrossRef]
- Didierjean, C.; Buntinx, G.; Poizat, O. Ultrafast intermolecular hydrogen abstraction by 4,4’-bipyridine from alcohols : time-resolved Raman analysis. J. Phys. Chem. A 1998, 102, 7938–7944. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
Poizat, O.; Buntinx, G. Probing the Dynamics of Solvation and Structure of the OH- Ion in Aqueous Solution from Picosecond Transient Absorption Measurements. Molecules 2010, 15, 3366-3377. https://doi.org/10.3390/molecules15053366
Poizat O, Buntinx G. Probing the Dynamics of Solvation and Structure of the OH- Ion in Aqueous Solution from Picosecond Transient Absorption Measurements. Molecules. 2010; 15(5):3366-3377. https://doi.org/10.3390/molecules15053366
Chicago/Turabian StylePoizat, Olivier, and Guy Buntinx. 2010. "Probing the Dynamics of Solvation and Structure of the OH- Ion in Aqueous Solution from Picosecond Transient Absorption Measurements" Molecules 15, no. 5: 3366-3377. https://doi.org/10.3390/molecules15053366
APA StylePoizat, O., & Buntinx, G. (2010). Probing the Dynamics of Solvation and Structure of the OH- Ion in Aqueous Solution from Picosecond Transient Absorption Measurements. Molecules, 15(5), 3366-3377. https://doi.org/10.3390/molecules15053366