Peptide Phage Display as a Tool for Drug Discovery: Targeting Membrane Receptors

Abstract
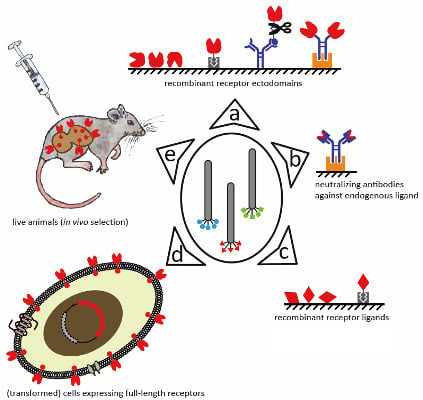
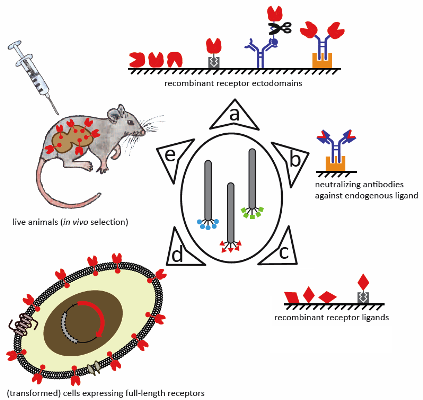
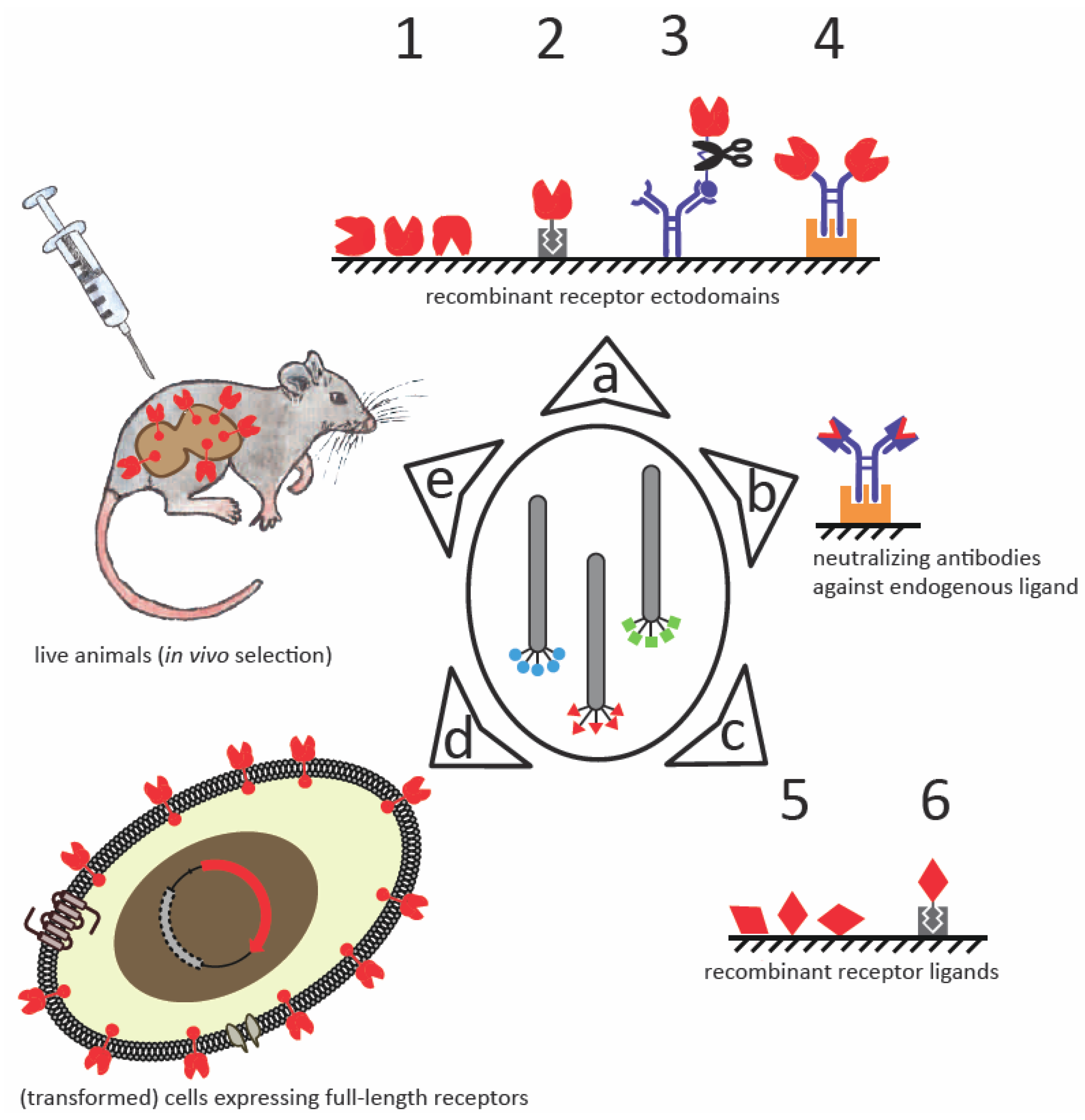
:
1. Introduction
2. General Considerations on Phage Display for Targeting Membrane Receptors
3. Biopanning Strategies on Membrane Receptors: From Single Molecules to Organisms
3.1. Recombinant Receptor Mimetics
3.1.1. Recombinant Receptor Fragments
3.1.2. Neutralizing Antibodies against Endogenous Binding Partners
3.2. Panning against Whole Cells
3.3. In Vivo Selection
3.4. Combining Selection Strategies
4. Further Optimization of Selected Peptides
4.1. Optimization of Amino Acid Sequence (Affinity Optimization)
4.2. Options for Improving the In Vivo Fate of Peptide Drug Candidates
5. Future Prospects
Acknowledgements
References and Notes
- Smith, G.P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Bratkovič, T. Progress in phage display: evolution of the technique and its applications. Cell. Mol. Life Sci. 2010, 67, 749–767. [Google Scholar] [CrossRef] [PubMed]
- Grøn, H.; Hyde-DeRuyscher, R. Peptides as tools in drug discovery. Curr. Opin. Drug Discov. Devel. 2000, 3, 636–645. [Google Scholar] [PubMed]
- Ja, W.W.; Roberts, R.W. G-protein-directed ligand discovery with peptide combinatorial libraries. Trends Biochem. Sci. 2005, 30, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, N.C.; Farrell, F.X.; Chang, R.; Kashyap, A.K.; Barbone, F.P.; Mulcahy, L.S.; Johnson, D.L.; Barrett, R.W.; Jolliffe, L.K.; Dower, W.J. Small peptides as potent mimetics of the protein hormone erythropoietin. Science 1996, 273, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Cwirla, S.E.; Balasubramanian, P.; Duffin, D.J.; Wagstrom, C.R.; Gates, C.M.; Singer, S.C.; Davis, A.M.; Tansik, R.L.; Mattheakis, L.C.; Boytos, C.M.; Schatz, P.J.; Baccanari, D.P.; Wrighton, N.C.; Barrett, R.W.; Dower, W.J. Peptide agonist of the thrombopoietin receptor as potent as the natural cytokine. Science 1997, 276, 1696–1699. [Google Scholar] [CrossRef] [PubMed]
- Su, J.L.; Lai, K.P.; Chen, C.A.; Yang, C.Y.; Chen, P.S.; Chang, C.C.; Chou, C.H.; Hu, C.L.; Kuo, M.L.; Hsieh, C.Y.; Wei, L.H. A novel peptide specifically binding to interleukin-6 receptor (gp80) inhibits angiogenesis and tumor growth. Cancer Res. 2005, 65, 4827–4835. [Google Scholar] [CrossRef] [PubMed]
- Hetian, L.; Ping, A.; Shumei, S.; Xiaoying, L.; Luowen, H.; Jian, W.; Lin, M.; Meisheng, L.; Junshan, Y.; Chengchao, S. A novel peptide isolated from a phage display library inhibits tumor growth and metastasis by blocking the binding of vascular endothelial growth factor to its kinase domain receptor. J. Biol. Chem. 2002, 277, 43137–43142. [Google Scholar] [CrossRef] [PubMed]
- Schooltink, H.; Rose-John, S. Designing cytokine variants by phage-display. Comb. Chem. High Throughput Screen. 2005, 8, 173–179. [Google Scholar] [CrossRef] [PubMed]
- McConnell, S.J.; Dinh, T.; Le, M.H.; Brown, S.J.; Becherer, K.; Blumeyer, K.; Kautzer, C.; Axelrod, F.; Spinella, D.G. Isolation of erythropoietin receptor agonist peptides using evolved phage libraries. Biol. Chem. 1998, 379, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Tipps, M.E.; Lawshe, J.E.; Ellington, A.D.; Mihic, S.J. Identification of novel specific allosteric modulators of the glycine receptor using phage display. J. Biol. Chem. 2010, 285, 22840–22845. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Wendler, P.; Connelly, G.; Lim, A.; Zhang, J.; King, M.; Li, T.; Silverman, J.A.; Schimmel, P.R.; Tally, F.P. Drug target validation: Lethal infection blocked by inducible peptide. Proc. Natl. Acad. Sci. USA 2000, 97, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.Y.; Lee, H.Y.; Kwak, W.; Yoo, J.; Na, M.H.; So, I.S.; Kwon, T.H.; Park, H.S.; Huh, S.; Oh, G.T.; Kwon, I.C.; Kim, I.S.; Lee, B.H. Phage display selection of peptides that home to atherosclerotic plaques: IL-4 receptor as a candidate target in atherosclerosis. J. Cell. Mol. Med. 2008, 12, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- De Berardinis, P.; Haigwood, N. New recombinant vaccines based on the use of prokaryotic antigen-display systems. Expert Rev. Vaccines 2004, 3, 673. [Google Scholar] [CrossRef] [PubMed]
- Skerra, A. Alternative non-antibody scaffolds for molecular recognition. Curr. Opin. Biotechnol. 2007, 18, 295. [Google Scholar] [CrossRef] [PubMed]
