3.2. Chemistry
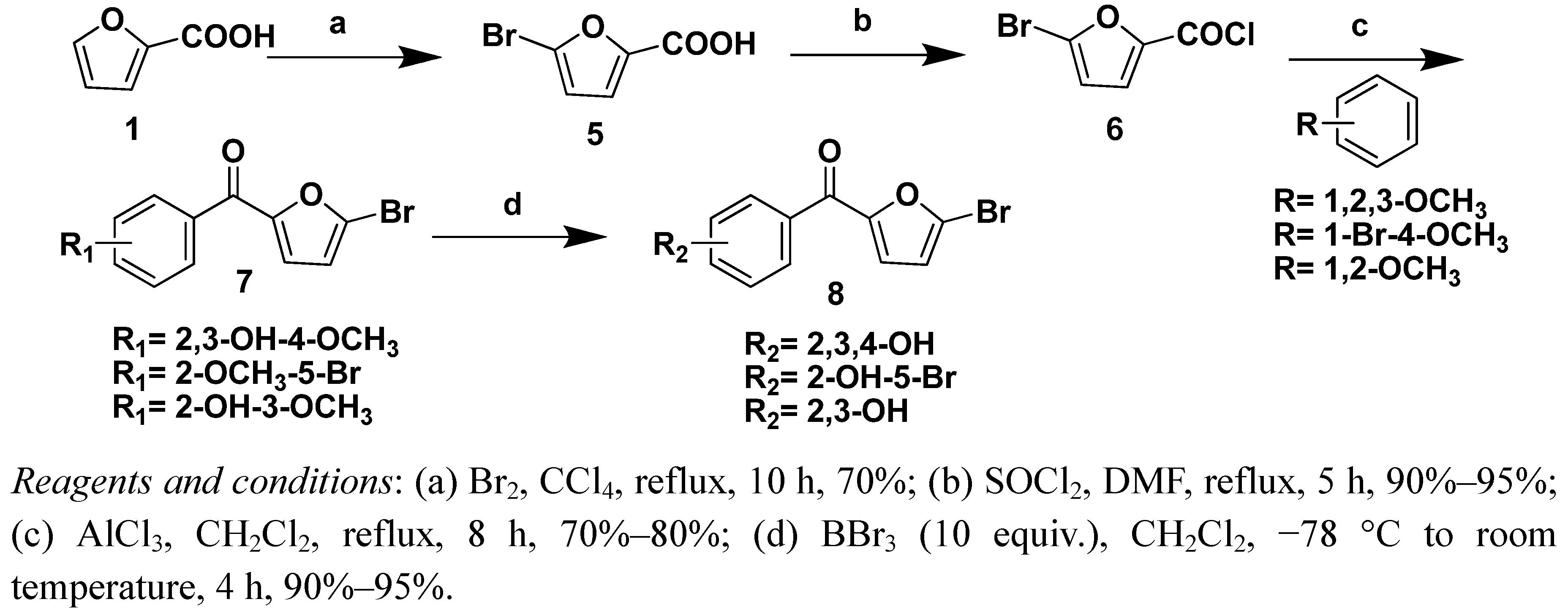
3.2.1. Procedure for the synthesis of 5-bromofuran-2-carboxylic acid 5
Br2 (8 mL) was slowly added to a solution of 2-furancarboxylic acid (1, 14.0 g) in CCl4 (60 mL). The reaction mixture was stirred at 45–50 °C for 24 h. The solvent was then removed under reduced pressure to yield a red solid which was recrystallized from boiling water to give the compound 5.
3.2.2. Procedure for the synthesis of 4,5-dibromofuran-2-carboxylic acid 11
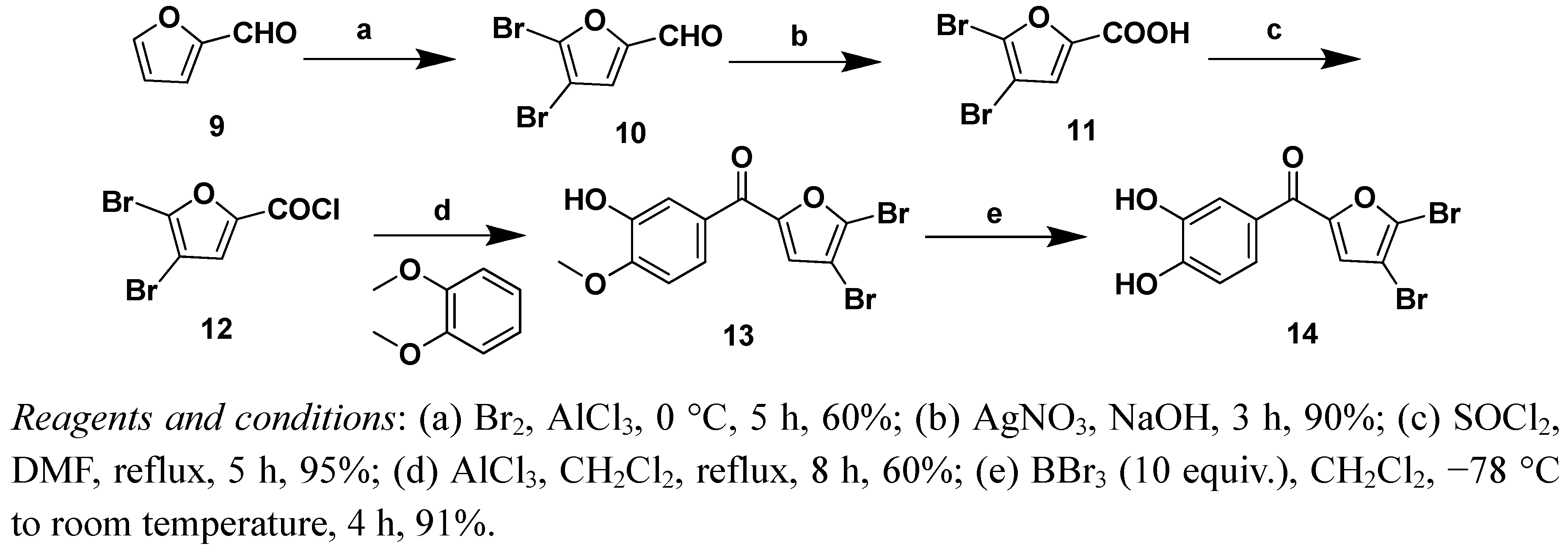
Freshly distilled furfural (10.0 g) was added dropwise with mechanical stirring at 0 °C to aluminum chloride (32.0 g) over a two hour period. Bromine (37.0 g) was then added dropwise at 0 °C over a two hours period, after which stirring was discontinued and the reaction mixture allowed to stand overnight. The reaction was quenched by carefully pouring the mixture into ice (800 mL) and then extracting the aqueous layer three times with ether. The combined organics were washed twice with saturated sodium bicarbonate, once with brine, and dried with anhydrous MgSO4. The solvent was removed under reduced pressure to yield a red oil. Purification using column chromatography on silica (hexanes/ethyl acetate V/V = 1:1 as eluent) yielded compound 10 (13.2 g, Rf = 0.45, 20% ethyl acetate in hexanes) as