Selection of Peptides Targeting Helix 31 of Bacterial 16S Ribosomal RNA by Screening M13 Phage-Display Libraries

Abstract
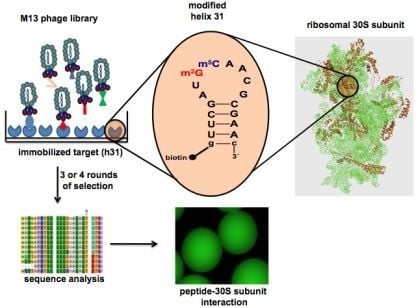
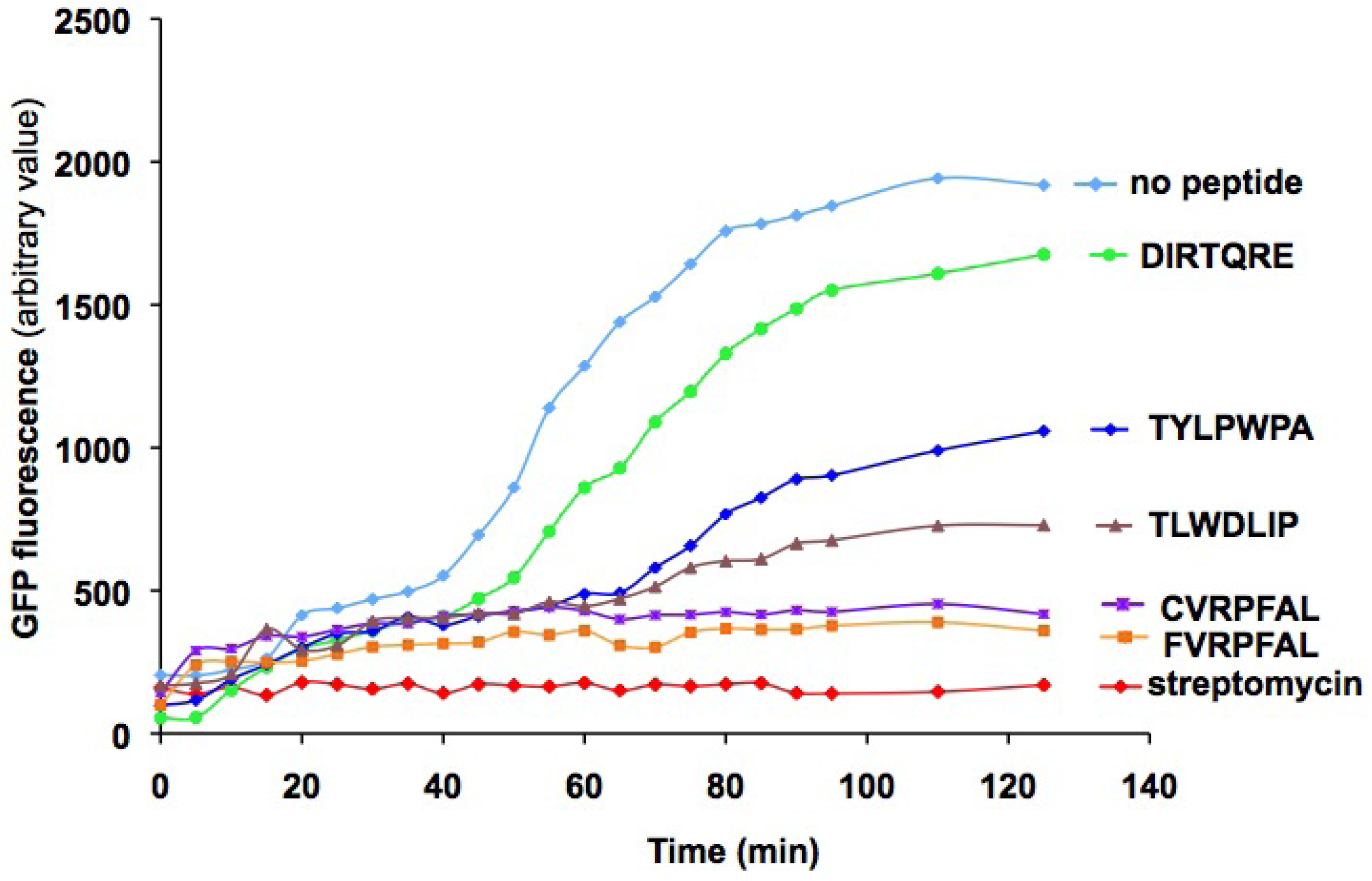
:
1. Introduction

2. Results and Discussion
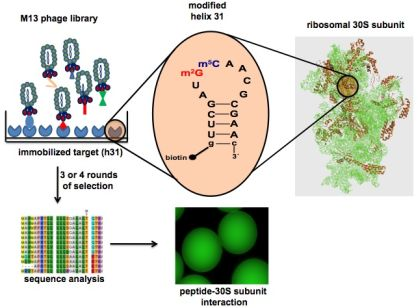
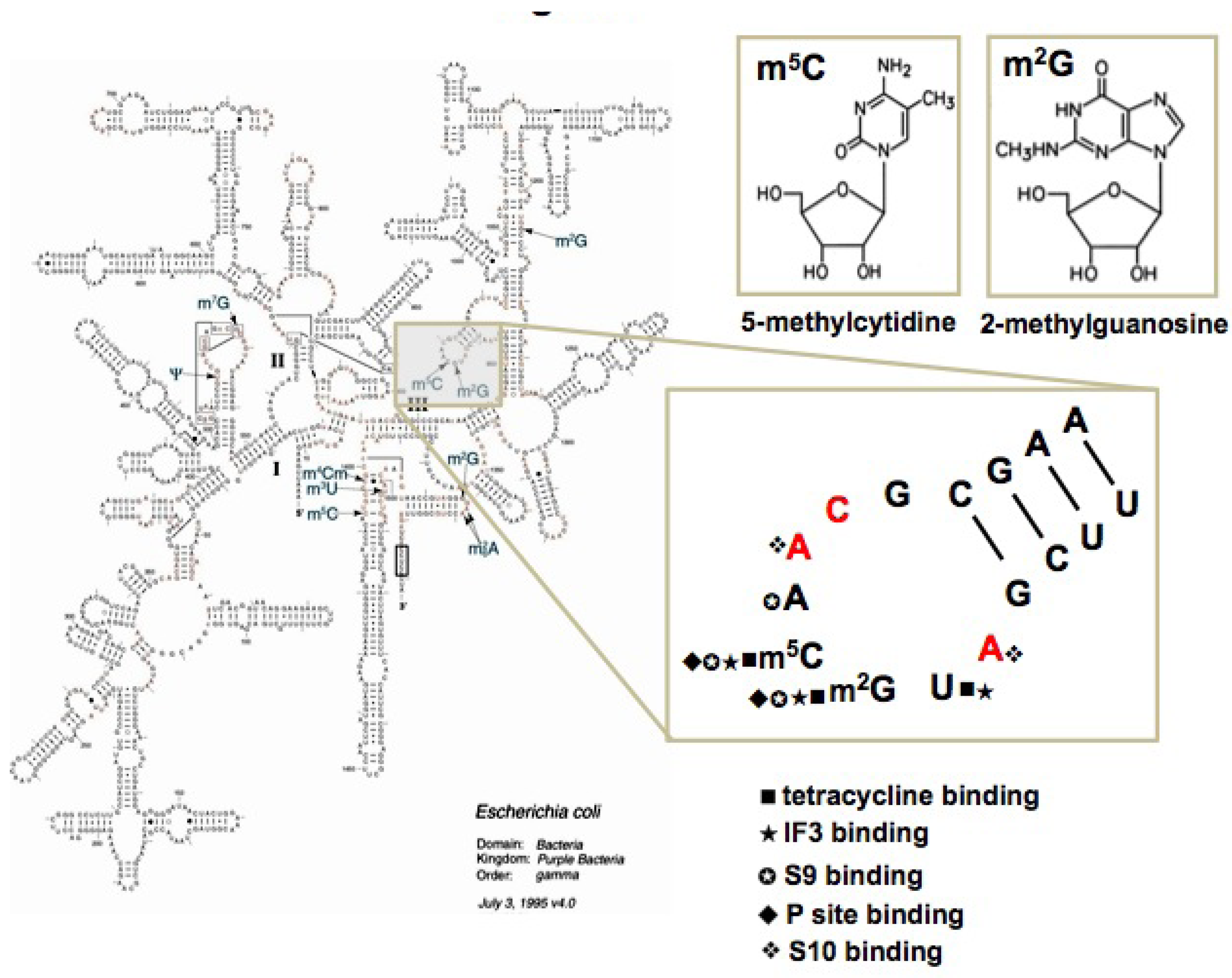
2.1. Target preparation

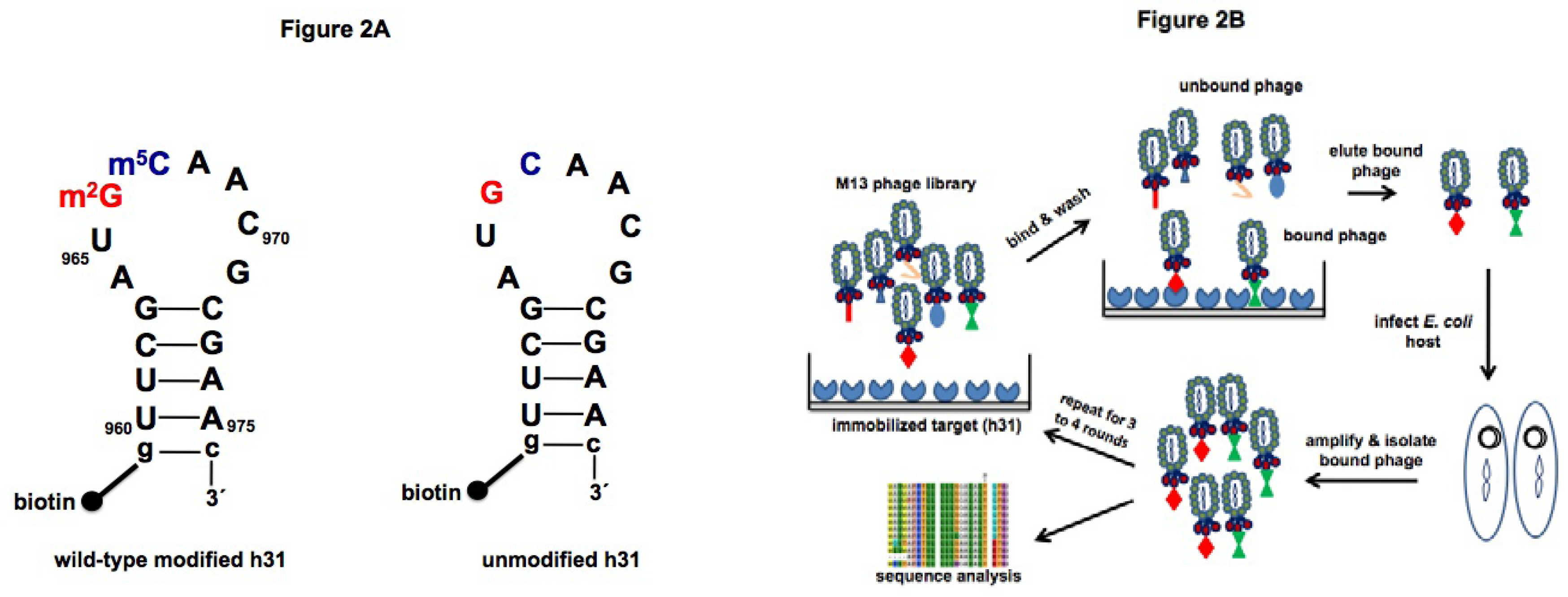
2.2. Screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Round no. | Round 1 | Round 2 | Round 3 | Round 4 |
|---|---|---|---|---|
| No. of washes (200 µL/wash) | 12 | 18 | 24 | 24 |
| Tween-20 | 0.1% | 0.1% | 0.3% | 0.5% |
| Target RNA (h31) | 50 pmoles | 50 pmoles | 30 pmoles | 30 pmoles |
| Competitor tRNA | 30 pmoles | 30 pmoles | 30 pmoles | 30 pmoles |
| Peptides | Frequency | ||||||
|---|---|---|---|---|---|---|---|
| F | V | R | P | F | P | L | 6 |
| F | V | R | P | F | A | L | 4 |
| F | V | R | P | Y | A | P | 2 |
| F | P | R | T | I | A | P | 1 |
| D | I | R | T | Q | R | E | 4 |
| D | I | R | T | Q | T | R | 2 |
| D | I | R | A | T | Q | A | 2 |
| A | T | P | L | W | L | K | 5 |
| A | T | P | T | Q | R | E | 3 |
| A | T | P | L | Y | L | R | 2 |
| T | L | W | D | L | I | P | 5 |
| T | L | W | S | F | M | P | 3 |
| T | L | W | V | P | S | R | 2 |
| T | L | T | T | L | T | N | 2 |
| T | L | T | F | F | H | R | 2 |
| T | Y | L | P | W | P | A | 5 |
| T | Y | L | P | W | P | P | 2 |
| T | Y | L | R | A | R | L | 2 |
| T | Y | P | F | A | P | W | 2 |
| T | Y | L | R | A | R | L | 1 |
| Peptides | Frequency | ||||||
|---|---|---|---|---|---|---|---|
| C | V | R | P | F | A | L | 5 |
| C | V | R | A | P | T | L | 2 |
| T | V | R | P | F | T | L | 2 |
| T | L | W | D | L | I | P | 2 |
| T | L | W | P | L | S | P | 2 |
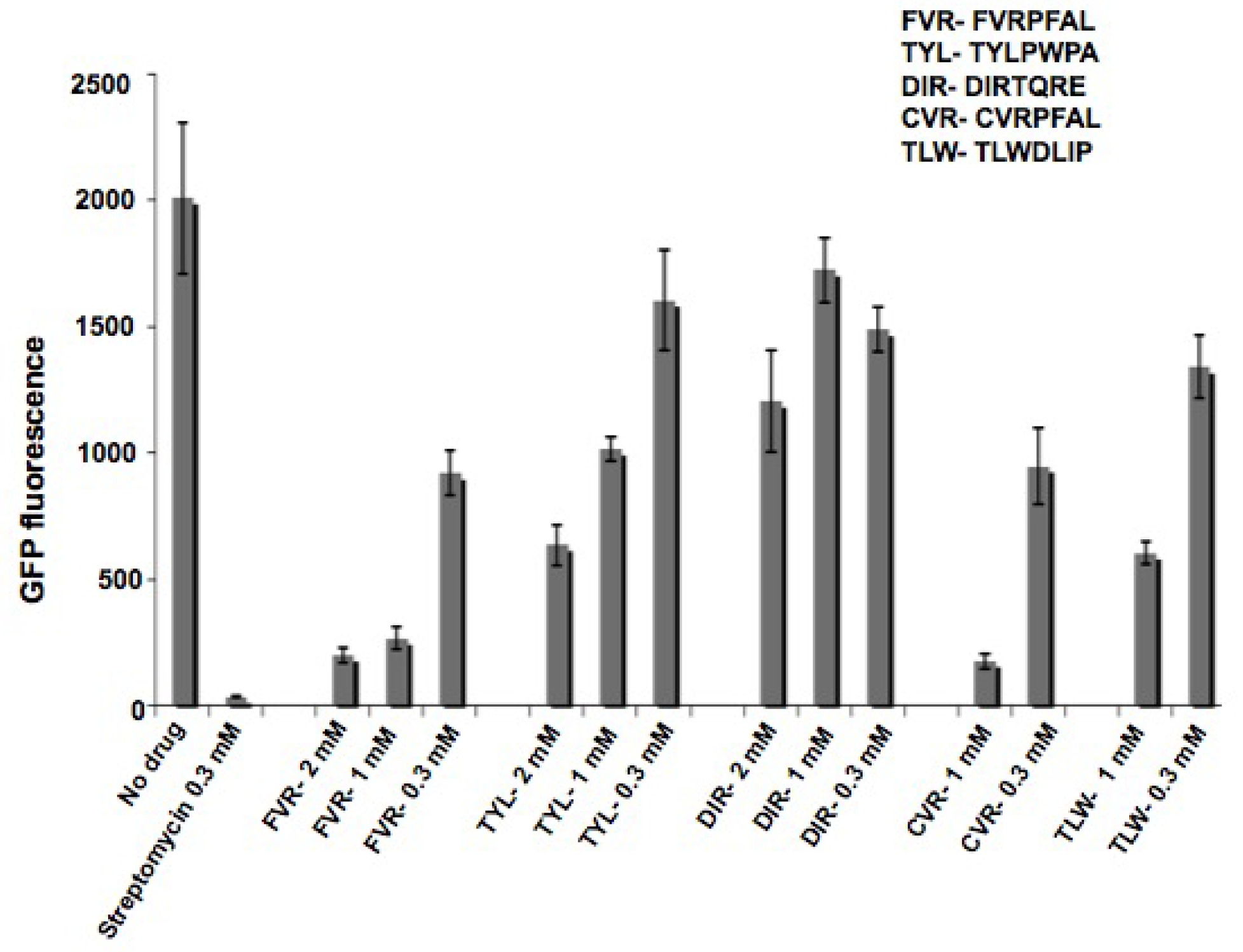
2.3. Characterization of peptides with a coupled in vitro transcription-translation assay

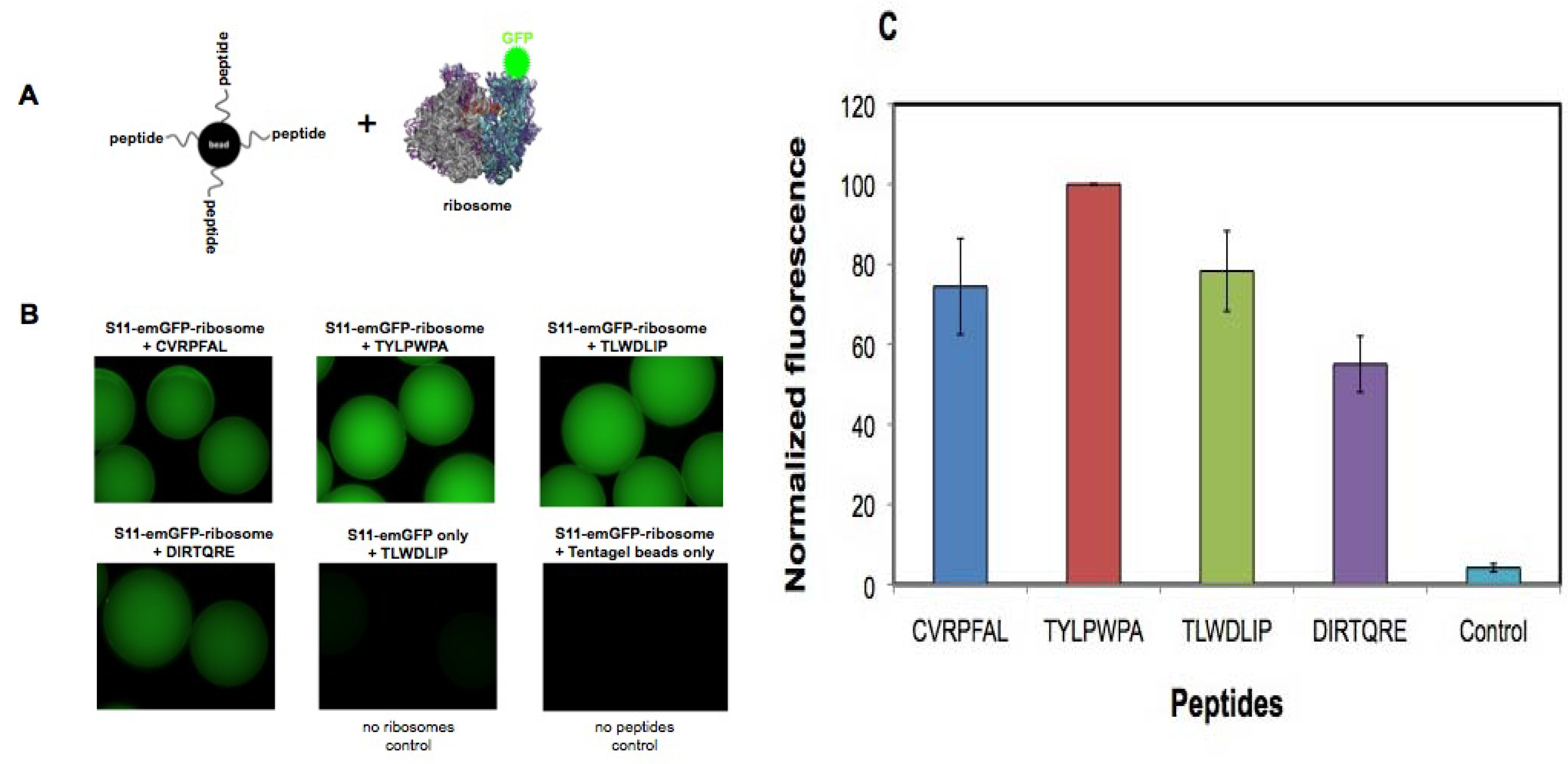
2.4. In vitro binding to bacterial 30S ribosomes

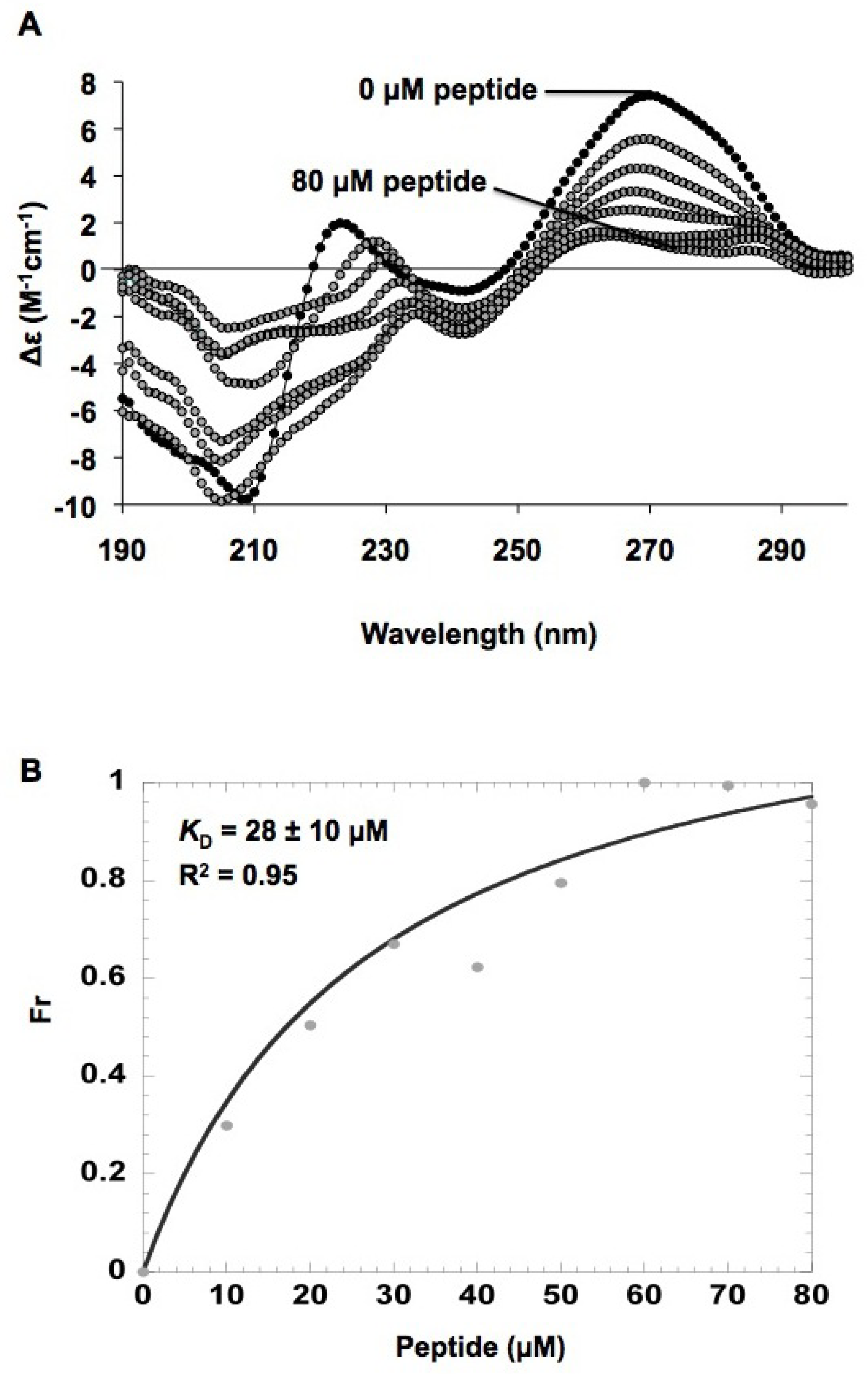
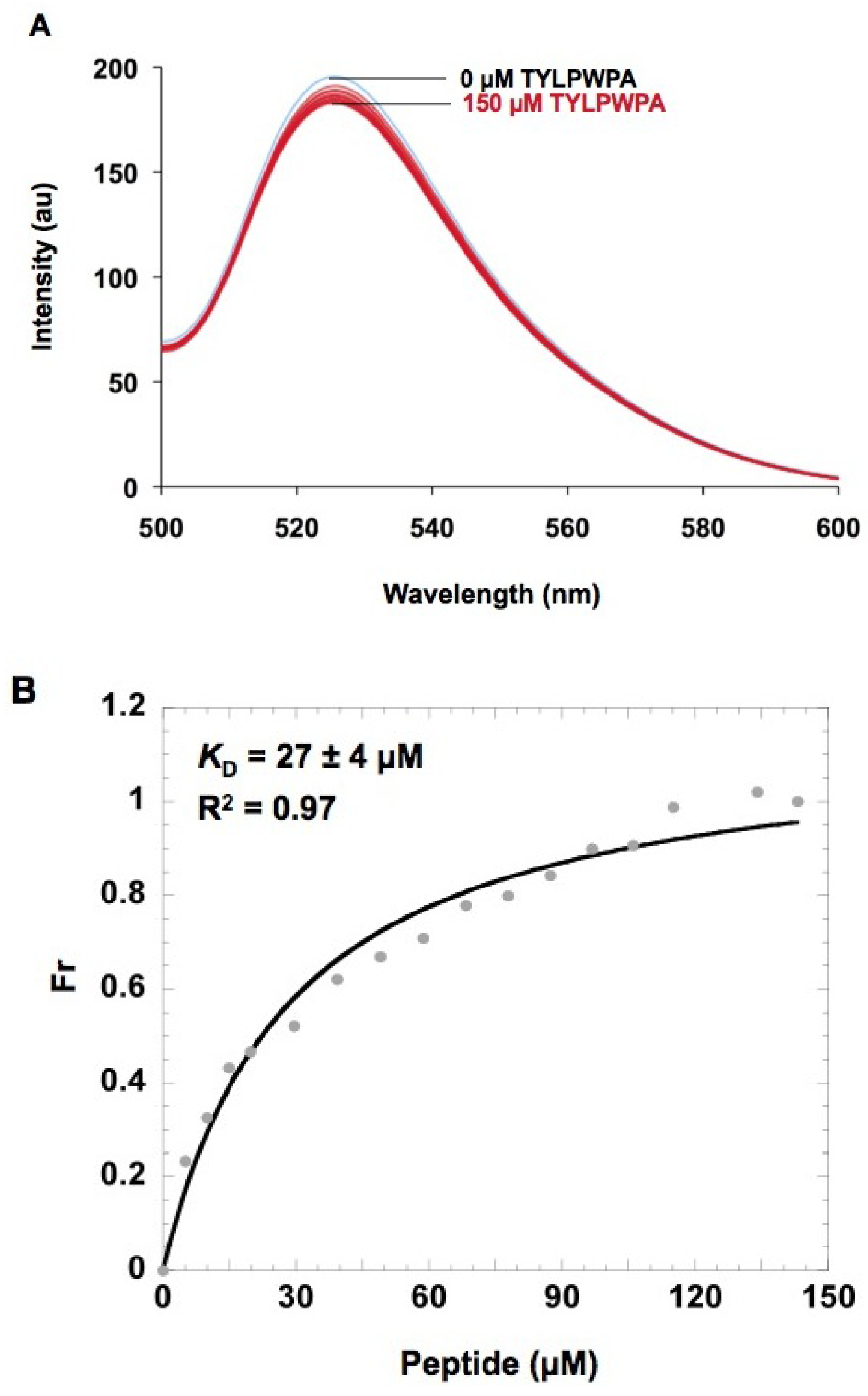
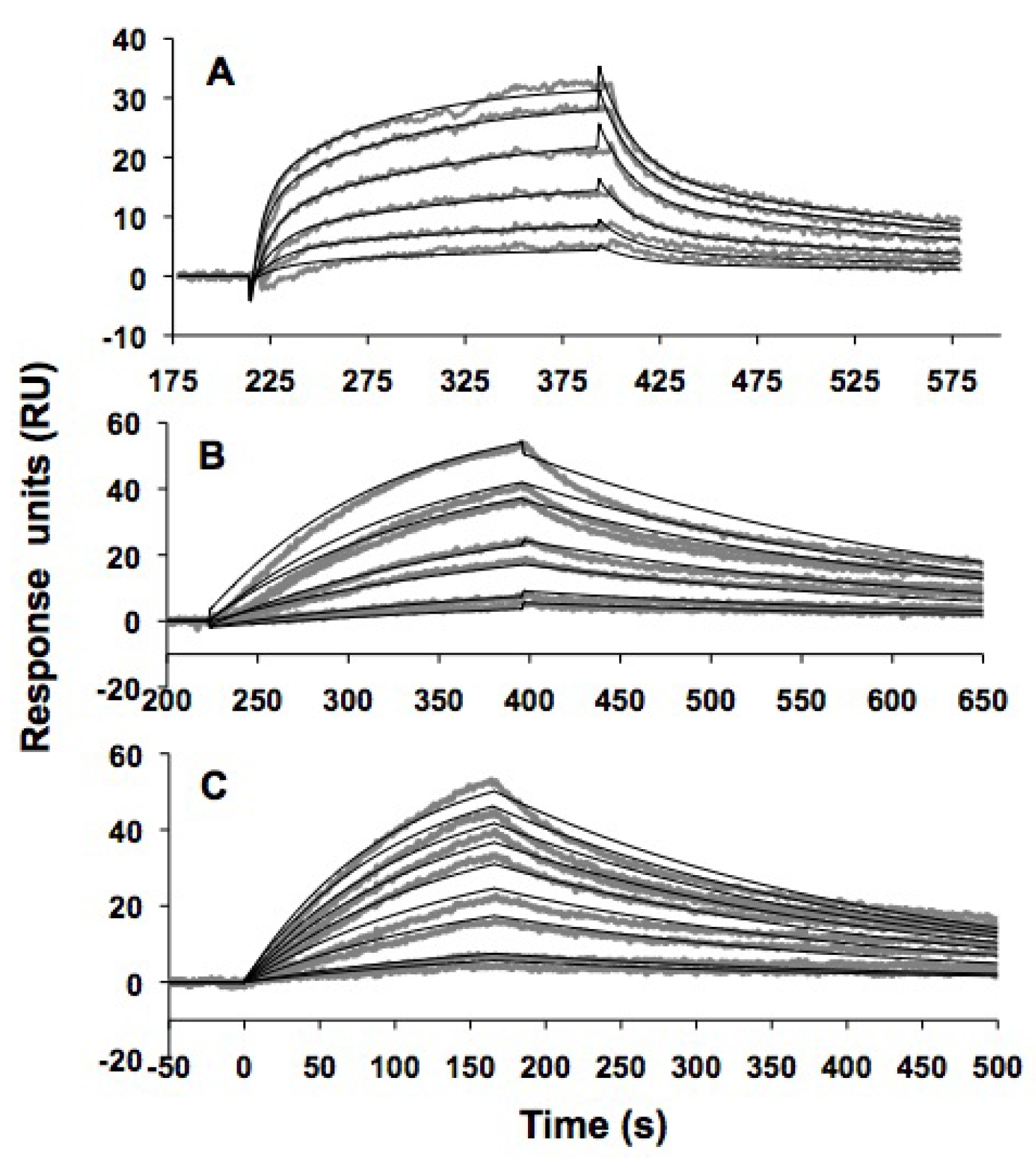
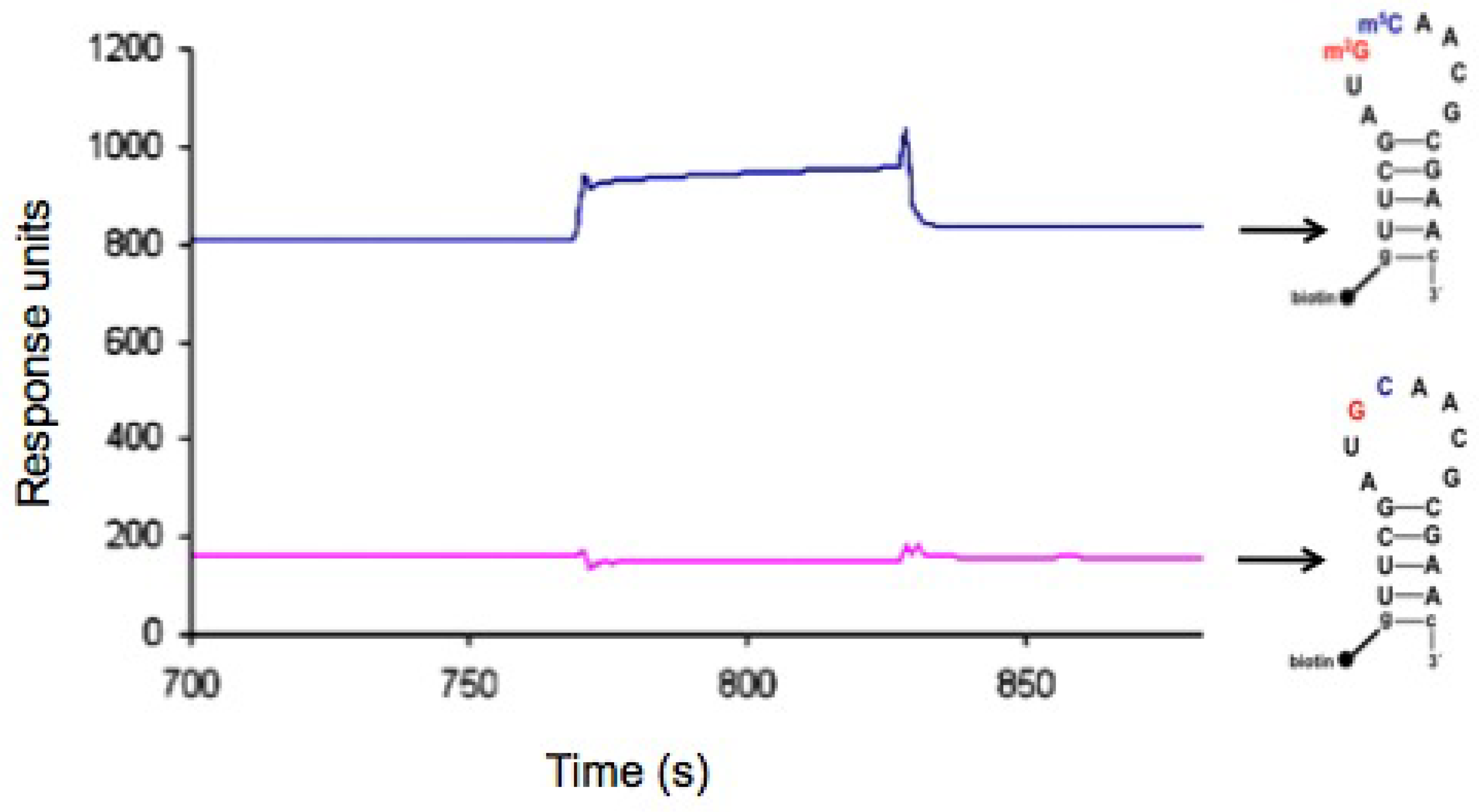
2.5. In vitro h31 binding studies


| TYLPWPA-GFP | TLWDLIP-GFP | CVRPFAL-GFP | GFP | |
|---|---|---|---|---|
| ka(M-1s-1) | (1.50 ± 0.02) × 102 | (93 ± 2) × 102 | (120 ± 1) × 102 | (2.22 ± 0.03) × 102 |
| kd (s-1) | (4.2 ± 0.04) × 10-3 | (3.1 ± 0.3) × 10-3 | (2.6 ± 0.02) × 10-3 | (4.4 ± 0.04) × 10-3 |
| KD | 28 ± 1 μM | 330 ± 7 nM | 230 ± 3 nM | 20 ± 1 μM |
| χ2 | 0.5 | 2.1 | 1.4 | 1.3 |

2.6. Discussion
3. Materials and Methods
3.1. Media and bacterial strains
3.2. RNA sample preparation
3.3. Biopanning of h31 target
3.4. General procedure for the Fmoc heptapeptide synthesis
3.5. HPLC purification of peptides
3.6. Quantification and analysis of peptides
