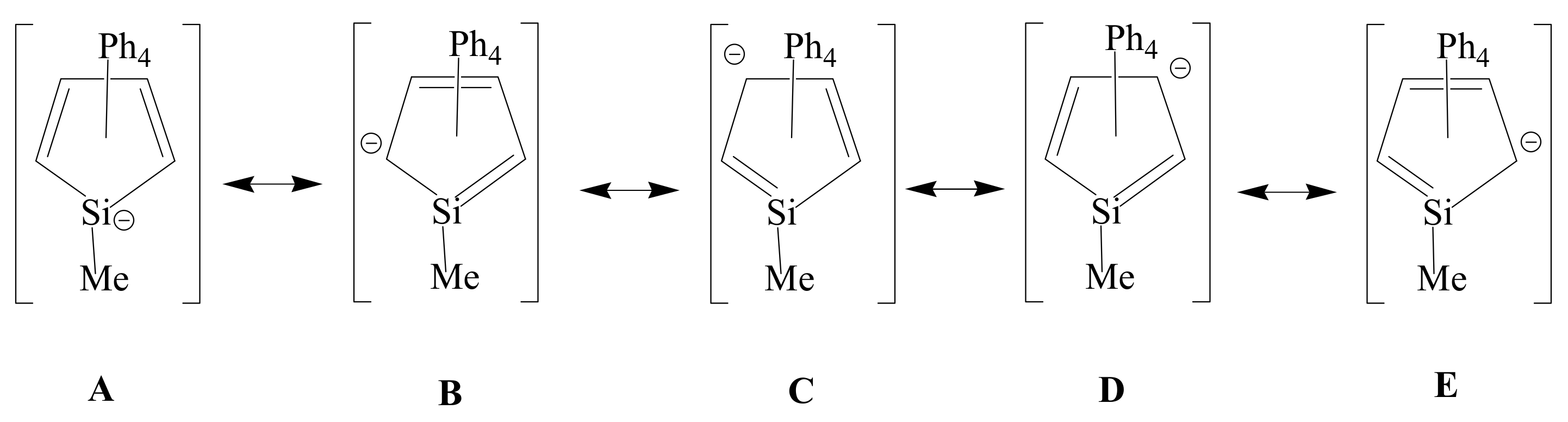
Dissociation of the Disilatricyclic Diallylic Dianion [(C4Ph4SiMe)2]−2 to the Silole Anion [MeSiC4Ph4]− by Halide Ion Coordination or Halide Ion Nucleophilic Substitution at the Silicon Atom

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
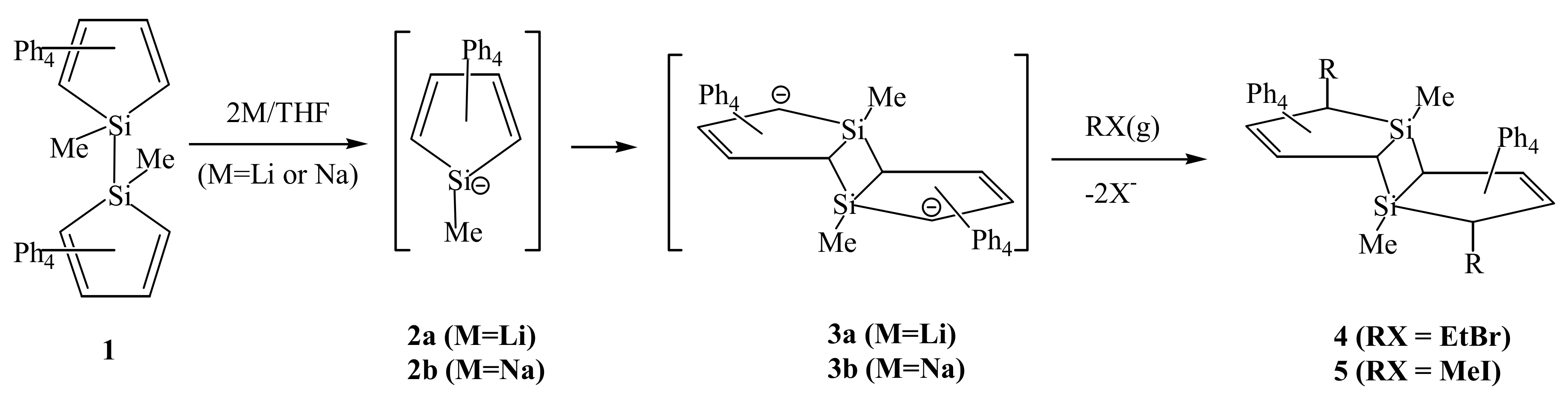
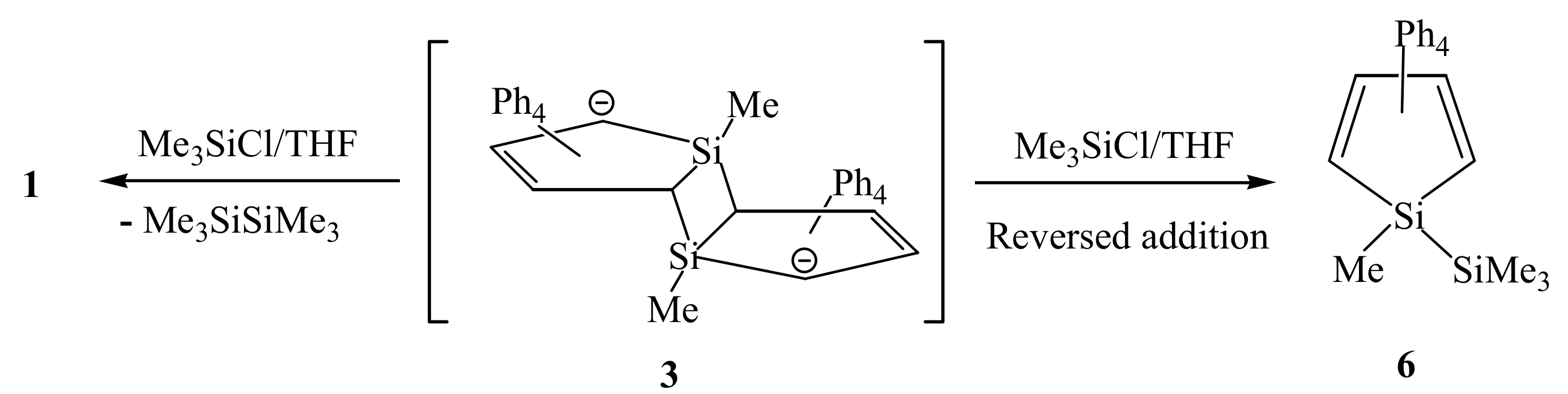
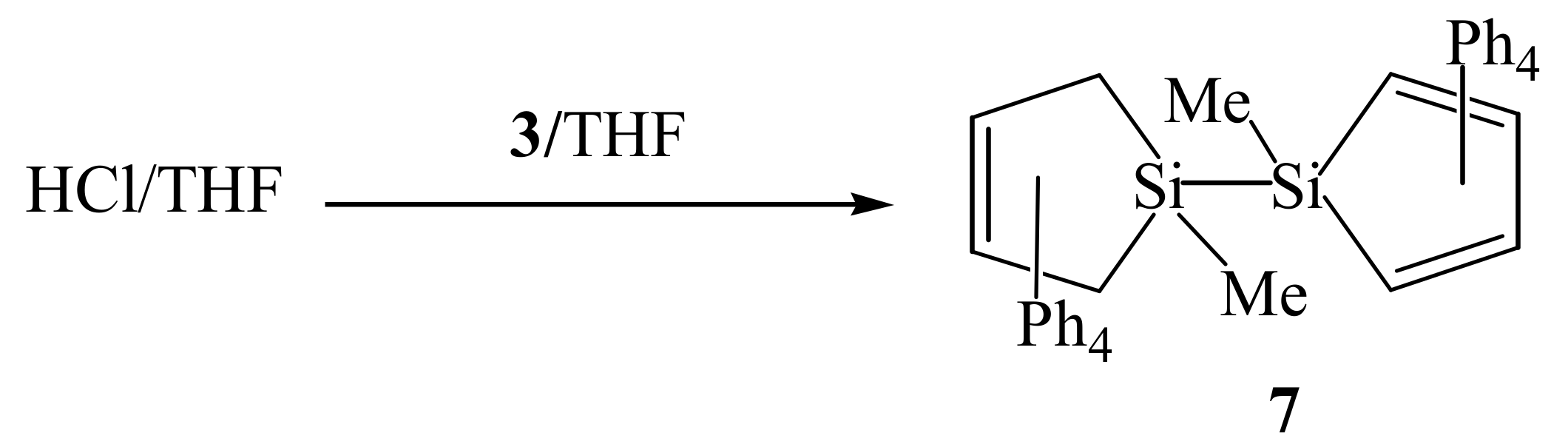
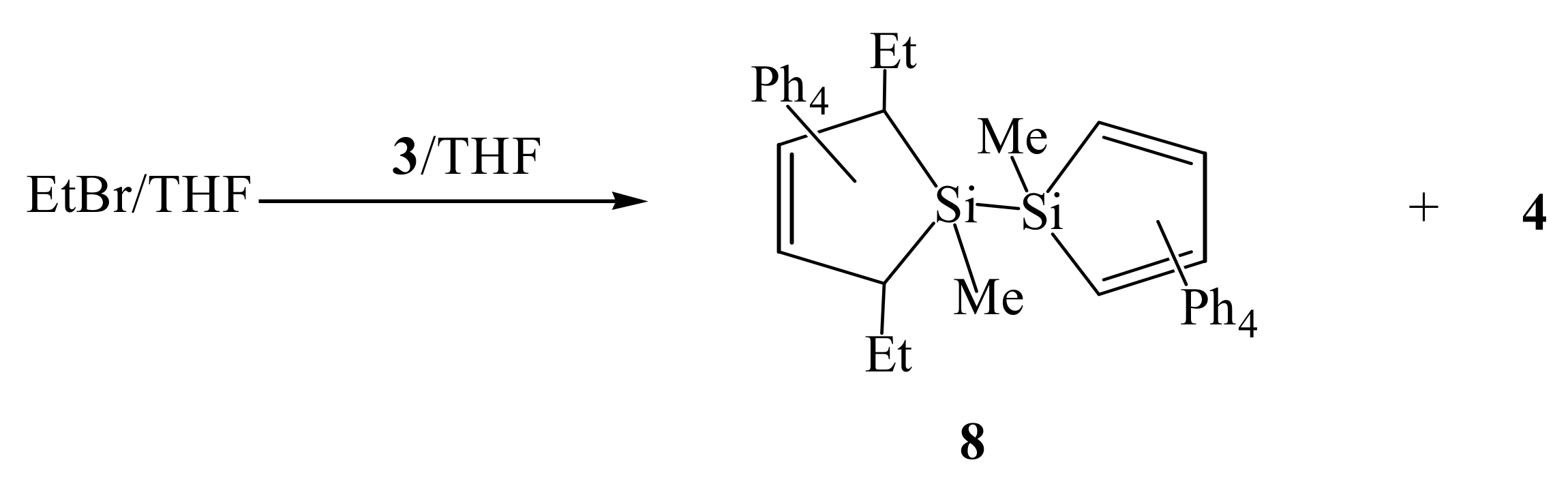
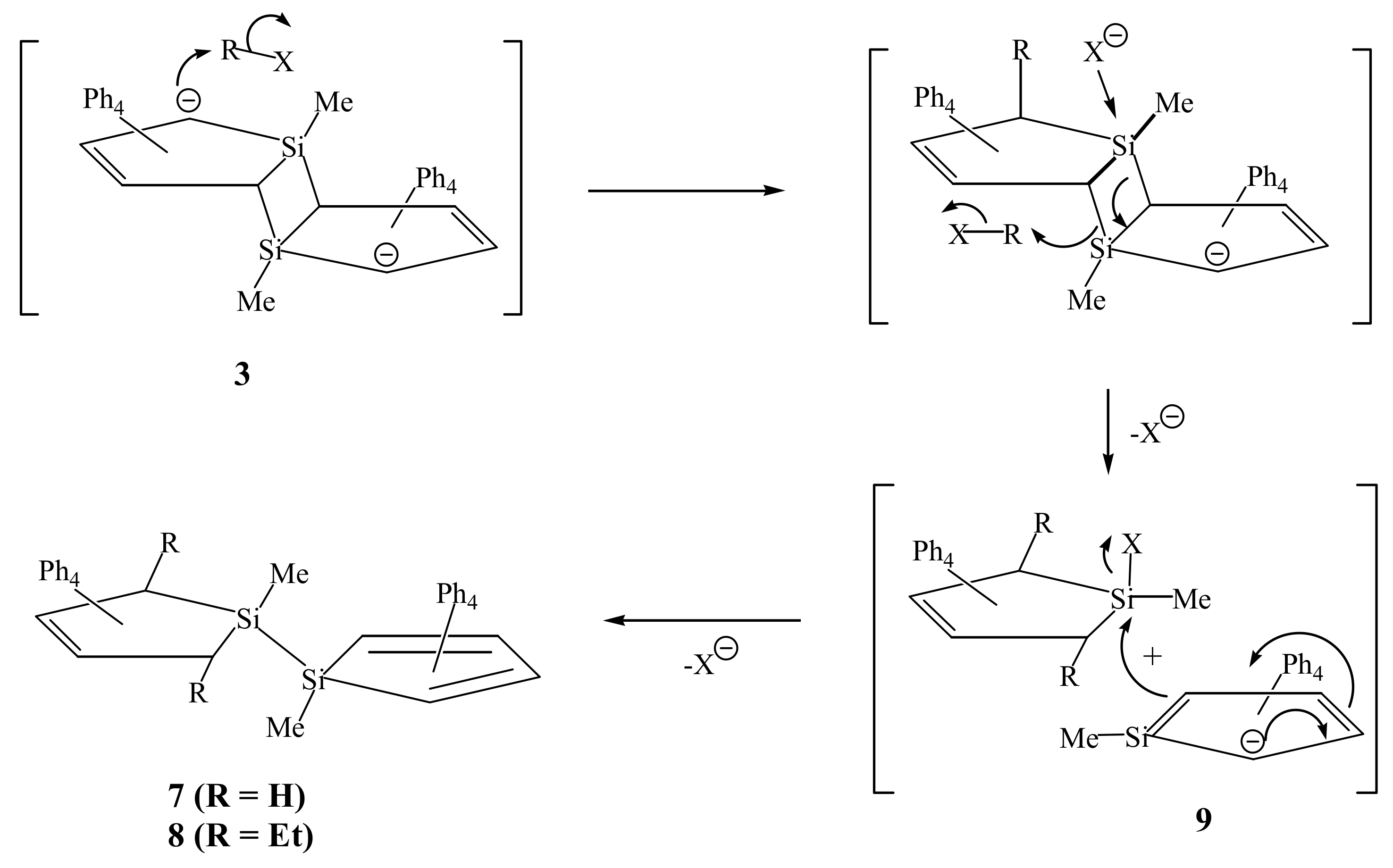
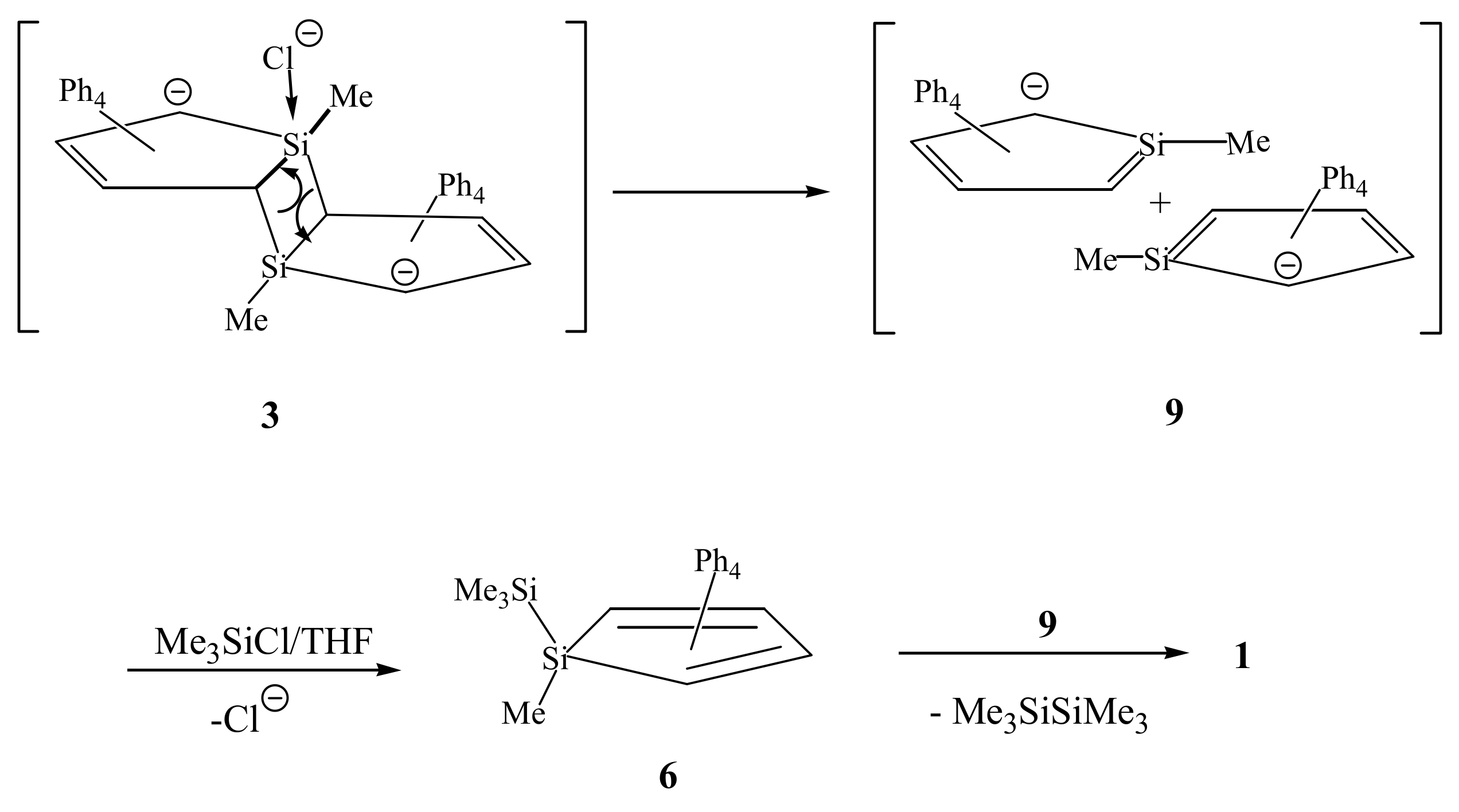
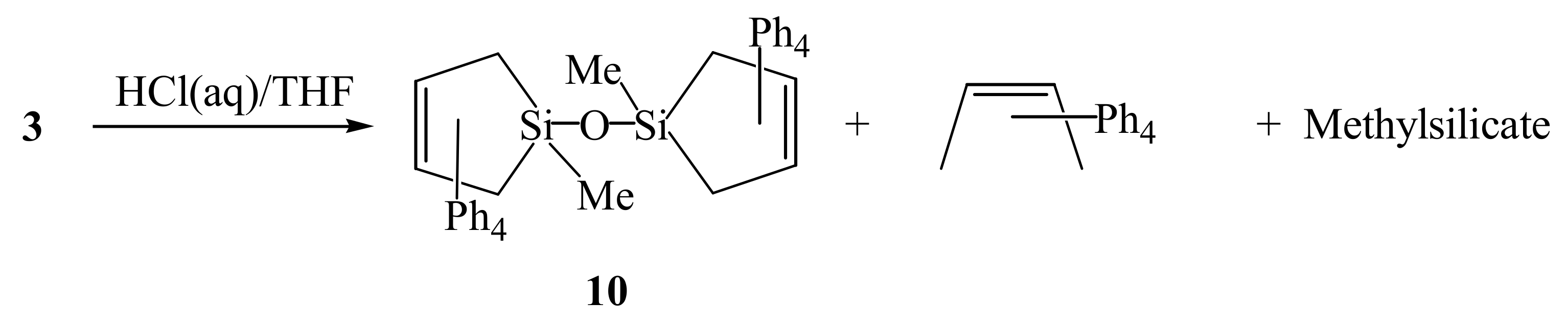
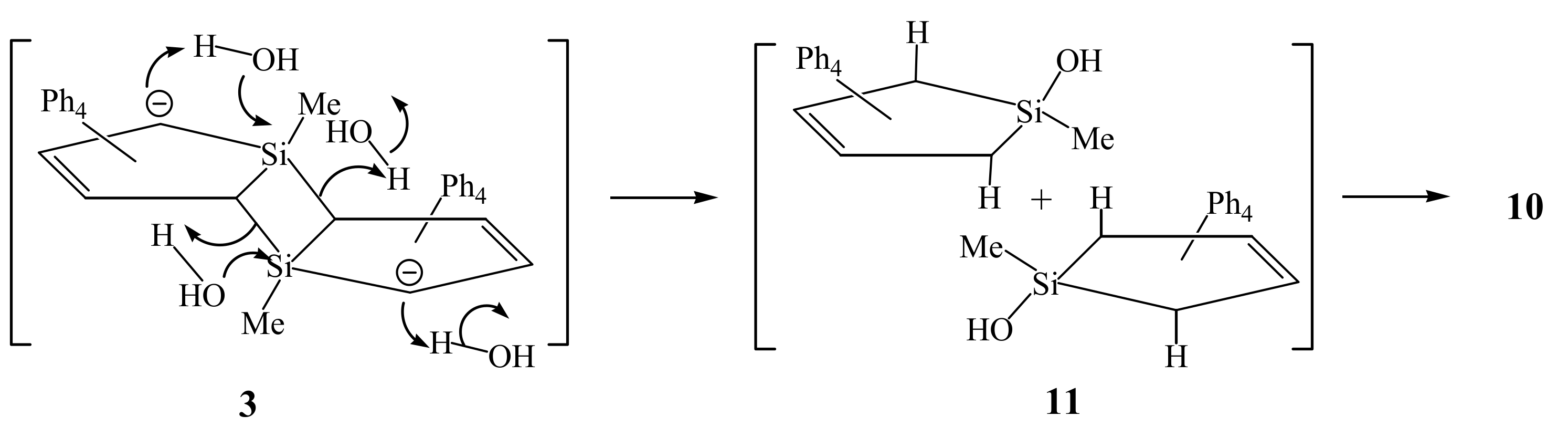
2. Results and Discussion
3. Experimental
General
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Gilman, H.; Gorsich, R.D. Cyclic Organosilicon Compounds. I. Synthesis of Compounds Containing the Dibenzosilole Nucleus. J. Am. Chem. Soc. 1958, 80, 1883–1886. [Google Scholar] [CrossRef]
- Corey, J.Y. Organometallic Benzheterocycles. Adv. Organomet. Chem. 1975, 13, 139–271. [Google Scholar]
- Barton, T.J. Carbacyclic Silanes. In Comprehensive Organometallic Chemistry; Wilkinson, G., Stone, F.G.A., Abel, E.W., Eds.; Pergamon Press: Oxford, UK, 1982; Volume 2, pp. 205, 250–261. [Google Scholar]
- Dubac, J.; Laporterie, A.; Manuel, G. Group 14 metalloles. 1. Synthesis, organic chemistry, and physicochemical data. Chem. Rev. 1990, 90, 215–282. [Google Scholar] [CrossRef]
- Colomer, E.; Corriu, R.J.P.; Lheureux, M. Group 14 metalloles. 2. Ionic species and coordination compounds. Chem. Rev. 1990, 90, 265–282. [Google Scholar] [CrossRef]
