Specific Radioactivity of Neutron Induced Radioisotopes: Assessment Methods and Application for Medically Useful 177Lu Production as a Case

Abstract
:1. Introduction
1.1. Units of specific radioactivity, their conversion and SA of carrier-free radionuclide



2. Theoretical Approach and Assessment Methods



- S1,A is the target stable isotope of element A in the target; Sg,A (with g ≥ 2) is the impure stable isotope of element A originally presented or produced in the target.
- S1,B is the target stable isotope of element B in the target; S2,B is the stable isotope of element B in the target.
- Ri,A or Ri is the wanted radioisotope of element A produced in the target from stable isotope S1,A.
- Rx and Ry are the radioisotopes of element B produced in the target.
- The particle emitted from reaction (n, particle) may be proton or alpha.
- σ(th), σ(epi) and σ(fast) are reaction cross sections for thermal, epi-thermal and fast neutrons, respectively.
- σ1,i(th), σ2,x(th), σ2,y(th),. are cross sections of thermal neutrons for the formation of isotopes i, x, y,. from stable isotope 1, 2, 2, respectively.
- λ is the decay constant.
2.1. The specific radioactivity of radionuclide Ri in the simple target system for the (n, γ) reaction based radioisotope production
2.1.1. Main characteristics of the simple target system
2.1.1.1. The target burn-up for each isotope in simple target system








2.1.1.2. Reaction yield of radioisotope Ri in the simple target system









2.1.2. The SA assessment of radionuclide Ri in the simple target for (n, γ) reaction based radioisotope production
2.1.2.1. General formula of SA calculation for the simple multi-isotope target