- Grönwall, C.; Stahl, S. Engineered affinity proteins - generation and applications. J. Biotechnol. 2009, 140, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, A.; Kolonin, M.G.; Molldrem, J.J.; Pasqualini, R.; Arap, W. Display technologies: application for the discovery of drug and gene delivery agents. Adv. Drug Deliv. Rev. 2006, 58, 1622–1654. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Kolonin, M.G.; Trepel, M.; Lahdenranta, J.; Cardo-Vila, M.; Giordano, R.J.; Mintz, P.J.; Ardelt, P.U.; Yao, V.J.; Vidal, C.I.; Chen, L.; Flamm, A.; Valtanen, H.; Weavind, L.M.; Hicks, M.E.; Pollock, R.E.; Botz, G.H.; Bucana, C.D.; Koivunen, E.; Cahill, D.; Troncoso, P.; Baggerly, K.A.; Pentz, R.D.; Do, K.A.; Logothetis, C.J.; Pasqualini, R. Steps toward mapping the human vasculature by phage display. Nat. Med. 2002, 8, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Hertveldt, K.; Belien, T.; Volckaert, G. General M13 phage display: M13 phage display in identification and characterization of protein-protein interactions. Methods Mol. Biol. 2009, 502, 321–339. [Google Scholar] [PubMed]
- Rowley, M.J.; O’Connor, K.; Wijeyewickrema, L. Phage display for epitope determination: A paradigm for identifying receptor-ligand interactions. Biotechnol. Annu. Rev. 2004, 10, 151–188. [Google Scholar] [PubMed]
- Sedlacek, R.; Chen, E. Screening for protease substrate by polyvalent phage display. Comb. Chem. High Throughput Screen. 2005, 8, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Kay, B.K.; Hamilton, P.T. Identification of enzyme inhibitors from phage-displayed combinatorial peptide libraries. Comb. Chem. High Throughput Screen. 2001, 4, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Honegger, A.; Pluckthun, A. Selection for improved protein stability by phage display. J. Mol. Biol. 1999, 294, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gacio, A.; Uguen, M.; Fastrez, J. Phage display as a tool for the directed evolution of enzymes. Trends Biotechnol. 2003, 21, 408–414. [Google Scholar] [CrossRef]
- Rosenberg, A.; Griffin, K.; Studier, F.; McCormick, M.; Berg, J.; Novy, R.; Mierendorf, R.; Cloning, P. T7Select® Phage Display System: A powerful new protein display system based on bacteriophage T7. Innovations 1996, 1–6. [Google Scholar]
- Hoess, R.H. Bacteriophage lambda as a vehicle for peptide and protein display. Curr. Pharm. Biotechnol. 2002, 3, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Krumpe, L.R.; Atkinson, A.J.; Smythers, G.W.; Kandel, A.; Schumacher, K.M.; McMahon, J.B.; Makowski, L.; Mori, T. T7 lytic phage-displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries. Proteomics 2006, 6, 4210–4222. [Google Scholar] [CrossRef] [PubMed]
- Gram, H.; Marconi, L.A.; Barbas, C.F., 3rd; Collet, T.A.; Lerner, R.A.; Kang, A.S. In vitro selection and affinity maturation of antibodies from a naive combinatorial immunoglobulin library. Proc. Natl. Acad. Sci. USA 1992, 89, 3576–3580. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, V.; Sorokulova, I.B.; Samoylov, A.M.; Simonian, A.L.; Petrenko, V.A.; Vodyanoy, V. Phage as a molecular recognition element in biosensors immobilized by physical adsorption. Biosens. Bioelectron. 2007, 22, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Skerra, A. Engineered protein scaffolds for molecular recognition. J. Mol. Recognit. 2000, 13, 167–187. [Google Scholar] [CrossRef]
- Nygren, P.A.; Skerra, A. Binding proteins from alternative scaffolds. J. Immunol. Methods 2004, 290, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, F.; Tanaka, Y.; Minari, Y.; Tokui, N. Designing scaffolds of peptides for phage display libraries. J. Biosci. Bioeng. 2005, 99, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Yanofsky, S.D.; Baldwin, D.N.; Butler, J.H.; Holden, F.R.; Jacobs, J.W.; Balasubramanian, P.; Chinn, J.P.; Cwirla, S.E.; Peters-Bhatt, E.; Whitehorn, E.A.; Tate, E.H.; Akeson, A.; Bowlin, T.L.; Dower, W.J.; Barrett, R.W. High affinity type I interleukin 1 receptor antagonists discovered by screening recombinant peptide libraries. Proc. Natl. Acad. Sci. USA 1996, 93, 7381–7386. [Google Scholar] [CrossRef] [PubMed]
- Giordano, R.J.; Cardo-Vila, M.; Lahdenranta, J.; Pasqualini, R.; Arap, W. Biopanning and rapid analysis of selective interactive ligands. Nat. Med. 2001, 7, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Ladner, R.C.; Sato, A.K.; Gorzelany, J.; de Souza, M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov. Today 2004, 9, 525–529. [Google Scholar] [CrossRef]
- Vrielink, J.; Heins, M.S.; Setroikromo, R.; Szegezdi, E.; Mullally, M.M.; Samali, A.; Quax, W.J. Synthetic constrained peptide selectively binds and antagonizes death receptor 5. FEBS J. 2010, 277, 1653–1665. [Google Scholar] [CrossRef] [PubMed]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv. 2010, 28, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Arinaminpathy, Y.; Khurana, E.; Engelman, D.M.; Gerstein, M.B. Computational analysis of membrane proteins: the largest class of drug targets. Drug Discov. Today 2009, 14, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Deller, M.C.; Yvonne Jones, E. Cell surface receptors. Curr. Opin. Struct. Biol. 2000, 10, 213–219. [Google Scholar] [CrossRef]