- Dubac, J.; Guerin, C.; Meunier, P. The Chemistry of Organosilicon Compounds; Patai, S., Rappoport, Z., Eds.; Willey-Interscience: Chichester, UK, 1998; Volume 2, p. 1961. [Google Scholar]
- Hissler, M.; Dyer, P.W.; Reau, R. Linear organic π-conjugated systems featuring the heavy Group 14 and 15 elements. Coord. Chem. Rev. 2003, 244, 1–44. [Google Scholar] [CrossRef]
- Saito, M.; Yoshioka, M. The anions and dianions of group 14 metalloles. Coord. Chem. Rev. 2005, 249, 765–780. [Google Scholar] [CrossRef]
- Joo, W.-C.; Hong, J.-H.; Choi, S.-B.; Son, H.-E. Synthesis and reactivity of 1,1,-disodio-2,3,4,5-tetraphenyl-1-silacyclopentadiene. J. Organomet. Chem. 1990, 391, 27–36. [Google Scholar] [CrossRef]
- Hong, J.-H.; Boudjouk, P. Synthesis and characterization of a delocalized germanium-containing dianion: dilithio-2,3,4,5-tetraphenyl-germole. Bull. Soc. Chim. Fr. 1995, 132, 495–498. [Google Scholar]
- Hong, J.-H.; Boudjouk, P.; Castellino, S. Synthesis and Characterization of Two Aromatic Silicon-Containing Dianions: The 2,3,4,5-Tetraphenylsilole Dianion and the 1,1’-Disila-2,2’,3,3’,4,4’, 5,5’-octaphenylfulvalene Dianion. Organometallics 1994, 13, 3387–3389. [Google Scholar] [CrossRef]
- Hong, J.-H. Synthesis and NMR-Study of the 2,3,4,5-Tetraethylsilole Dianion [SiC4Et4]2−•2[Li]+. Molecules 2011, 16, 8033–8040. [Google Scholar] [CrossRef] [PubMed]
- West, R.; Sohn, H.; Bankwitz, U.; Calabrese, J.; Apeloig, T.; Mueller, T. Dilithium Derivative of Tetraphenylsilole: An η1- η5 Dilithium. J. Am. Chem. Soc. 1995, 117, 11608–11609. [Google Scholar] [CrossRef]