an orange oil. Next compound 10 (13.2 g), H2O (40.0 mL) and AgNO3 (8.0 g) were added into a flask and the mixture was stirred at room temperature for 4 h. Then the reaction was completed, 6 M hydrochloric acid adjusted to PH to 2~3, and extracting the aqueous layer three times with ethyl acetate. The combined organics were dried with Na2SO4. The solvent was removed under reduced pressure, the solid separated was recrystallized from boiling water to give compound 11.
3.2.3. General procedure for synthesis of compounds 3a–3d, 7a–7c and 13
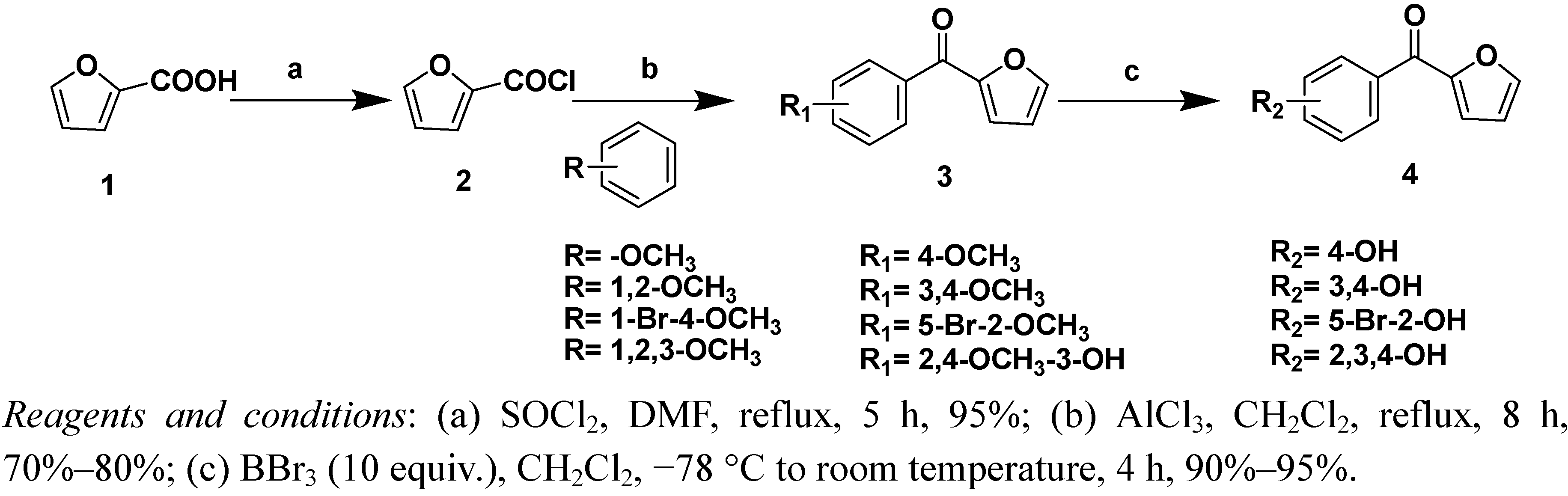
A solution of 2-furancarboxylic acid (1, 1.0 g) and a catalytic amount of DMF in thionyl chloride (5 mL) was stirred at 80–90 °C for 5 h. After concentration under reduced pressure, furoyl chloride 2 was dissolved in dried CH2Cl2 and reacted with 1,2-dimethoxybenzene (1 mL) catalyzed by AlCl3. The reaction mixture was heated to 50–60 °C for 12 h. After the reaction was completed, the reaction was quenched by carefully pouring the mixture into iced water (100 mL), extracting the aqueous layer three times with CH2Cl2, and drying with anhydrous Na2SO4. The solvent was removed under reduced pressure to yield a yellow oil. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc 80:20, v/v) to give compound 3a as a light yellow solid (1.66 g, 80% yield).