- Bantscheff, M.; Eberhard, D.; Abraham, Y.; Bastuck, S.; Boesche, M.; Hobson, S.; Mathieson, T.; Perrin, J.; Raida, M.; Rau, C.; Reader, V.; Sweetman, G.; Bauer, A.; Bouwmeester, T.; Hopf, C.; Kruse, U.; Neubauer, G.; Ramsden, N.; Rick, J.; Kuster, B.; Drewes, G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, F.; Schermuly, R.T.; Ghofrani, H.A. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat. Rev. Drug Discov. 2010, 9, 956–970. [Google Scholar] [CrossRef] [PubMed]
- Goubaeva, F.; Ghosh, M.; Malik, S.; Yang, J.; Hinkle, P.M.; Griendling, K.K.; Neubig, R.R.; Smrcka, A.V. Stimulation of cellular signaling and G protein subunit dissociation by G protein betagamma subunit-binding peptides. J. Biol. Chem. 2003, 278, 19634–19641. [Google Scholar] [CrossRef] [PubMed]
- Lavery, K.S.; King, T.H. Antisense and RNAi: Powerful tools in drug target discovery and validation. Curr. Opin. Drug Discov. Devel. 2003, 6, 561–569. [Google Scholar] [PubMed]
- Zhu, X.; Wu, H.; Luo, S.; Xianyu, Z.; Zhu, D. Screening and identification of a novel hepatocellular carcinoma cell binding peptide by using a phage display library. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2008, 28, 299. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Liu, W.; Jiang, A.; Zhang, K.; Chen, M. A novel peptide, selected from phage display library of random peptides, can efficiently target into human breast cancer cell. Chin. Sci. Bullet. 2008, 53, 860–867. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, E.J.; Hong, H.Y.; Kwon, M.K.; Kwon, T.H.; Choi, J.Y.; Park, R.W.; Kwon, T.G.; Yoo, E.S.; Yoon, G.S.; Kim, I.S.; Ruoslahti, E.; Lee, B.H. Targeting bladder tumor cells in vivo and in the urine with a peptide identified by phage display. Mol. Cancer Res. 2007, 5, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug and gene targeting to the brain with molecular Trojan horses. Nat. Rev. Drug Discov. 2002, 1, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Y.; Zhang, T.Y.; Luo, J.X.; He, G.A.; Gu, Q.L.; Xiao, F. Selection of CC chemokine receptor 5-binding peptide from a phage display peptide library. Biosci. Biotechnol. Biochem. 2006, 70, 2035–2041. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, F.; Tarli, L.; Viti, F.; Neri, D. The use of phage display for the development of tumour targeting agents. Adv. Drug Deliv. Rev. 2000, 43, 165. [Google Scholar] [CrossRef]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 8, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.C. Peptidic tumor targeting agents: The road from phage display peptide selections to clinical applications. Curr. Pharm. Des. 2010, 16, 1040–1054. [Google Scholar] [CrossRef] [PubMed]
- Fievez, V.; Plapied, L.; Plaideau, C.; Legendre, D.; des Rieux, A.; Pourcelle, V.; Freichels, H.; Jerome, C.; Marchand, J.; Preat, V.; Schneider, Y.J. In vitro identification of targeting ligands of human M cells by phage display. Int. J. Pharm. 2010, 394, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.K.; Kang, S.K.; Choi, J.H.; Park, I.K.; Na, H.S.; Lee, H.C.; Kim, E.B.; Lee, N.K.; Nah, J.W.; Choi, Y.J.; Cho, C.S. Targeted delivery of chitosan nanoparticles to Peyer’s patch using M cell-homing peptide selected by phage display technique. Biomaterials 2010, 31, 7738–7747. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, V.V.; Felici, F.; Menon, A.G. Targeted delivery of multivalent phage display vectors into mammalian cells. Biochim. Biophys. Acta 1999, 1448, 463–472. [Google Scholar] [CrossRef]
- Vives, E.; Schmidt, J.; Pelegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 2008, 1786, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Lin, C.T.; Kuo, S.Y.; Chang, D.K.; Wu, H.C. Peptide-mediated targeting to tumor blood vessels of lung cancer for drug delivery. Cancer Res. 2007, 67, 10958–10965. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, J.; Zhao, R.; Xu, Y.; Gu, J. Preparation of peptide-targeted phagemid particles using a protein III-modified helper phage. Biotechniques 2005, 39, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Larocca, D.; Burg, M.A.; Jensen-Pergakes, K.; Ravey, E.P.; Gonzalez, A.M.; Baird, A. Evolving phage vectors for cell targeted gene delivery. Curr. Pharm. Biotechnol. 2002, 3, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, R.; Koivunen, E.; Ruoslahti, E. Alpha v integrins as receptors for tumor targeting by circulating ligands. Nat. Biotechnol. 1997, 15, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, G.; Walter, M.R. Cytokine-receptor interactions as drug targets. Curr. Opin. Chem. Biol. 2010, 14, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Kaushansky, K. Small molecule mimics of hematopoietic growth factors: Improving on Mother Nature? Leukemia 2001, 15, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Kallen, K.J.; Grotzinger, J.; Rose-John, S. New perspectives on the design of cytokines and growth factors. Trends Biotechnol. 2000, 18, 455–461. [Google Scholar] [CrossRef]