3.7. In vitro transcription-translation inhibition assay
3.8. Ribosome-binding assay
3.9. Circular dichroism (CD) studies
3.10. Fluorescence binding methods
3.11. SPR methods
4. Conclusions
Acknowledgements
References
- Mattaj, I.W.; Nagai, K. Recruiting proteins to the RNA world. Nat. Struct. Biol. 1995, 2, 518–522. [Google Scholar] [CrossRef]
- Agris, P.F. The importance of being modified: roles of modified nucleosides and Mg2+ in RNA structure and function. Prog. Nucl. Acid Res. Mol. Biol. 1996, 53, 79–129. [Google Scholar] [CrossRef]
- Xu, Z.; O'Farrell, H.C.; Rife, J.P.; Culver, G.M. A conserved rRNA methyltransferase regulates ribosome biogenesis. Nat. Struct. Mol. Biol. 2008, 15, 534–536. [Google Scholar]
- Demirci, H.; Murphy, F., 4th.; Belardinelli, R.; Kelley, A.C.; Ramakrishnan, V.; Gregory, S.T.; Dahlberg, A.E.; Jogl, G. Modification of 16S ribosomal RNA by the KsgA methyltransferase restructures the 30S subunit to optimize ribosome function. RNA 2010, 16, 2319–2324. [Google Scholar] [CrossRef]
- Olejniczak, M.; Dale, T.; Fahlman, R.P.; Uhlenbeck, O.C. Idiosyncratic tuning of tRNAs to achieve uniform ribosome binding. Nat. Struct. Mol. Biol. 2005, 12, 788–793. [Google Scholar] [CrossRef]
- Chow, C.S.; Lamichhane, T.N.; Mahto, S.K. Expanding the nucleotide repertoire of the ribosome with post-transcriptional modifications. ACS Chem. Biol. 2007, 2, 610–619. [Google Scholar] [CrossRef]
- Decatur, W.A.; Fournier, M.J. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002, 27, 344–351. [Google Scholar] [CrossRef]
- Cunningham, P.R.; Richard, R.B.; Weitzmann, C.J.; Nurse, K.; Ofengand, J. The absence of modified nucleotides affects both in vitro assembly and in vitro function of the 30S ribosomal subunit of Escherichia coli. Biochimie 1991, 73, 789–796. [Google Scholar] [CrossRef]
- Basturea, G.N.; Rudd, K.E.; Deutscher, M.P. Identification and characterization of RsmE, the founding member of a new RNA base methyltransferase family. RNA 2006, 12, 426–434. [Google Scholar] [CrossRef]
- Green, R.; Noller, H.F. Reconstitution of functional 50S ribosomes from in vitro transcripts of Bacillus stearothermophilus 23S rRNA. Biochemistry 1999, 38, 1772–1779. [Google Scholar] [CrossRef]
- Green, R.; Noller, H.F. In vitro complementation analysis localizes 23S rRNA posttranscriptional modifications that are required for Escherichia coli 50S ribosomal subunit assembly and function. RNA 1996, 2, 1011–1021. [Google Scholar]
- Baxter-Roshek, J.L.; Petrov, A.N.; Dinman, J.D. Optimization of ribosome structure and function by rRNA base modification. PLoS One 2007, 2, e174. [Google Scholar] [CrossRef]
- Kehrenberg, C.; Schwarz, S.; Jacobsen, L.; Hansen, L.H.; Vester, B. A new mechanism for chloramphenicol, florfenicol and clindamycin resistance: methylation of 23S ribosomal RNA at A2503. Mol. Microbiol. 2005, 57, 1064–1073. [Google Scholar]