2.1.2.2. SA of radioisotope Ri in the simple two-isotope target






2.1.2.3. SA of radioisotope Ri in the simple one-isotope target system


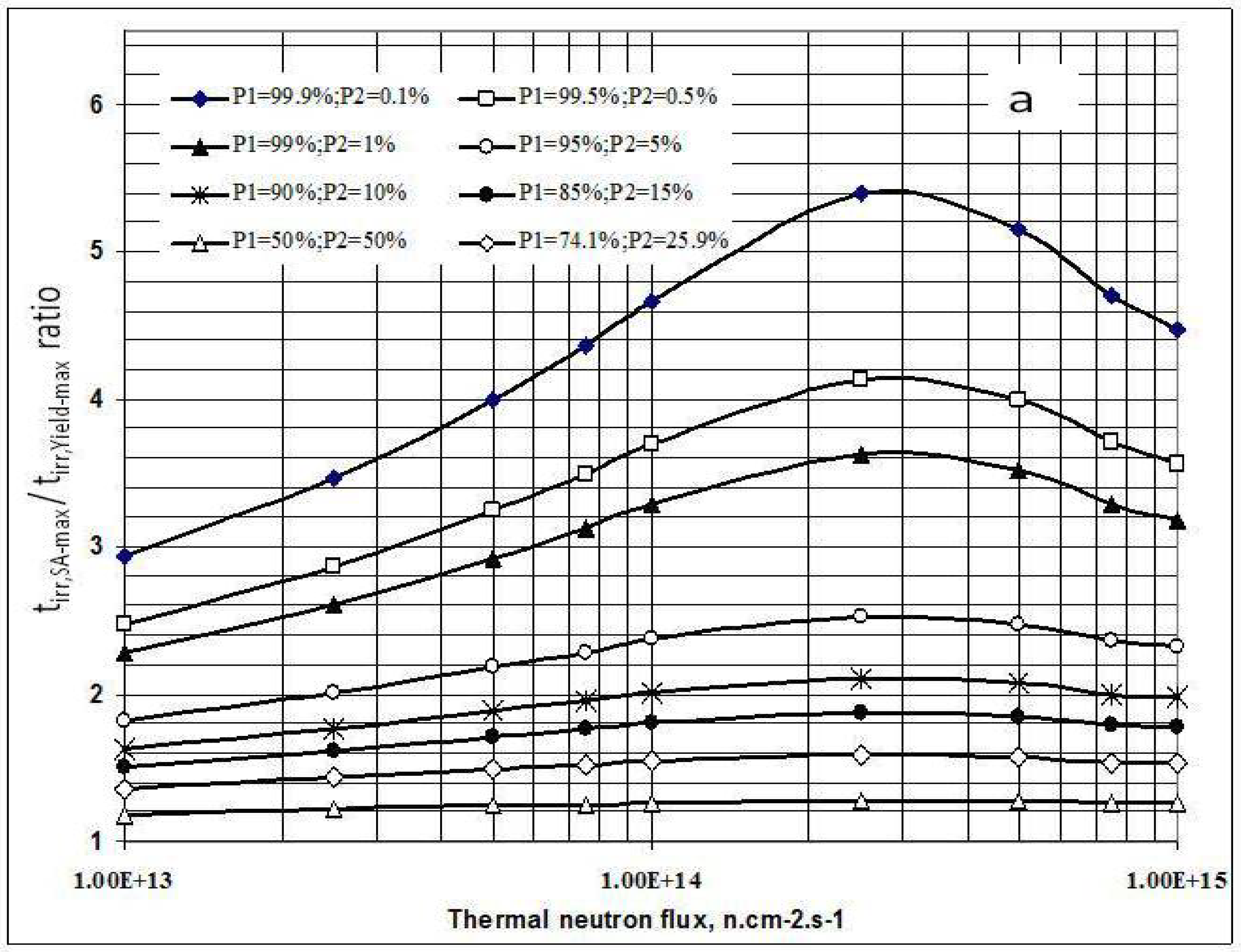
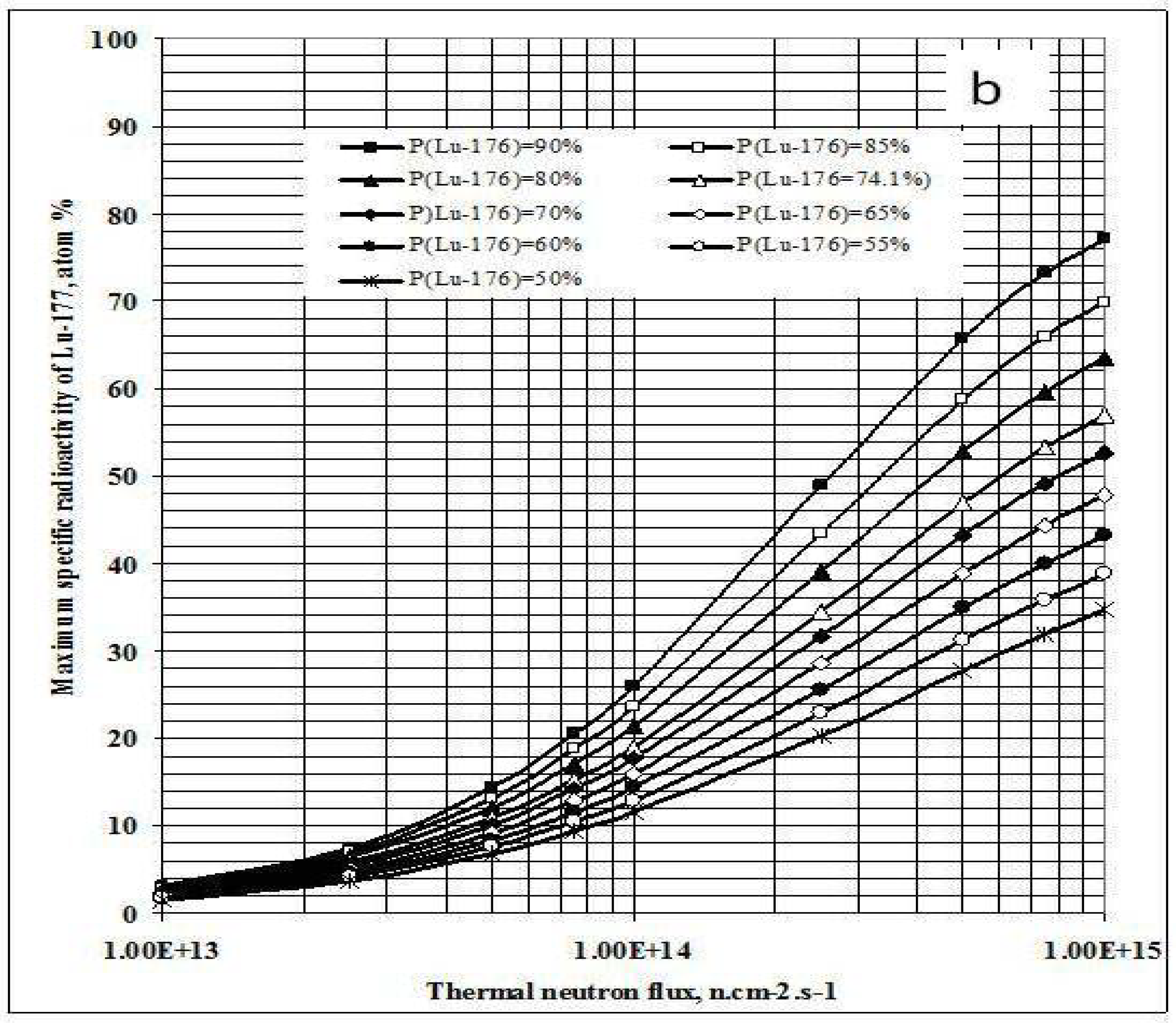
2.2. The specific radioactivity of radionuclide Ri in a complex target system for the S(n, γ) Rx (β- decay) Ri reaction based radioisotope production
2.2.1. Main characteristics of the complex target system
2.2.1.1. The yield of S1B(n, γ) Rx (β- decay) Ri reaction