- Sato, A.; Sone, S. A peptide mimetic of human interferon (IFN)-beta. Biochem. J. 2003, 371, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Zurita, A.J.; Troncoso, P.; Cardo-Vila, M.; Logothetis, C.J.; Pasqualini, R.; Arap, W. Combinatorial screenings in patients: The interleukin-11 receptor alpha as a candidate target in the progression of human prostate cancer. Cancer Res. 2004, 64, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Bai, G.; Li, Z.H.; Yang, W.B. Antagonist peptides of human interferon-alpha2b isolated from phage display library inhibit interferon induced antiviral activity. Acta Pharmacol. Sin. 2006, 27, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Yayon, A.; Aviezer, D.; Safran, M.; Gross, J.L.; Heldman, Y.; Cabilly, S.; Givol, D.; Katchalski-Katzir, E. Isolation of peptides that inhibit binding of basic fibroblast growth factor to its receptor from a random phage-epitope library. Proc. Natl. Acad. Sci. USA 1993, 90, 10643–10647. [Google Scholar] [CrossRef] [PubMed]
- Livnah, O.; Johnson, D.L.; Stura, E.A.; Farrell, F.X.; Barbone, F.P.; You, Y.; Liu, K.D.; Goldsmith, M.A.; He, W.; Krause, C.D.; Pestka, S.; Jolliffe, L.K.; Wilson, I.A. An antagonist peptide-EPO receptor complex suggests that receptor dimerization is not sufficient for activation. Nat. Struct. Biol. 1998, 5, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.; Al-Lazikani, B.; Hopkins, A. How many drug targets are there? Nat. Rev. Drug Disc. 2006, 5, 993. [Google Scholar] [CrossRef] [PubMed]
- Heins, M.; Quax, W. Implications of a Newly Discovered DR5 Specific Antagonistic Peptide for Neurodegenerative Disorders. Mol. Cell. Pharmacol. 2010, 2, 97. [Google Scholar]
- Stephen, C.W.; Helminen, P.; Lane, D.P. Characterisation of epitopes on human p53 using phage-displayed peptide libraries: insights into antibody-peptide interactions. J. Mol. Biol. 1995, 248, 58–78. [Google Scholar] [CrossRef] [PubMed]
- Cardo-Vila, M.; Zurita, A.J.; Giordano, R.J.; Sun, J.; Rangel, R.; Guzman-Rojas, L.; Anobom, C.D.; Valente, A.P.; Almeida, F.C.; Lahdenranta, J.; Kolonin, M.G.; Arap, W.; Pasqualini, R. A ligand peptide motif selected from a cancer patient is a receptor-interacting site within human interleukin-11. PLoS One 2008, 3, e3452. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Russell, S.J.; Compaan, D.M.; Totpal, K.; Marsters, S.A.; Ashkenazi, A.; Cochran, A.G.; Hymowitz, S.G.; Sidhu, S.S. Activation of the proapoptotic death receptor DR5 by oligomeric peptide and antibody agonists. J. Mol. Biol. 2006, 361, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Monne, M.; Chan, K.W.; Slotboom, D.J.; Kunji, E.R. Functional expression of eukaryotic membrane proteins in Lactococcus lactis. Protein Sci. 2005, 14, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Tate, C.G. Overexpression of mammalian integral membrane proteins for structural studies. FEBS Lett. 2001, 504, 94–98. [Google Scholar] [CrossRef]
- Majumdar, S.; Hajduczki, A.; Mendez, A.S.; Weiss, G.A. Phage display of functional, full-length human and viral membrane proteins. Bioorg. Med. Chem. Lett. 2008, 18, 5937–5940. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Goodson, B.; Winter, J. T7 displayed peptides as targets for selecting peptide specific scFvs from M13 scFv display libraries. J. Immunol. Methods 2001, 257, 117–122. [Google Scholar] [CrossRef]
- Karasseva, N.G.; Glinsky, V.V.; Chen, N.X.; Komatireddy, R.; Quinn, T.P. Identification and characterization of peptides that bind human ErbB-2 selected from a bacteriophage display library. J. Protein Chem. 2002, 21, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Binetruy-Tournaire, R.; Demangel, C.; Malavaud, B.; Vassy, R.; Rouyre, S.; Kraemer, M.; Plouet, J.; Derbin, C.; Perret, G.; Mazie, J.C. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J. 2000, 19, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, S.; Carlavan, I.; Baty, D. Isolation and characterization of antagonist and agonist peptides to the human melanocortin 1 receptor. Peptides 2005, 26, 2302–2313. [Google Scholar] [CrossRef] [PubMed]
- Cardo-Vila, M.; Giordano, R.J.; Sidman, R.L.; Bronk, L.F.; Fan, Z.; Mendelsohn, J.; Arap, W.; Pasqualini, R. From combinatorial peptide selection to drug prototype (II): Targeting the epidermal growth factor receptor pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 5118–5123. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N.C.; Lien, S.; Johnson, J.; Wallweber, H.J.; Tran, T.; Currell, B.; Mathieu, M.; Quan, C.; Starovasnik, M.A.; Hymowitz, S.G.; Kelley, R.F. Multiple novel classes of APRIL-specific receptor-blocking peptides isolated by phage display. J. Mol. Biol. 2010, 396, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Erdag, B.; Balcioglu, K.B.; Kumbasar, A.; Celikbicak, O.; Zeder-Lutz, G.; Altschuh, D.; Salih, B.; Baysal, K. Novel short peptides isolated from phage display library inhibit vascular endothelial growth factor activity. Mol. Biotechnol. 2007, 35, 51–63. [Google Scholar] [CrossRef]