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2500–2505. [Google Scholar] [CrossRef]
- Wright, G.D. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007, 5, 175–186. [Google Scholar] [CrossRef]
- Anderson, A.D.; Nelson, J.M.; Rossiter, S.; Angulo, F.J. Public health consequences of use of antimicrobial agents in food animals in the United States. Microb. Drug Resist. 2003, 9, 373–379. [Google Scholar]
- Tomasz, A. Multiple-antibiotic-resistant pathogenic bacteria. A report on the Rockefeller University Workshop. N. Engl. J. Med. 1994, 330, 1247–1251. [Google Scholar] [CrossRef]
- Gregory, S.T.; Carr, J.F.; Dahlberg, A.E. A mutation in the decoding center of Thermus thermophilus 16S rRNA suggests a novel mechanism of streptomycin resistance. J. Bacteriol. 2005, 187, 2200–2202. [Google Scholar] [CrossRef]
- Thompson, J.; Skeggs, P.A.; Cundliffe, E. Methylation of 16S ribosomal RNA and resistance to the aminoglycoside antibiotics gentamicin and kanamycin determined by DNA from the gentamicin-producer, Micromonospora purpurea. Mol. Gen. Genet. 1985, 201, 168–173. [Google Scholar] [CrossRef]
- Pfister, P.; Hobbie, S.; Vicens, Q.; Böttger, E.C.; Westhof, E. The molecular basis for A-site mutations conferring aminoglycoside resistance: relationship between ribosomal susceptibility and X-ray crystal structures. ChemBioChem 2003, 4, 1078–1088. [Google Scholar] [CrossRef]
- Cannone, J.J.; Subramanian, S.; Schnare, M.N.; Collett, J.R.; D'Souza, L.M.; Du, Y.; Feng, B.; Lin, N.; Madabusi, L.V.; Müller, K.M.; Pande, N.; Shang, Z.; Yu, N.; Gutell, R.R. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 2002, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Kowalak, J.A.; Bruenger, E.; Crain, P.F.; McCloskey, J.A. Identities and phylogenetic comparisons of posttranscriptional modifications in 16S ribosomal RNA from Haloferax volcanii. J. Biol. Chem. 2000, 275, 24484–24489. [Google Scholar]
- Maden, B.E. The numerous modified nucleotides in eukaryotic ribosomal RNA. Prog. Nucl. Acid Res. Mol. Biol. 1990, 39, 241–303. [Google Scholar] [CrossRef]
- Woese, C.R.; Gupta, R.; Hahn, C.M.; Zillig, W.; Tu, J. The phylogenetic relationships of three sulfur dependent archaebacteria. Syst. Appl. Microbiol. 1984, 5, 97–105. [Google Scholar]
- Youvan, D.C.; Hearst, J.E. A sequence from Drosophila melanogaster 18S rRNA bearing the conserved hypermodified nucleoside am psi: analysis by reverse transcription and high-performance liquid chromatography. Nucl. Acid. Res. 1981, 9, 1723–1741. [Google Scholar] [CrossRef]