- Freeman, W.P.; Tilley, T.D.; Yap, G.P.A.; Rheingold, A.L. Siloyl Anions and Silole Dianions: Structure of [K([18]crown-6)+]2[C4Me4Si2-]. Angew. Chem. Int. Ed. 1996, 35, 882–884. [Google Scholar] [CrossRef]
- West, R.; Sohn, H.; Powell, D.R.; Mueller, T.; Apeloig, Y. Dianion of Tetraphenylgermole is Aromatic. Angw. Chem., Int. Ed. 1996, 35, 1002–1004. [Google Scholar] [CrossRef]
- Freeeman, W.P.; Tilley, T.D.; Liable-Sands, L.M.; Rheingold, A.L. Synthesis and Study of Cyclic π-Systems Containing Silicon and Germanium. The Question of Aromaticity in Cyclopentadienyl Analogues. J. Am. Chem. Soc. 1996, 118, 10457–10468. [Google Scholar] [CrossRef]
- Choi, S.-B.; Boudjouk, P.; Hong, J.-H. Unique Bis-η5/η1 Bonding in a Dianionic Germole. Synthesis and Structural Characterization of the Dilithium Salt of the 2,3,4,5-Tetraethyl Germole Dianion. Organometallics 1999, 18, 2919–2921. [Google Scholar] [CrossRef]
- Choi, S.-B.; Boudjouk, P. Synthesis and characterization of dibenzannulated silole dianions. The 1,1-dilithiosilafluorene and 1,1’-dilithiobis(silafluorene) dianions. Tetrahedron Lett. 2000, 41, 6685–6688. [Google Scholar] [CrossRef]
- Liu, Y.L.; Stringfellow, C.; Ballweg, D.; Guzei, I.A.; West, R. Structural and chemistry of 1-silafluorenyl dianion. Its derivatives, and an organosilicon diradical dianion. J. Am. Chem. Soc. 2002, 124, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-B.; Boudjouk, P.; Wei, P. Aromatic benzannulated silole dianions. The dilithio and disodio salts of a silaindenyl dianion. J. Am. Chem. Soc. 1998, 120, 5814–5815. [Google Scholar] [CrossRef]