- Fairbrother, W.J.; Christinger, H.W.; Cochran, A.G.; Fuh, G.; Keenan, C.J.; Quan, C.; Shriver, S.K.; Tom, J.Y.; Wells, J.A.; Cunningham, B.C. Novel peptides selected to bind vascular endothelial growth factor target the receptor-binding site. Biochemistry 1998, 37, 17754–17764. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Hajdin, K.; Iljin, K.; Greiner, O.; Niggli, F.K.; Schafer, B.W.; Bernasconi, M. Identification of a rhabdomyosarcoma targeting peptide by phage display with sequence similarities to the tumour lymphatic-homing peptide LyP-1. Int. J. Cancer 2009, 124, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, Y.; Wang, J.; Chen, J.; Pan, Y.; Ren, L.; Hu, Z.; Zhao, J.; Liao, M.; Wang, S. Screening and identification of a targeting peptide to hepatocarcinoma from a phage display peptide library. Mol. Med. 2007, 13, 246–254. [Google Scholar] [PubMed]
- Giordano, R.J.; Cardo-Vila, M.; Salameh, A.; Anobom, C.D.; Zeitlin, B.D.; Hawke, D.H.; Valente, A.P.; Almeida, F.C.; Nor, J.E.; Sidman, R.L.; Pasqualini, R.; Arap, W. From combinatorial peptide selection to drug prototype (I): Targeting the vascular endothelial growth factor receptor pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 5112–5117. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.R.; Quinn, T.P.; Deutscher, S.L. Evaluation of an 111In-radiolabeled peptide as a targeting and imaging agent for ErbB-2 receptor expressing breast carcinomas. Clin. Cancer Res. 2007, 13, 6070–6079. [Google Scholar] [CrossRef] [PubMed]
- Giordano, R.J.; Anobom, C.D.; Cardo-Vila, M.; Kalil, J.; Valente, A.P.; Pasqualini, R.; Almeida, F.C.; Arap, W. Structural basis for the interaction of a vascular endothelial growth factor mimic peptide motif and its corresponding receptors. Chem. Biol. 2005, 12, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Pande, J.; Mallhi, K.K.; Grover, A.K. A novel plasma membrane Ca(2+)-pump inhibitor: caloxin 1A1. Eur. J. Pharmacol. 2005, 508, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, J.; Walia, M.; Matharu, J.; Escher, E.; Grover, A.K. Caloxin: A novel plasma membrane Ca2+ pump inhibitor. Am. J. Physiol. Cell Physiol. 2001, 280, C1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Pande, J.; Mallhi, K.K.; Sawh, A.; Szewczyk, M.M.; Simpson, F.; Grover, A.K. Aortic smooth muscle and endothelial plasma membrane Ca2+ pump isoforms are inhibited differently by the extracellular inhibitor caloxin 1b1. Am. J. Physiol. Cell Physiol. 2006, 290, C1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.; Shi, L.; Guo, J.; Pan, Q.; Wu, W.; Pan, X.; Wang, J. Screening and identification of a peptide specifically targeted to NCI-H1299 from a phage display peptide library. Cancer Lett. 2009, 281, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.S. Making the most of affinity tags. Trends Biotechnol. 2005, 23, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Lichty, J.J.; Malecki, J.L.; Agnew, H.D.; Michelson-Horowitz, D.J.; Tan, S. Comparison of affinity tags for protein purification. Protein Expr. Purif. 2005, 41, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Koolpe, M.; Burgess, R.; Dail, M.; Pasquale, E.B. EphB receptor-binding peptides identified by phage display enable design of an antagonist with ephrin-like affinity. J. Biol. Chem. 2005, 280, 17301–17311. [Google Scholar] [CrossRef] [PubMed]
- El-Mousawi, M.; Tchistiakova, L.; Yurchenko, L.; Pietrzynski, G.; Moreno, M.; Stanimirovic, D.; Ahmad, D.; Alakhov, V. A vascular endothelial growth factor high affinity receptor 1-specific peptide with antiangiogenic activity identified using a phage display peptide library. J. Biol. Chem. 2003, 278, 46681–46691. [Google Scholar] [CrossRef] [PubMed]
- Azzazy, H.M.; Highsmith, W.E., Jr. Phage display technology: clinical applications and recent innovations. Clin. Biochem. 2002, 35, 425–445. [Google Scholar] [CrossRef]
- Lee, C.V.; Sidhu, S.S.; Fuh, G. Bivalent antibody phage display mimics natural immunoglobulin. J. Immunol. Methods 2004, 284, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Fagerlund, A.; Myrset, A.H.; Kulseth, M.A. Construction and characterization of a 9-mer phage display pVIII-library with regulated peptide density. Appl. Microbiol. Biotechnol. 2008, 80, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Choi, H.J.; Lim, J.S.; Earm, J.H.; Lee, B.H.; Kim, I.S.; Frokiaer, J.; Nielsen, S.; Kwon, T.H. A novel method of ligand peptidomics to identify peptide ligands binding to AQP2-expressing plasma membranes and intracellular vesicles of rat kidney. Am. J. Physiol. Renal Physiol. 2008, 295, F300–309. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, F.; Howard, C.R. An improved selection procedure for the screening of phage display peptide libraries. J. Immunol. Methods 2001, 247, 191–203. [Google Scholar] [CrossRef]