2.2.1.2. SA-degradation effect of impure stable isotope generated from S2,B(n, γ) Ry(β- decay) Sg,Areaction










2.2.1.3. SA-degradation effect of impure isotopes of the chemical element A
2.2.2. The SA assessment of radionuclide Ri in a complex target system

2.3. Notes on formalism









3. Experimental
3.1. Reagents and materials
3.2. Targets, reactor irradiations, chemical separation, elemental analysis and radioactivity calibration
4. Results and Discussion
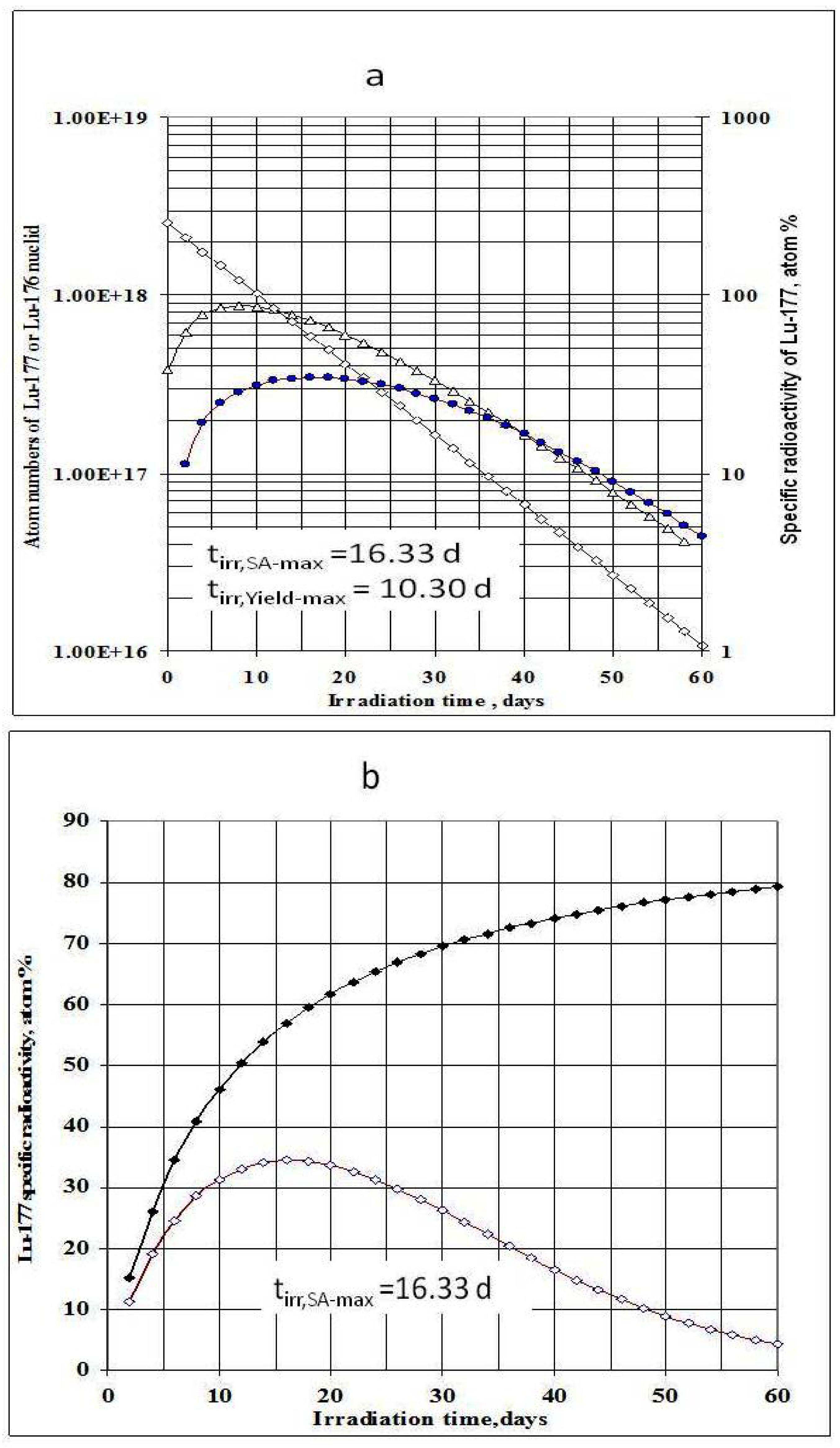
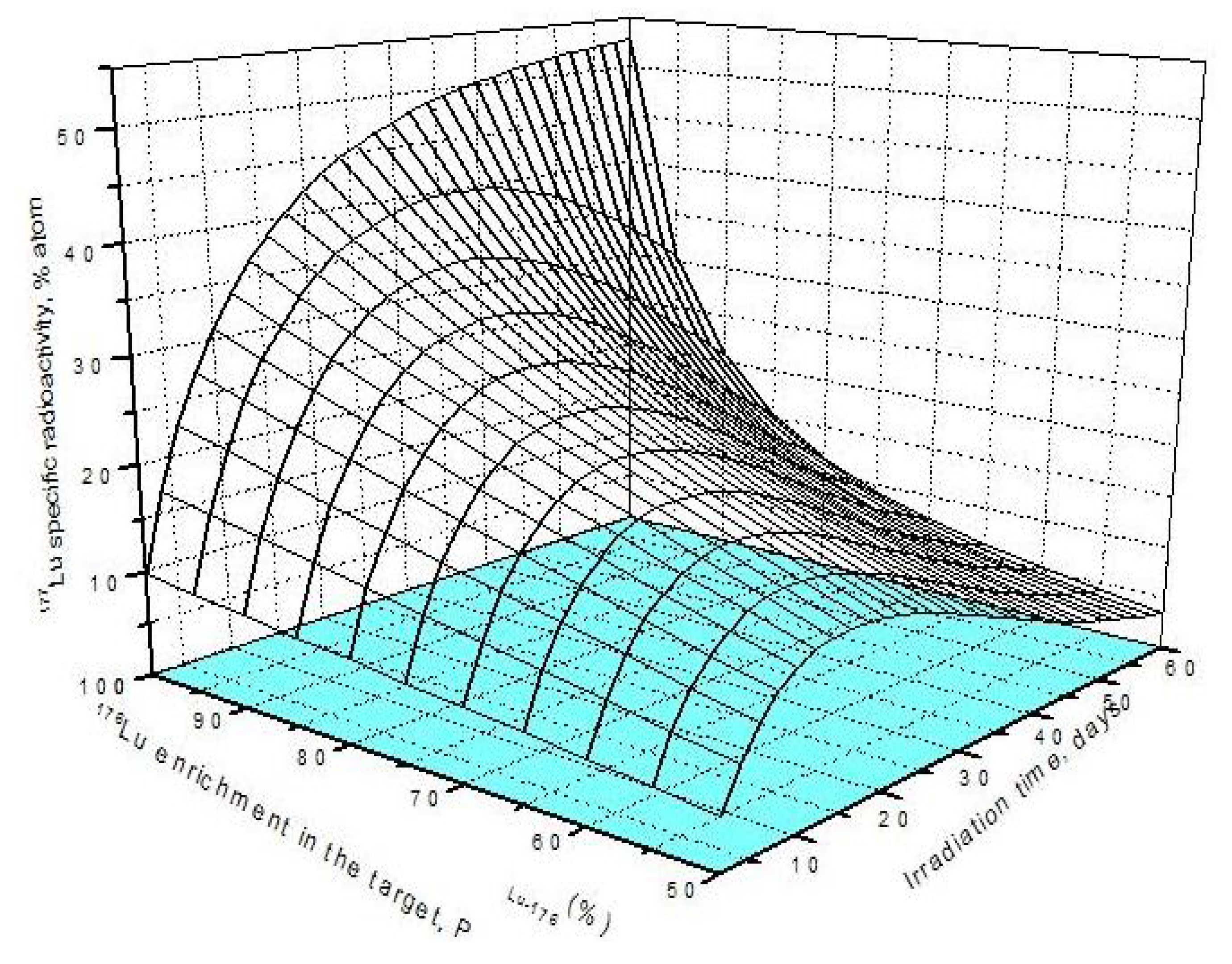