- Lesnyak, D.V.; Osipiuk, J.; Skarina, T.; Sergiev, P.V.; Bogdanov, A.A.; Edwards, A.; Savchenko, A.; Joachimiak, A.; Dontsova, O.A. Methyltransferase that modifies guanine 966 of the 16S rRNA: functional identification and tertiary structure. J. Biol. Chem. 2007, 282, 5880–5887. [Google Scholar]
- Gu, X.R.; Gustafsson, C.; Ku, J.; Yu, M.; Santi, D.V. Identification of the 16S rRNA m5C967 methyltransferase from Escherichia coli. Biochemistry 1999, 38, 4053–4057. [Google Scholar] [CrossRef]
- Saraiya, A.A.; Lamichhane, T.N.; Chow, C.S.; SantaLucia, J., Jr.; Cunningham, P.R. Identification and role of functionally important motifs in the 970 loop of Escherichia coli 16S ribosomal RNA. J. Mol. Biol. 2008, 376, 645–657. [Google Scholar] [CrossRef]
- Schuwirth, B.S.; Borovinskaya, M.A.; Hau, C.W.; Zhang, W.; Vila-Sanjurjo, A.; Holton, J.M.; Cate, J.H. Structures of the bacterial ribosome at 3.5 Å resolution. Science 2005, 310, 827–834. [Google Scholar] [CrossRef]
- Yusupov, M.M.; Yusupova, G.Z.; Baucom, A.; Lieberman, K.; Earnest, T.N.; Cate, J.H.; Noller, H.F. Crystal structure of the ribosome at 5.5 Å resolution. Science 2001, 292, 883–896. [Google Scholar] [CrossRef]
- Johnson, R.M.; Harrison, S.D.; Maclean, D. Therapeutic applications of cell-penetrating peptides. Methods Mol. Biol. 2011, 683, 535–551. [Google Scholar] [CrossRef]
- Huther, A.; Dietrich, U. The emergence of peptides as therapeutic drugs for the inhibition of HIV-1. AIDS Rev. 2007, 9, 208–217. [Google Scholar]
- Sato, A.K.; Viswanathan, M.; Kent, R.B.; Wood, C.R. Therapeutic peptides: technological advances driving peptides into development. Curr. Opin. Biotechnol. 2006, 17, 638–642. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Adessi, C.; Soto, C. Converting a peptide into a drug: strategies to improve stability and bioavailability. Curr. Med. Chem. 2002, 9, 963–978. [Google Scholar]
- Smith, G.P.; Scott, J.K. Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol. 1993, 217, 228–257. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef]
- Calcutt, M.J.; Kremer, M.T.; Giblin, M.F.; Quinn, T.P.; Deutscher, S.L. Isolation and characterization of nucleic acid-binding antibody fragments from autoimmune mice-derived bacteriophage display libraries. Gene 1993, 137, 77–83. [Google Scholar] [CrossRef]
- Laird-Offringa, I.A.; Belasco, J.G. In vitro genetic analysis of RNA-binding proteins using phage display libraries. Methods Enzymol. 1996, 267, 149–168. [Google Scholar] [CrossRef]
- Frankel, A.D. Peptide models of the Tat-TAR protein-RNA interaction. Protein Sci. 1992, 1, 1539–1542. [Google Scholar] [CrossRef]
- Grate, D.; Wilson, C. Role REVersal: understanding how RRE RNA binds its peptide ligand. Structure 1997, 5, 7–11. [Google Scholar] [CrossRef]
- Patel, D.J. Adaptive recognition in RNA complexes with peptides and protein modules. Curr. Opin. Struct. Biol. 1999, 9, 74–87. [Google Scholar] [CrossRef]