- Choi, S.-B.; Boudjouk, P.; Qin, K. Aromatic Benzannulated Germole Dianions. The Dilithio and Disodio Salts of a Germaindenyl Dianion. Organometallics 2000, 19, 1806–1809. [Google Scholar] [CrossRef]
- Liu, Y.; Ballweg, D.; Müller, T.; Guzei, I.A.; Clark, R.W.; West, R. Chemistry of the Aromatic 9-Germafluorenyl Dianion and Some Related Silicon and Carbon Species. J. Am. Chem.Soc. 2002, 124, 12174–12181. [Google Scholar] [CrossRef] [PubMed]
- Goldfuss, B.; Schleyer, P.v.R.; Hampel, F. Aromaticity in Group 14 Metalloles: Structural, Energetic, and Magnetic Criteria. Organometallics 1996, 15, 1755–1757. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N.J.R.v.E. Nucleus-Independent Chemical shifts: A simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.-H.; Boudjouk, P. A Stable Aromatic Species Containing Silicon. Synthesis and Characterization of the 1-tert-Butyl-2,3,4,5-tetraphenyl-1-silacyclopentadienide Anion. J. Am. Chem. Soc. 1993, 115, 5883–5884. [Google Scholar] [CrossRef]
- Freeeman, W.P.; Tilley, T.D.; Rheingold, A.L. Stable Silacyclopentadienyl Complexes of Ruthenium: (η5-C5Me5)Ru[η5-Me4C4SiSi(SiMe3)3] and X-ray Structure of Its Protonated Form. J. Am. Chem. Soc. 1994, 116, 8428–8429. [Google Scholar] [CrossRef]
- Dysard, J.M.; Tilley, T.D. η5-Silolyl and η5-Germolyl Complexes of d0 Hafnium. Structural Characterization of an η5-Silolyl Complex. J. Am. Chem. Soc. 1998, 120, 8245–8246. [Google Scholar] [CrossRef]
- Dysard, J.M.; Tilley, T.D. Synthesis and reactivity of η5-Silolyl, η5-Germolyl, and η5-Germolyl dianion complexes of zirconium and hafnium. J. Am. Chem. Soc. 2000, 122, 3097–3105. [Google Scholar] [CrossRef]