- Noppe, W.; Plieva, F.; Galaev, I.Y.; Pottel, H.; Deckmyn, H.; Mattiasson, B. Chromato-panning: An efficient new mode of identifying suitable ligands from phage display libraries. BMC Biotechnol. 2009, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Plieva, F.M.; Galaev, I.Y.; Noppe, W.; Mattiasson, B. Cryogel applications in microbiology. Trends Microbiol. 2008, 16, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Otrock, Z.K.; Makarem, J.A.; Shamseddine, A.I. Vascular endothelial growth factor family of ligands and receptors: review. Blood Cells Mol. Dis. 2007, 38, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Livnah, O.; Stura, E.A.; Middleton, S.A.; Johnson, D.L.; Jolliffe, L.K.; Wilson, I.A. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 1999, 283, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Dower, W.J. Targeting growth factor and cytokine receptors with recombinant peptide libraries. Curr. Opin. Chem. Biol. 1998, 2, 328–334. [Google Scholar] [CrossRef]
- Livnah, O.; Stura, E.A.; Johnson, D.L.; Middleton, S.A.; Mulcahy, L.S.; Wrighton, N.C.; Dower, W.J.; Jolliffe, L.K.; Wilson, I.A. Functional mimicry of a protein hormone by a peptide agonist: the EPO receptor complex at 2.8 A. Science 1996, 273, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.L.; Farrell, F.X.; Barbone, F.P.; McMahon, F.J.; Tullai, J.; Hoey, K.; Livnah, O.; Wrighton, N.C.; Middleton, S.A.; Loughney, D.A.; Stura, E.A.; Dower, W.J.; Mulcahy, L.S.; Wilson, I.A.; Jolliffe, L.K. Identification of a 13 amino acid peptide mimetic of erythropoietin and description of amino acids critical for the mimetic activity of EMP1. Biochemistry 1998, 37, 3699–3710. [Google Scholar] [CrossRef] [PubMed]
- McGregor, D.P. Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Hematide™ Injection for Anemia in Chronic Hemodialysis (HD) Patients (EMERALD 1). http://clinicaltrials.gov/ct2/show/NCT00597753?term=hematide&rank=5 (accessed on 15 November 2010).
- Hematide™ Injection for Anemia in Chronic Hemodialysis (HD) Patients (EMERALD 2). http://clinicaltrials.gov/ct2/show/NCT00597584?term=hematide&rank=6 (accessed on 15 November 2010).
- Safety & Efficacy of Hematide™ for the Correction of Anemia in Patients With Chronic Renal Failure (PEARL 1). http://clinicaltrials.gov/ct2/show/NCT00598273?term=hematide&rank=8 (accessed on 15 November 2010).
- Safety and Efficacy of Hematide™ for the Correction of Anemia in Patients With Chronic Renal Failure (PEARL 2). http://clinicaltrials.gov/ct2/show/NCT00598442?term=hematide&rank=11 (accessed on 15 November 2010).
- Cines, D.B.; Yasothan, U.; Kirkpatrick, P. Romiplostim. Nat. Rev. Drug Discov. 2008, 7, 887–888. [Google Scholar] [CrossRef] [PubMed]
- Newland, A. Romiplostim: A breakthrough treatment for the management of immune thrombocytopenic purpura. Eur. J. Haematol. Suppl. 2009, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Bahadur, R.P.; Chakrabarti, P. Protein-protein interaction and quaternary structure. Q. Rev. Biophys. 2008, 41, 133–180. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, D.; Rahat, O.; Cohen, M.; Neuvirth, H.; Schreiber, G. The molecular architecture of protein-protein binding sites. Curr. Opin. Struct. Biol. 2007, 17, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.A.; Laakkonen, P.; Porkka, K.; Bernasconi, M.; Ruoslahti, E. In vivo and ex vivo selections using phage-displayed libraries. In Phage Display: A Practical Approach; Lowman, H.B., Clackson, T., Eds.; Oxford University Press: New York, NY, USA, 2004; pp. 171–192. [Google Scholar]
- Ribeiro, S.; Horuk, R. The clinical potential of chemokine receptor antagonists. Pharmacol. Ther. 2005, 107, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Kromdijk, W.; Huitema, A.; Mulder, J. Treatment of HIV infection with the CCR5 antagonist maraviroc. Expert Opin. Pharmacother. 2010, 11, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.I.; Lynch, J.W. Molecular pharmacology of the glycine receptor chloride channel. Curr. Pharm. Des. 2007, 13, 2350–2367. [Google Scholar] [CrossRef] [PubMed]
- Bagri, A.; Tessier-Lavigne, M.; Watts, R.J. Neuropilins in tumor biology. Clin. Cancer Res. 2009, 15, 1860–1864. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, R.; Ruoslahti, E. Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380, 364. [Google Scholar] [CrossRef] [PubMed]
- Gaumet, M.; Vargas, A.; Gurny, R.; Delie, F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur. J. Pharm. Biopharm. 2008, 69, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Passarella, R.J.; Zhou, L.; Phillips, J.G.; Wu, H.; Hallahan, D.E.; Diaz, R. Recombinant peptides as biomarkers for tumor response to molecular targeted therapy. Clin. Cancer Res. 2009, 15, 6421–6429. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, M.M.; Pande, J.; Grover, A.K. Caloxins: a novel class of selective plasma membrane Ca2+ pump inhibitors obtained using biotechnology. Pflugers Arch. 2008, 456, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Lowman, H.B. Therapeutic peptides. Trends Biotechnol. 2003, 21, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Funke, S.A.; Willbold, D. Mirror image phage display - a method to generate D-peptide ligands for use in diagnostic or therapeutical applications. Mol. Biosyst. 2009, 5, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Russel, M.; Lowman, H.; Clackson, T. Introduction to phage biology and phage display. In Phage Display: A Practical Approach; Lowman, H.B., Clackson, T., Eds.; Oxford University Press: New York, NY, USA, 2004; pp. 1–26. [Google Scholar]