- Calnan, B.J.; Biancalana, S.; Hudson, D.; Frankel, A.D. Analysis of arginine-rich peptides from the HIV Tat protein reveals unusual features of RNA-protein recognition. Genes Dev. 1991, 5, 201–210. [Google Scholar] [CrossRef]
- Powers, J.E.; Marchbank, M.T.; Deutscher, S.L. The isolation of U1 RNA-binding antibody fragments from autoimmune human-derived bacteriophage display libraries. Nucl. Acid. Symp. Ser. 1995, 240–243. [Google Scholar]
- Hoet, R.M.; Pieffers, M.; Stassen, M.H.; Raats, J.; de Wildt, R.; Pruijn, G.J.; van den Hoogen, F.; van Venrooij, W.J. The importance of the light chain for the epitope specificity of human anti-U1 small nuclear RNA autoantibodies present in systemic lupus erythematosus patients. J. Immunol. 1999, 163, 3304–3312. [Google Scholar]
- Pustowka, A.; Dietz, J.; Ferner, J.; Baumann, M.; Landersz, M.; Königs, C.; Schwalbe, H.; Dietrich, U. Identification of peptide ligands for target RNA structures derived from the HIV-1 packaging signal psi by screening phage-displayed peptide libraries. ChemBioChem 2003, 4, 1093–1097. [Google Scholar] [CrossRef]
- Dietz, J.; Koch, J.; Kaur, A.; Raja, C.; Stein, S.; Grez, M.; Pustowka, A.; Mensch, S.; Ferner, J.; Möller, L.; Bannert, N.; Tampé, R.; Divita, G.; Mély, Y.; Schwalbe, H.; Dietrich, U. Inhibition of HIV-1 by a peptide ligand of the genomic RNA packaging signal Psi. ChemMedChem 2008, 3, 749–755. [Google Scholar] [CrossRef]
- Li, M.; Duc, A.C.; Klosi, E.; Pattabiraman, S.; Spaller, M.R.; Chow, C.S. Selection of peptides that target the aminoacyl-tRNA site of bacterial 16S ribosomal RNA. Biochemistry 2009, 48, 8299–8311. [Google Scholar] [CrossRef]
- Llano-Sotelo, B.; Klepacki, D.; Mankin, A.S. Selection of small peptides, inhibitors of translation. J. Mol. Biol. 2009, 391, 813–819. [Google Scholar] [CrossRef]
- Mucha, P.; Szyk, A.; Rekowski, P.; Agris, P.F. Sequence-altered peptide adopts optimum conformation for modification-dependent binding of the yeast tRNAPhe anticodon domain. Protein J. 2004, 23, 33–38. [Google Scholar]
- Mucha, P.; Szyk, A.; Rekowski, P.; Weiss, P.A.; Agris, P.F. Anticodon domain methylated nucleosides of yeast tRNA(Phe) are significant recognition determinants in the binding of a phage display selected peptide. Biochemistry 2001, 40, 14191–14199. [Google Scholar]
- Eshete, M.; Marchbank, M.T.; Deutscher, S.L.; Sproat, B.; Leszczynska, G.; Malkiewicz, A.; Agris, P.F. Specificity of phage display selected peptides for modified anticodon stem and loop domains of tRNA. Protein J. 2007, 26, 61–73. [Google Scholar]
- Agris, P.F.; Marchbank, M.T.; Newman, W.; Guenther, R.; Ingram, P.; Swallow, J.; Mucha, P.; Szyk, A.; Rekowski, P.; Peletskaya, E.; Deutscher, S.L. Experimental models of protein-RNA interaction: isolation and analyses of tRNA(Phe) and U1 snRNA-binding peptides from bacteriophage display libraries. J. Protein Chem. 1999, 18, 425–435. [Google Scholar] [CrossRef]