- Sohn, H.; Powel, R.; West, R.; Hong, J.-H.; Joo, W.-C. Dimerization of the Silole Anion [C4Ph4SiMe]− to a Tricyclic Diallylic Dianion. Organometallics 1997, 16, 2770–2772 and references therein. [Google Scholar] [CrossRef]
- Gordon, M.S.; Boudjouk, P.; Anwari, F. Are the silacyclopentadienyl anion and the silacyclopropenyl cation aromatic? J. Am. Chem. Soc. 1983, 105, 4972–4976. [Google Scholar] [CrossRef]
- Damewood, J.R. Pyramidal inversion and electron delocalization in the silacyclopentadienyl anion. J. Org. Chem. 1986, 51, 5028–5029. [Google Scholar] [CrossRef]
- Goldfuss, B.; Schleyer, P.v.R. The Siloyl Anion C4H4SiH- is Aromatic and the Lithium Silolide C4H4SiHLi Even More So. Organometallics 1995, 14, 1553–1555. [Google Scholar] [CrossRef]
- Goldfuss, B.; Schleyer, P.v.R. Aromaticity in Group 14 Metalloles: Structural, Energetic, and Magnetic Criteria. Organometallics 1997, 16, 1543–1552. [Google Scholar] [CrossRef]
- Lee, V.Y.; Kato, R.; Ichinohe, M.; Sekiguchi, A. The Heavy Analogue of CpLi: Lithium 1,2-Disila-3-germacyclopentadienide, a 6π-Electron Aromatic System. J. Am. Chem. Soc. 2005, 127, 13142–13143. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.-H; Boudjouk, P. Synthesis and Characterization of a Novel Pentavalent Silane: 1-Methyl-1,1-dihydro-2,3,4,5-tetraphenyl-1-silacyclopentadiene Silicate, [Ph4C4SiMeH2−]•[K+]. Organometallics 1995, 14, 574–576. [Google Scholar] [CrossRef]
- Cowley, A.H.; Brown, D.S.; Decken, A.; Kamepalli, S. Novel dimeric ring systems containing gallium. J. Chem. Soc., Chem. Commun. 1996, 2425–2426. [Google Scholar] [CrossRef]