- Dennis, M.S.; Lowman, H.B. Phage selection strategies for improved affinity and specificity of proteins and peptides. In Phage Display: A Practical Approach; Lowman, H.B., Clackson, T., Eds.; Oxford University Press: New York, NY, USA, 2004; pp. 66–68. [Google Scholar]
- Schumacher, T.N.; Mayr, L.M.; Minor, D.L., Jr.; Milhollen, M.A.; Burgess, M.W.; Kim, P.S. Identification of D-peptide ligands through mirror-image phage display. Science 1996, 271, 1854–1857. [Google Scholar] [CrossRef] [PubMed]
- Dintzis, H.M.; Symer, D.E.; Dintzis, R.Z.; Zawadzke, L.E.; Berg, J.M. A comparison of the immunogenicity of a pair of enantiomeric proteins. Proteins 1993, 16, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Dower, W.J.; Cwirla, S.E.; Balasubramanian, P.; Schatz, P.J.; Baccanari, D.P.; Barrett, R.W. Peptide agonists of the thrombopoietin receptor. Stem Cells 1998, 16 (Suppl. 2), 21–29. [Google Scholar] [CrossRef]
- Macdougall, I.C.; Rossert, J.; Casadevall, N.; Stead, R.B.; Duliege, A.M.; Froissart, M.; Eckardt, K.U. A peptide-based erythropoietin-receptor agonist for pure red-cell aplasia. N. Engl. J. Med. 2009, 361, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Zobel, K.; Koehler, M.F.; Beresini, M.H.; Caris, L.D.; Combs, D. Phosphate ester serum albumin affinity tags greatly improve peptide half-life in vivo. Bioorg. Med. Chem. Lett. 2003, 13, 1513–1515. [Google Scholar] [CrossRef]
- Koehler, M.F.; Zobel, K.; Beresini, M.H.; Caris, L.D.; Combs, D.; Paasch, B.D.; Lazarus, R.A. Albumin affinity tags increase peptide half-life in vivo. Bioorg. Med. Chem. Lett. 2002, 12, 2883–2886. [Google Scholar] [CrossRef]
- Vaccaro, C.; Zhou, J.; Ober, R.J.; Ward, E.S. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat. Biotechnol. 2005, 23, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Jespers, L.S.; Roberts, A.; Mahler, S.M.; Winter, G.; Hoogenboom, H.R. Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen. Biotechnology (NY) 1994, 12, 899–903. [Google Scholar] [CrossRef]
- Levy, J.H.; O’Donnell, P.S. The therapeutic potential of a kallikrein inhibitor for treating hereditary angioedema. Expert Opin. Investig. Drugs 2006, 15, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Tsao, M.L.; Schultz, P.G. A phage display system with unnatural amino acids. J. Am. Chem. Soc. 2004, 126, 15962–15963. [Google Scholar] [CrossRef] [PubMed]
- Sandman, K.; Benner, J.; Noren, C. Phage display of selenopeptides. J. Am. Chem. Soc. 2000, 122, 960–961. [Google Scholar] [CrossRef]
- Woiwode, T.F.; Haggerty, J.E.; Katz, R.; Gallop, M.A.; Barrett, R.W.; Dower, W.J.; Cwirla, S.E. Synthetic compound libraries displayed on the surface of encoded bacteriophage. Chem. Biol. 2003, 10, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Schulz, P.; Liu, R.; Sierks, M.R. Improved affinity selection using phage display technology and off-rate based selection. Electron. J. Biotechnol. 2006, 9, 171–175. [Google Scholar] [CrossRef]
- Takakusagi, Y.; Takakusagi, K.; Kuramochi, K.; Kobayashi, S.; Sugawara, F.; Sakaguchi, K. Identification of C10 biotinylated camptothecin (CPT-10-B) binding peptides using T7 phage display screen on a QCM device. Bioorg. Med. Chem. 2007, 15, 7590–7598. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |


{kind=link}
{kind=link}
| Targeted protein a | Selection strategy b | Library type c | Selected peptide(s) d | Biological activity | Affinity | Potential applications (Biological effects) | Ref. |
|---|---|---|---|---|---|---|---|
| IL-1R type I | a3 | 3 and 8 | peptides with C-terminal motif YWQPYALPL | antagonists | IC50 2-500 nM | therapy of autoimmune and inflammatory disorders (anti-inflammatory effects) | [34] |
| IL-6Rα | a1 | 3 | LSLITRL | antagonist | IC50 >30 μM | cancer therapy (preventing the anti-apoptotic and angiogenic effects of IL-6) | [8] |
| IL-11Rα | e | 3 | CGRRAGGSC | agonist | N/A | cancer therapy (targeted delivery of therapeutic or diagnostic agents to prostate tumors; prevention of chemotherapy-induced thrombocytopenia) | [19,66,73] |
| FGFR | b | 3 | KRTGQYKL | antagonist | IC50 ~5 nM | cancer therapy (inhibition of angiogenesis and tumor progression) | [68] |
| EGFR2 (ErbB-2) | a2 | 3 | KCCYSL | N/A | Kd ~30 μM | cancer therapy (targeted delivery of therapeutic or diagnostic agents to tumors) | [79,89] |
| EGFR | c5 | 3 | N/A | indirect antagonist (decoy receptor) | N/A | cancer therapy (inhibition of tumor cell proliferation) | [82] |