- Abeydeera, N.D.; Chow, C.S. Synthesis and characterization of modified nucleotides in the 970 hairpin loop of Escherichia coli 16S ribosomal RNA. Bioorg. Med. Chem. 2009, 17, 5887–5893. [Google Scholar] [CrossRef]
- Devlin, J.J.; Panganiban, L.C.; Devlin, P.E. Random peptide libraries: a source of specific protein binding molecules. Science 1990, 249, 404–406. [Google Scholar]
- Mandava, S.; Makowski, L.; Devarapalli, S.; Uzubell, J.; Rodi, D.J. RELIC-a bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics 2004, 4, 1439–1460. [Google Scholar] [CrossRef]
- Korostelev, A.; Trakhanov, S.; Laurberg, M.; Noller, H.F. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell 2006, 126, 1065–1077. [Google Scholar] [CrossRef]
- Berk, V.; Zhang, W.; Pai, R.D.; Cate, J.H. Structural basis for mRNA and tRNA positioning on the ribosome. Proc. Natl. Acad. Sci. USA 2006, 103, 15830–15834. [Google Scholar] [CrossRef]
- Berk, V.; Cate, J.H. Insights into protein biosynthesis from structures of bacterial ribosomes. Curr. Opin. Struct. Biol. 2007, 17, 302–309. [Google Scholar] [CrossRef]
- Sidhu, S.S.; Koide, S. Phage display for engineering and analyzing protein interaction interfaces. Curr. Opin. Struct. Biol. 2007, 17, 481–487. [Google Scholar] [CrossRef]
- Park, H.Y.; Kim, J.; Cho, J.H.; Moon, J.Y.; Lee, S.J.; Yoon, M.Y. Phage display screen for peptides that bind bcl-2 protein. J. Biomol. Screen. 2011, 16, 82–89. [Google Scholar] [CrossRef]
- Llano-Sotelo, B.; Azucena, E.F., Jr.; Kotra, L.P.; Mobashery, S.; Chow, C.S. Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol. 2002, 9, 455–463. [Google Scholar] [CrossRef]
- Cooper, J.P.; Hagerman, P.J. Analysis of fluorescence energy transfer in duplex and branched DNA molecules. Biochemistry 1990, 29, 9261–9268. [Google Scholar] [CrossRef]
- Maruyama, T.; Shinohara, T.; Ichinose, H.; Kitaoka, M.; Okamura, N.; Kamiya, N.; Goto, M. Mutation detection in DNA oligonucleotides based on a guanine quenching method coupled with enzymatic digestion of single-stranded DNA. Biotechnol. Lett. 2005, 27, 1349–1354. [Google Scholar] [CrossRef]
- Selmer, M.; Dunham, C.M.; Murphy, F.V., 4th.; Weixlbaumer, A.; Petry, S.; Kelley, A.C.; Weir, J.R.; Ramakrishnan, V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science 2006, 313, 1935–1942. [Google Scholar] [CrossRef]
- Fourmy, D.; Recht, M.I.; Blanchard, S.C.; Puglisi, J.D. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 1996, 274, 1367–1371. [Google Scholar]
- Fourmy, D.; Yoshizawa, S.; Puglisi, J.D. Paromomycin binding induces a local conformational change in the A-site of 16S rRNA. J. Mol. Biol. 1998, 277, 333–345. [Google Scholar]
- Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983, 166, 557–580. [Google Scholar] [CrossRef]
- Luria, S.E.; Burrous, J.W. Hybridization between Escherichia coli and Shigella. J. Bacteriol. 1957, 74, 461–476. [Google Scholar]
- Dower, W.J.; Miller, J.F.; Ragsdale, C.W. High efficiency transformation of E. coli by high voltage electroporation. Nucl. Acid. Res. 1988, 16, 6127–6145. [Google Scholar] [CrossRef]
- Porath, J.; Carlsson, J.; Olsson, I.; Belfrage, G. Metal chelate affinity chromatography, a new approach to protein fractionation. Nature 1975, 258, 598–599. [Google Scholar]
- Gorman, J.J. Fluorescent labeling of cysteinyl residues to facilitate electrophoretic isolation of proteins suitable for amino-terminal sequence analysis. Anal. Biochem. 1987, 160, 376–387. [Google Scholar] [CrossRef]
- Qin, P.Z.; Pyle, A.M. Site-specific labeling of RNA with fluorophores and other structural probes. Methods 1999, 18, 60–70. [Google Scholar] [CrossRef]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Mac, F.D.; Fowler, N. On the mechanism of the reaction of ninhydrin with amino acids. II. A spectrophotometric study of hydrindantin reactions. J. Biol. Chem. 1950, 186, 13–22. [Google Scholar]
- Custom peptide synthesis: concentration calculation. Available online: http://www.sigmaaldrich.com/life-science/ custom-oligos/custom-peptides/learning-center/concentration-calculation.html Accessed on 26 January 2011.
- Protein assays: Micro BCA Protein Assay Kit. Available online: http://www.piercenet.com/products /browse.cfm?fldID=02020102 Accessed on 26 January 2011.
- Carpino, L.A.; Han, G.Y. The 9-fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group. J. Am. Chem. Soc. 1970, 92, 5748–5749. [Google Scholar] [CrossRef]
- Rink, H. Solid-phase synthesis of protected peptide fragments using a trialkoxy-diphenyl-methyl ester resin. Tetrahedron Lett. 1987, 28, 3787–3790. [Google Scholar] [CrossRef]
- Greenfield, L.; Boone, T.; Wilcox, G. DNA sequence of the araBAD promoter in Escherichia coli B/r. Proc. Natl. Acad. Sci. USA 1978, 75, 4724–4728. [Google Scholar] [CrossRef]
- Kaplan, D.A.; Greenfield, L.; Boone, T.; Wilcox, G. Hybrid plasmids containing the araBAD genes of Escherichia coli B/r. Gene 1978, 3, 177–189. [Google Scholar] [CrossRef]
- Moazed, D.; Stern, S.; Noller, H.F. Rapid chemical probing of conformation in 16S ribosomal RNA and 30S ribosomal subunits using primer extension. J. Mol. Biol. 1986, 187, 399–416. [Google Scholar]
- Davis, T.M.; Wilson, W.D. Surface plasmon resonance biosensor analysis of RNA-small molecule interactions. Methods Enzymol. 2001, 340, 22–51. [Google Scholar]
- Marquart, A. SPR pages. Access online. Available online: http://www.sprpages.nl/Index.php. Accessed on 26 January 2011.