- Campion, B.K.; Heyn, R.K.; Tilley, T.D. Preparation, isolation, and characterization of transition-metal η2-silene complexes. X-ray crystal structure of (η5-C5Me5)[P(iso-Pr)3]Ru(H)(η2 -CH2SiPh2). J. Am. Chem. Soc. 1988, 110, 7558–7560. [Google Scholar] [CrossRef]
- Campion, B.K.; Heyn, R.H.; Tilley, T.D.; Rheingold, A.L. Synthesis and study of the ruthenium-silene complexes (η5- C5Me5)(PR3)RuH(η2-CH2=SiR’2) (R = iPr, Cy; R’ = Me, Ph). J. Am. Chem. Soc. 1993, 115, 5527–5537. [Google Scholar] [CrossRef]
- Dioumaev, V.K.; Plössl, K.; Carrol, P.J.; Berry, D.H. Formation and Interconversion of Ruthenium−Silene and 16-Electron Ruthenium Silyl Complexes. J. Am. Chem. Soc. 1999, 121, 8391–8392. [Google Scholar] [CrossRef]
- Ishikawa, M.; Tabohashi, T.; Sugisawa, H.; Nishimura, K.; Kumada, M. Chemistry of siloles. The ractions of siloles with organolithium reagents. J. Organomet. Chem. 1983, 250, 109–119. [Google Scholar] [CrossRef]
- Ishikawa, M.; Tabohashi, T.; Sugisawa, H.; Nishimura, K.; Kumada, M. Unexpected behavior of siloles toward organolithium regagents. J. Organomet. Chem. 1981, 218, C21–C24. [Google Scholar] [CrossRef]
- Ishikawa, M.; Tabohashi, T.; Ohashi, H.; Kumada, M.; Iyoda, J. Chemsitry of siloles. 1-Methyldibenzosilacyclopentadienide anion. Organometallics 1983, 2, 351–352. [Google Scholar] [CrossRef]
- Pan, Y.; Hong, J.-H.; Choi, S.-B.; Boudjouk, P. Surprising Reaction of Non-Nucleophilic Bases with 1-Hydrosiloles: Addition and Not Deprotonation. Organometallics 1997, 16, 1445–1451. [Google Scholar] [CrossRef]