| VEGFR (KDR) | a1 | 3 | HTMYYHHYQHHL | antagonist | IC50 >30 μM | cancer therapy, treatment of diabetic retinopathy (inhibition of angiogenesis and cellular proliferation) | [9] |
| VEGFR (KDR) | b, d | 3 | ATWLPPR | antagonist | IC50 ~80 μM | cancer therapy, treatment of diabetic retinopathy (inhibition of angiogenesis and cellular proliferation) | [80] |
| VEGFR (KDR and Flt-1) | c6 | 8 + 8 and 3 + 3 | GERWCFDGPRAW-VCGWEI, GGNECDIARMWE-WECFERL, RGWVEICAADDY-GRCLTEAQ | indirect antagonists (decoy receptors) | IC50 ~0.7-7 μM | cancer therapy, treatment of diabetic retinopathy (inhibition of angiogenesis and cellular proliferation) | [85] |
| VEGFR | c5 | 3 | WHLPFKC, WHKPFRF | indirect antagonists (decoy receptors) | Kd ~2.7 μM | cancer therapy, treatment of diabetic retinopathy (inhibition of angiogenesis and cellular proliferation) | [84] |
| VEGFR1 and NRP1 | d | 3 | CPQPRPLC | antagonist | N/A | cancer therapy, treatment of diabetic retinopathy (inhibition of angiogenesis and cellular proliferation) | [35,88,90] |
| IFNAR | a4, b | 3 | SVQARWEAAFDL-DLY | agonist | IC50 ~50 μM | study of the mechanism of IFNAR activation | [65] |
| IFNAR | d, b | 3 | SLSPGLP, FSAPVRY | antagonists | N/A | treatment of autoimmune diseases, study of ligand-receptor interactions | [67] |
| EpoR | a3 | 8 + 8 and 3 + 3 | GGTYSCHFGPLT-WVCKPQGG | agonist | IC50 ~0.2 μM | treatment of anemia, pure red cell aplasia resulting from anti-Epo antibodies (stimulation of erythropoiesis) | [6] |
| TpoR | a3 | 8 + 8 (and non-phage libraries) | GGCADGPTLREW-ISFCGG | agonist | IC50 ~60 nM | treatment of idiopathic thrombocytopenic purpura, thrombocytopenia (stimulation of thrombopoiesis) | [7] |
| GlyRα1 | d | 3 | YESIRIGVAPSQ (and others) | pos./neg. allosteric modulators | N/A | treatment of alcoholism, leads for developing anesthetics (inhibition/enhancement of ethanol activity) | [12] |
| CCR5 | d | 3 | AFDWTFVPSLIL | antagonist | IC50 ~2.6 μM | treatment of multiple sclerosis, rheumatoid arthritis, HCV and HIV infections, prevention of renal allograft rejection (anti-inflammatory effects, prevention of HIV-1 entry to CD4+ cells) | [49] |
| DR5 | a4 | 3 | CKVILTHRC | antagonist | Kd ~272 nM | therapy of neurodegenerative disorders (inhibition of TRAIL-induced apoptosis in neuronal cells) | [37] |
| DR5 | a1 | 8 + 8 and 3 + 3 | QEVCMTSCDKLM-KCNWMAAM | agonist | IC50 ~6 nM | cancer therapy (triggering of apoptosis in tumor cells) | [74] |
| PMCA4 | a1, a2 | 3 | TAWSEVLDLLRR | allosteric inhibitor | Ki ~2.3 μM | study of physiological PMCA4 function, study of arterial hypertension mechanisms and retinopathies, development of new class of contraceptives | [38,91,92,93] |
| B-cell maturation antigen | c5, c6 | 8 + 8 | SSCESPEVDYLE-CLY, LQCRYDQLIEEW-RCEY (and others) | indirect antagonists (decoy receptors) | IC50 0.49-27 μM | cancer therapy (inhibition of APRIL (a proliferation-inducing ligand)-stimulated proliferation) | [83] |
| αVβ3 integrin, unknown [RD cell line] | d | T7Select 415-1b | CQQSNRGDRKRC, CMGNKRSAKRPC | N/A | N/A | cancer therapy (targeted delivery of therapeutic or diagnostic agents to rhabdomiosarcoma) | [86] |
| unknown [HT-1376 cells from human bladder carcinoma xenografts] | d | T7Select 415-1b | CSNRDARRC | N/A | N/A | cancer therapy (targeted delivery of therapeutic or diagnostic agents to bladder cancer) | [47] |
| unknown [NCI-H1299 cell line] | d | 3 | EHMALTYPFRPP | N/A | N/A | cancer therapy (targeted delivery of therapeutic or diagnostic agents to NSCLC e cells) | [94] |
| unknown [HepG2 cell line] | d | 3 | FLLEPHLMDTSM | N/A | N/A | cancer therapy (targeted delivery of therapeutic or diagnostic agents to hepatocellular carcinoma) | [45] |
| unknown [MDA-MB-435 cells from breast cancer xenograft] | d, e | T7Select 415-1b | CGNKRTRGC | N/A | N/A | cancer therapy (targeted delivery of therapeutic or diagnostic agents to tumor lymphatics) | [51] |
| unknown [MDA-MB-231 cell line] | d | 8 + 8 | CASPSGALRSC | N/A | N/A | cancer therapy (targeted delivery of therapeutic or diagnostic agents to breast cancer) | [46] |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Molek, P.; Strukelj, B.; Bratkovic, T. Peptide Phage Display as a Tool for Drug Discovery: Targeting Membrane Receptors. Molecules 2011, 16, 857-887. https://doi.org/10.3390/molecules16010857
Molek P, Strukelj B, Bratkovic T. Peptide Phage Display as a Tool for Drug Discovery: Targeting Membrane Receptors. Molecules. 2011; 16(1):857-887. https://doi.org/10.3390/molecules16010857
Chicago/Turabian StyleMolek, Peter, Borut Strukelj, and Tomaz Bratkovic. 2011. "Peptide Phage Display as a Tool for Drug Discovery: Targeting Membrane Receptors" Molecules 16, no. 1: 857-887. https://doi.org/10.3390/molecules16010857
APA StyleMolek, P., Strukelj, B., & Bratkovic, T. (2011). Peptide Phage Display as a Tool for Drug Discovery: Targeting Membrane Receptors. Molecules, 16(1), 857-887. https://doi.org/10.3390/molecules16010857