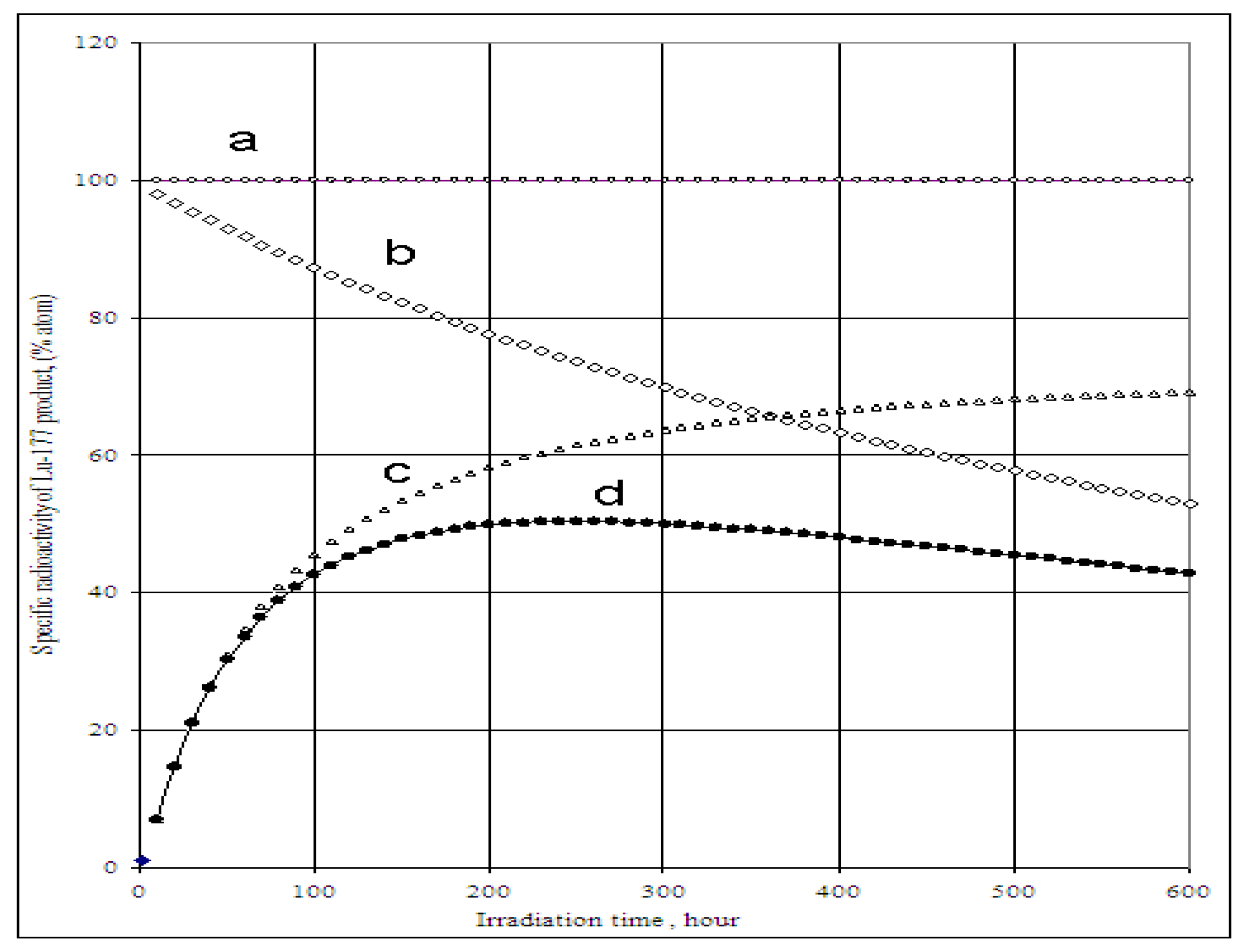
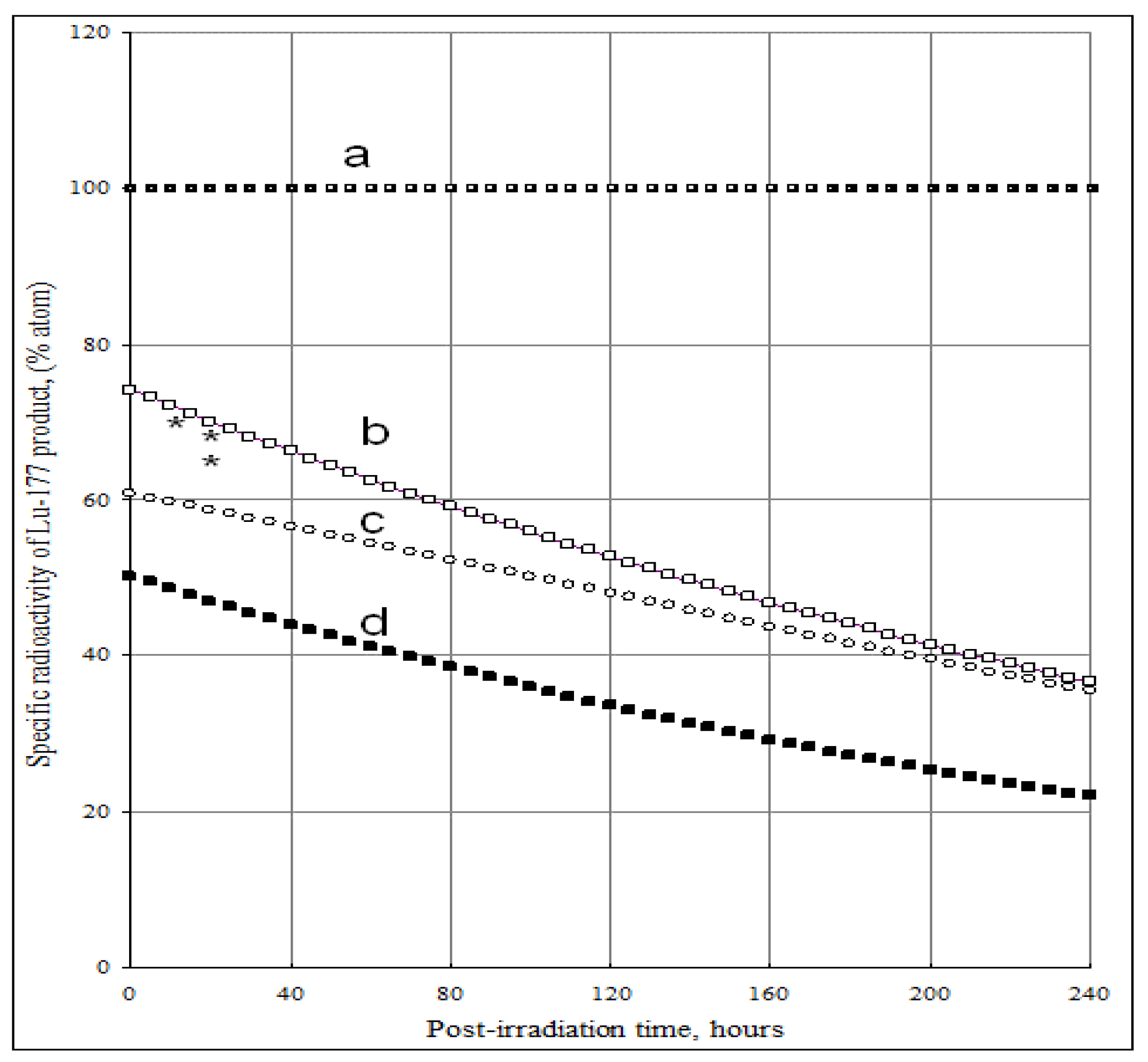
4.1. The specific radioactivity of 177Lu radioisotope produced via 176Lu (n, γ)177Lu reaction
4.2. The specific radioactivity of 177Lu radioisotope produced via neutron- capture- followed- by- radioactive transformation, 176Yb (n, γ) 177Yb (β- decay) 177Lu
4.2.1. 177Lu radioactive source 1 (S1): Radioactivity (A1-Lu--177) and specific radioactivity (SA1-Lu--177) of 177Lu isotope in the elementally pure (Lu impurity-free) Yb target