- Tamao, K.; Asahara, M.; Kawachi, A. The differing modes of reaction of 1-(8-dimethylamino-1-naphthyl)-1-hydrodisilane and 1-(1-naphthyl)-1-hydrodisilane in nickel-catalyzed reactions with acetylene: formation of a pseudo-pentacoordinate silole via Si-Si bond cleavage vs. hydrosilation without Si-Si bond cleavage. J. Organomet. Chem. 1996, 521, 325–334. [Google Scholar]
- Choi, S.-B.; Boudjouk, P.; Pan, Y. Mechanistic Studies of the Reaction of Hydrosiloles with Nucleophiles. Crystal Structures of the Enantiomers of 1-Methyl-2-(dimethylsilyl)-2,3,4,5-tetraphenyl-1-silacyclo- 3-pentene and 1-(Bis(trimethylsilyl)amino)-5-methyl- 2,3,4,5-tetraphenyl-1-silacyclo-3-pentene. Organometallics 1999, 18, 3813–3817. [Google Scholar]
- Jutzi, P.; Karl, A. Synthesis and reactions of some 1-R-1-R’-2,3,4,5-tetraphenyl-1-silacyclopentadienes. J. Organometa. Chem. 1981, 214, 289–302. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Geroge, M.V. Alkali metal, alkaline and acidic decomposition of silacyclopentadienes. J. Organomet. Chem. 1975, 85, 311–316. [Google Scholar] [CrossRef]
- Kalikhman, I.; Girshberg, O.; Lameyer, L.; Stalke, D.; Kost, D. Tautomeric Equilibrium between Penta- and Hexacoordinate Silicon Chelates. A Chloride Bridge between Two Pentacoordinate Silicons. J. Am. Chem. Soc. 2001, 123, 4709–4716. [Google Scholar] [CrossRef] [PubMed]
- Brook, A.G.; Brook, M.A. The Chemistry of Silenes. Adv. Organomet. Chem. 1996, 39, 71–158. [Google Scholar]
- Sekiguchi, A.; Zigler, S.S.; West, R. A synthon for the silicon-silicon triple bond. J. Am. Chem. Soc. 1986, 108, 4241–4242. [Google Scholar] [CrossRef]