- Sample Availability: The plasmids to generate peptide-GFP constructs are available from the authors.
Supplementary Materials
| Rounds | Input PFU | Output PFU | % yield |
|---|---|---|---|
| Pre-screen with beads | 2.0 × 1011 | 2.5 × 1010 | 0.0125 |
| I | 2.8 × 1011 | 2.2 × 106 | 0.08 × 10-4 |
| II | 2.5 × 1011 | 4.3 × 106 | 0.17 × 10-4 |
| III | 2.2 × 1011 | 3.1 × 106 | 0.14 × 10-4 |
| Peptides | ||||||
|---|---|---|---|---|---|---|
| T | L | Q | P | G | G | A |
| S | L | L | A | H | P | H |
| H | L | E | N | H | P | M |
| S | L | V | S | H | P | M |
| H | L | E | N | H | P | M |
| G | L | D | H | H | P | P |
| I | P | E | W | H | P | Q |
| S | L | L | S | H | P | Q |
| T | L | L | A | H | P | Q |
| N | L | V | S | H | P | Q |
| I | P | E | W | H | P | Q |
| H | L | A | N | H | P | Q |
| S | L | L | A | H | P | Q |
| T | L | I | A | H | P | Q |
| S | L | I | A | H | P | Q |
| N | L | V | N | H | P | Q |
| N | L | L | N | H | P | Q |
| S | L | I | A | H | P | Q |
| I | S | S | T | H | P | Q |
| T | L | L | A | H | P | Q |
| I | P | E | W | H | P | Q |
| N | L | V | N | H | P | Q |
| T | L | L | A | H | P | Q |
| T | L | L | N | H | P | Q |
| T | L | L | A | H | P | Q |
| N | L | L | N | H | P | Q |
| T | L | I | S | H | P | Q |
| H | F | T | N | H | P | Q |
| I | A | P | N | H | P | Q |
| I | P | E | W | H | P | Q |
| T | L | L | N | H | P | Q |
| S | L | L | A | H | P | Q |
| T | L | I | S | H | P | Q |
| S | L | L | A | H | P | Q |
| N | L | L | N | H | P | Q |
| S | L | L | A | H | P | Q |
| I | P | E | W | H | P | Q |
| N | L | L | N | H | P | Q |
| N | L | L | N | H | P | Q |
| P | L | L | A | H | P | Q |
| S | L | L | A | H | P | Q |
| N | L | L | N | H | P | Q |
| S | L | L | A | H | P | Q |
| T | L | L | A | H | P | Q |
| I | P | E | W | H | P | Q |
| T | L | I | S | H | P | Q |
| T | L | I | S | H | P | Q |
| N | L | V | N | H | P | Q |
| S | L | I | A | H | P | Q |
| N | L | L | N | H | P | Q |
| G | Y | D | K | H | P | Q |
| S | L | I | A | H | P | Q |
| N | L | L | N | H | P | Q |
| I | P | E | W | H | P | Q |
| I | P | Y | W | H | P | Q |
| H | L | I | A | H | P | Q |
| S | L | L | S | H | P | Q |
| N | L | L | N | H | P | Q |
| T | L | L | A | H | P | Q |
| S | L | L | A | H | P | Q |
| N | L | V | N | H | P | Q |
| Y | L | V | N | H | P | Q |
| N | L | I | S | H | P | Q |
| N | L | L | N | H | P | Q |
| H | Y | E | G | H | P | Q |
| I | P | Y | W | H | P | Q |
| T | L | I | S | H | P | Q |
| I | P | Y | W | H | P | Q |
| T | L | L | A | H | P | Q |
| T | L | L | A | H | P | Q |
| I | A | P | N | H | P | Q |
| H | L | Y | A | H | P | Q |
| H | F | T | N | H | P | Q |
| N | L | L | N | H | P | Q |
| I | P | E | W | H | P | Q |
| H | L | Y | A | H | P | Q |
| N | P | T | K | H | Q | M |
| T | P | S | P | L | A | G |
| V | T | P | T | M | H | P |
| A | P | R | D | P | L | S |
| M | Q | S | H | Q | D | S |
| S | P | T | Y | Q | R | L |
| Y | M | E | H | S | R | V |
| H | R | I | S | W | P | S |
| D | P | F | F | Y | T | P |
| Peptides | Frequency | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H | H | H | P | P | L | A | 5 | ||||||
| H | H | P | P | F | P | P | 2 | ||||||
| H | P | P | S | W | G | D | 2 | ||||||
| H | P | P | H | F | P | N | 1 | ||||||
| H | P | T | G | L | F | R | 2 | ||||||
| H | N | H | L | G | V | H | 2 | ||||||
| K | P | F | H | N | S | T | 3 | ||||||
| K | P | F | H | N | S | T | 2 | ||||||
| K | P | G | Y | S | S | A | 2 | ||||||
| K | P | P | Q | V | P | L | 2 | ||||||
| K | P | H | A | P | H | R | 1 | ||||||
| K | P | P | H | H | P | R | 3 | ||||||
| K | P | P | H | P | V | Y | 1 | ||||||
| K | P | V | K | V | P | R | 2 | ||||||
| Sequences | Homology with | Position |
|---|---|---|
| TYLPWPA | ATP dependent RNA helicase, Dead/DeaH box family | 2 YLPWA 7 42 YLPWA 47 |
| FVRPFPL | Glycine tRNA synthetase, beta subunit | 1 FVRP 4 157 FVRP 160 |
| TLWDLIP | 23S rRNA m5U747 methyl transferase | 2 LWDL 5 237 LWDL 240 |
| Leucyl-tRNA synthetase | 1 TLW 3 771 TLW 773 | |
| CVRPFAL | Glycine tRNA synthetase, beta subunit | 4 PFAL 7 477 PFAL 480 |
| DIRTQRE | Predicted methyltransferase | 4 TQRE 7 224 TQRE 227 |
| Valyl tRNA synthetase | 2 IRTQR 7 63 IR - QR 67 | |
| ATPLWLK | Phenylalanine tRNA synthetase | 2 TPLWLK 7 230 TPLWLK 235 |
| HHHPPLA | Fused glutamine amidotransferase | 3 HPPLA 7 384 HPPLA 388 |
| KPFHNST | 16S rRNA m2G1207 methylase | 2 PFHN 5 270 PFHD 273 |


© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lamichhane, T.N.; Abeydeera, N.D.; Duc, A.-C.E.; Cunningham, P.R.; Chow, C.S. Selection of Peptides Targeting Helix 31 of Bacterial 16S Ribosomal RNA by Screening M13 Phage-Display Libraries. Molecules 2011, 16, 1211-1239. https://doi.org/10.3390/molecules16021211
Lamichhane TN, Abeydeera ND, Duc A-CE, Cunningham PR, Chow CS. Selection of Peptides Targeting Helix 31 of Bacterial 16S Ribosomal RNA by Screening M13 Phage-Display Libraries. Molecules. 2011; 16(2):1211-1239. https://doi.org/10.3390/molecules16021211
Chicago/Turabian StyleLamichhane, Tek N., N. Dinuka Abeydeera, Anne-Cécile E. Duc, Philip R. Cunningham, and Christine S. Chow. 2011. "Selection of Peptides Targeting Helix 31 of Bacterial 16S Ribosomal RNA by Screening M13 Phage-Display Libraries" Molecules 16, no. 2: 1211-1239. https://doi.org/10.3390/molecules16021211
APA StyleLamichhane, T. N., Abeydeera, N. D., Duc, A. -C. E., Cunningham, P. R., & Chow, C. S. (2011). Selection of Peptides Targeting Helix 31 of Bacterial 16S Ribosomal RNA by Screening M13 Phage-Display Libraries. Molecules, 16(2), 1211-1239. https://doi.org/10.3390/molecules16021211