4.2.2. 177Lu radioactive source 2 (S2): Radioactivity A2-Lu-177) and specific radioactivity SA2-Lu-177 of 177Lu generated from 176Lu (n, γ) 177Lu reaction of the elemental Lu-impurity in the Lu-contaminated 176Yb target

4.2.3. 177Lu specific radioactivity SALu-177 and 177Lu radioactivity ALu-177 in the Lu-contaminated 176Yb target as a combination of 177Lu radioactive source S1 and S2

5. Conclusions
Acknowledgments
References
- Knapp, F.F., Jr.; Mirzadeh, S.; Beets, A.L.; Du, M. Production of therapeutic radioisotopes in the ORNL High Flux Isotope Reactor (HFIR) for applications in nuclear medicine, oncology and interventional cardiology. J. Radioanal. Nucl. Chem. 2005, 263, 503–509. [Google Scholar] [CrossRef]
- Pillai, M.R.A.; Chakraborty, S.; Das, T.; Venkatesh, M.; Ramamoorthy, N. Production logistics of 177Lu for radionuclide therapy. Appl. Radiat. Isot. 2003, 59, 109–118. [Google Scholar] [CrossRef]
- Mirzadeh, S.; Walsh, P. Numerical evaluation of the production of radionuclides in a nuclear reactor (Part I). Appl. Radiat. Isot. 1998, 49, 379–382. [Google Scholar] [CrossRef]
- International Atomic Energy Agency. Manual for reactor produced radioisotopes, IAEA-TECDOC-1340; IAEA: Vienna, Austria, 2003; pp. 121–124. [Google Scholar]
- Dvorakova, Z.; Henkelmann, R.; Lin, X.; Turler, A.; Gerstenberg, H. Production of 177Lu at the new research reactor FRM-II: Irradiation yield of 176Lu (n, γ)177Lu. Appl. Radiat. Isot. 2008, 66, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Zhernosekov, K.P.; Perego, R.C.; Dvorakova, Z.; Henkelmann, R.; Turler, A. Target burn-up corrected specific activity of 177Lu produced via of 176Lu (n, γ)177Lu nuclear reactions. Appl. Radiat. Isot. 2008, 66, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczak, R.; Parus, J.L.; Pawlak, D.; Zakrzewska, E.; Michalak, W.; Sasinowska, I. Reactor produced 177Lu of specific activity and purity suitable for medical applications. J. Radioanal. Nucl. Chem. 2003, 257, 53–57. [Google Scholar] [CrossRef]
- Le, V.S.; Morcos, N.; Zaw, M.; Pellegrini, P.; Greguric, I. Alternative chromatographic processes for no-carrier added 177Lu radioisotope separation. Part I. Multi-column chromatographic process for clinically applicable. J. Radioanal. Nucl. Chem. 2008, 277, 663–673. [Google Scholar]
- Le, V.S.; Morcos, N.; Zaw, M.; Pellegrini, P.; Greguric, I.; Nevissi, A. Alternative chromatographic processes for no-carrier added 177Lu radioisotope separation. Part II. The conventional column chromatographic separation combined with HPLC for high purity. J. Radioanal. Nucl. Chem. 2008, 277, 675–683. [Google Scholar]
- Trace Sciences International Inc. Certificate of Analysis #1, Jan, 2003. Trace Sciences International Corp.: Richmond Hill, ON L4B 3N6, Canada.
- Parrington, J.R.; Knox, H.D.; Breneman, S.L.; Baum, E.M.; Feiner, F. Nuclides and Isotopes, 15th ed.; KAPL, Inc. and General Electric Co. Press: California, USA, 1996; pp. 7–51. [Google Scholar]
- Rubinson, W. The equations of radioactive transformation I a neutron flux. J. Chem. Phys. 1949, 17, 542–547. [Google Scholar] [CrossRef]
- Verheijke, M.L. Relation between the Hogdahl convention and the modified Westcott formalism for (n, γ) reactions with a pure 1/vn cross-section behavior. J. Radioanal. Nucl. Chem. 2008, 183, 293–299. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Stable Isotope (Denoted) | Conc. in target | Cross Sections, Barn | Nuclear reaction and product (T1/2) | Reaction No. (Reaction branch) | |
|---|---|---|---|---|---|
| σth or σ0 | σepi or I0 | ||||
| 176Lu (S1,Lu) | P1 =74.1 % | 2300 | 1200 | 176Lu (n,γ)177Lu (6.7d)(*) Hf (stable) | Lu-1 (i = 1) |
| 2 | 3 | 176Lu (n,γ)177mLu(160.7d) Lu(6.7d) Hf(s) | Lu-2 (i = 2) | ||
| <2.10−3 | - | 176Lu (n,α) 173Tm (8.2h) Yb ( Stable) | Lu-3 (i = 3) | ||
| 175Lu(S2,Lu) | Pimp,Lu =25.9 % | 16 | 550 | 175Lu (n,γ) 176mLu (3.7h) Lu ( Stable) | Lu-4 (y = 1) |
| 9 | 300 | 175Lu (n,γ) 176Lu ( Stable) | Lu-5 (y = 2) | ||
| <10−5 | 175Lu (n,α) 172Tm (2.6d ) Yb (Stable) | Lu-6 (y = 3) | |||
| Stable Isotope (Denoted) | Conc. in target(%) | Cross Section, Barns | Nuclear reactions and products (T1/2) | Reaction No. (Reaction branch) | |
|---|---|---|---|---|---|
| σth or σo | σepi or Io | ||||
| 176Yb (S1,B) | 97.6 | 3.0 | 8 | 176Yb(n,γ) 177Yb (1.9h) Lu (6.7d) Hf (stable) | Yb-1 (x = 1) |
| 174Yb (S2,B) | 1.93 | 63.0 | 60 | 174Yb (n,γ) 175Yb (4.2d) Lu (Stable) | Yb-2 (y = 1) |
| 173Yb (S3,B) | 0.18 | 17.4 | 400 | 173Yb (n,γ) 174Yb (Stable) | Yb-3 (z = 1) |
| 172Yb (S4,B) | 0.22 | 1.3 | 25 | 172Yb (n,γ) 173Yb (Stable) | Yb-4 (p = 1) |
| 171Yb (S5,B) | 0.07 | 50.0 | 320 | 171Yb (n,γ) 172Yb (Stable) | Yb-5 (q = 1) |
| 170Yb (S6,B) | <0.01 | 10.0 | 300 | 170Yb (n,γ) 171Yb (Stable) | Yb-6 (v = 1) |
| 168Yb (S7,B) | <0.01 | 2300 | 2100 | 168Yb(n,γ) 169Yb (32d) Tm (Stable) | Yb-7 (w = 1) |
| Lu (*) | 5.10-3 | Nuclear reactions and characteristics referred to Table 1 | |||
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Le, V.S. Specific Radioactivity of Neutron Induced Radioisotopes: Assessment Methods and Application for Medically Useful 177Lu Production as a Case. Molecules 2011, 16, 818-846. https://doi.org/10.3390/molecules16010818
Le VS. Specific Radioactivity of Neutron Induced Radioisotopes: Assessment Methods and Application for Medically Useful 177Lu Production as a Case. Molecules. 2011; 16(1):818-846. https://doi.org/10.3390/molecules16010818
Chicago/Turabian StyleLe, Van So. 2011. "Specific Radioactivity of Neutron Induced Radioisotopes: Assessment Methods and Application for Medically Useful 177Lu Production as a Case" Molecules 16, no. 1: 818-846. https://doi.org/10.3390/molecules16010818
APA StyleLe, V. S. (2011). Specific Radioactivity of Neutron Induced Radioisotopes: Assessment Methods and Application for Medically Useful 177Lu Production as a Case. Molecules, 16(1), 818-846. https://doi.org/10.3390/molecules16010818