- [Me(Me3Si)SiC4Ph4] (6) has been synthesized, however, the physical property and the spectral data were not provided: Wakahara, T.; Ando, W. Reaction of hydrosilanes with lithium. Formation of silole anions from 1-methylsilole via carbodianion. Chem. Letter. 1997, 11, 1179–1180. [Google Scholar] [CrossRef]
Sample Availability: Samples of the derivatives of the silole anion [MeSiC4Ph4]- are available from the author. |









© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hong, J.-H. Dissociation of the Disilatricyclic Diallylic Dianion [(C4Ph4SiMe)2]−2 to the Silole Anion [MeSiC4Ph4]− by Halide Ion Coordination or Halide Ion Nucleophilic Substitution at the Silicon Atom. Molecules 2011, 16, 8451-8462. https://doi.org/10.3390/molecules16108451
Hong J-H. Dissociation of the Disilatricyclic Diallylic Dianion [(C4Ph4SiMe)2]−2 to the Silole Anion [MeSiC4Ph4]− by Halide Ion Coordination or Halide Ion Nucleophilic Substitution at the Silicon Atom. Molecules. 2011; 16(10):8451-8462. https://doi.org/10.3390/molecules16108451
Chicago/Turabian StyleHong, Jang-Hwan. 2011. "Dissociation of the Disilatricyclic Diallylic Dianion [(C4Ph4SiMe)2]−2 to the Silole Anion [MeSiC4Ph4]− by Halide Ion Coordination or Halide Ion Nucleophilic Substitution at the Silicon Atom" Molecules 16, no. 10: 8451-8462. https://doi.org/10.3390/molecules16108451
APA StyleHong, J. -H. (2011). Dissociation of the Disilatricyclic Diallylic Dianion [(C4Ph4SiMe)2]−2 to the Silole Anion [MeSiC4Ph4]− by Halide Ion Coordination or Halide Ion Nucleophilic Substitution at the Silicon Atom. Molecules, 16(10), 8451-8462. https://doi.org/10.3390/molecules16108451