(3,4-Dimethoxyphenyl)(furan-2-yl)methanone (3a). Mol. formula (MW): C13H12O4 (232 g/mol); mp: 98–100 °C; 1H-NMR (CDCl3) δ 7.72 (d, J = 8.4 Hz, 1H, Ph-6-H), 7.67(s, 1H, Ph-2-H), 7.55 (d, J = 1.6 Hz, 1H, 5’-H), 7.22 (d, J = 3.6 Hz, 1H, 3’-H), 6.92 (d, J = 8.4 Hz, 1H, Ph-5-H), 6.57 (d, J = 2.0 Hz, 1H, 4’-H), 3.94 (s, 6H, CH3); 13C-NMR (CDCl3) δ 56.2, 56.2, 110.1, 111.8, 112.0, 119.6, 124.1, 129.9, 146.6, 148.6, 152.6, 153.1, 181.0; ESI-MS (%): m/z = 233 (100) [M+H]+, 255 (100) [M+Na]; HRMS (ESI): Calcd. for [M+H]+: 233.0808; Found: 233.0811.
Furan-2-yl(3-hydroxy-2,4-dimethoxyphenyl)methanone (3b). Mol. formula (MW): C13H12O5 (248 g/mol); light yellow solid; Yield: 75%; mp: 168–170 °C; 1H-NMR (DMSO-d6) δ 8.13 (d, J = 9.2 Hz, 1H, Ph-6-H), 7.73 (d, J =1.6 Hz, 1H, 5’-H), 7.38 (d, J = 3.6 Hz, 1H, 3’-H), 6.64 (dd, J = 3.6 Hz, J = 1.6 Hz, 1H, 4’-H), 6.58 (d, J =9.2 Hz, 1H, Ph-5-H), 3.97, (s, 3H, OCH3), 3.93 (s, 3H, OCH3); 13C-NMR (DMSO-d6) δ 56.1, 60.7, 103.1, 103.2, 112.4, 120.3, 128.2, 136.6, 146.8, 152.2, 158.3, 158.5, 183.9; ESI-MS (%): m/z = 249 (100) [M+H]+; HRMS (ESI): Calcd. for [M+H]+: 249.0758; Found: 249.0759.
(5-Bromo-2-methoxyphenyl)(furan-2-yl)methanone (3c). Mol. formula (MW): C12H9BrO3 (281 g/mol); light yellow solid; Yield: 70%; mp: 66–70 °C; 1H-NMR (CDCl3) δ 7.68 (s, 1H, 5’-H), 7.59 (dd, J = 8.8 Hz, J = 2.8 Hz, 1H, Ph-4-H), 7.54 (d, J = 2.8 Hz, 1H, Ph-6-H), 7.09 (d, J = 4.0 Hz, 1H, 3’-H), 6.91 (d, J = 8.8 Hz, 1H, Ph-3-H), 6.58 (dd, J = 4.0 Hz, J = 2.0 Hz, 1H, 4’-H), 3.98 (s, 3H, OCH3); 13C-NMR (CDCl3) δ 56.1, 112.4, 112.5, 113.4, 120.8, 129.6 , 131.9, 134.7, 147.6, 152.5, 156.5, 181.2; ESI-MS (%): m/z = 281(100) 283 (98.7) [M+H]+ 303 (100) 305 (98.7%) [M+Na].
Furan-2-yl(4-methoxyphenyl)methanone (3d). Mol. formula (MW): C12H10O3 (202 g/mol); light yellow solid;Yield: 80%; mp: 58–60 °C; 1H-NMR (CDCl3) δ 8.06 (s, 1H, Ph-2-H), 8.04 (s, 1H, Ph-6-H), 7.70 (s, 1H, 5’-H), 7.25 (d, J = 3.6 Hz, 1H, 3’-H), 7.01 (s, 1H, Ph-3-H), 6.99 (s, 1H, Ph-5-H), 6.61 (d, J = 3.6 Hz, 1H, 4’-H), 3.90 (s, 3H, OCH3); 13C-NMR (CDCl3) δ 55.5, 112.1, 113.7, 114.1, 119.7, 129.8, 131.7, 132.2, 146.6, 152.7, 163.3, 181.2; ESI-MS (%): m/z = 203 (100) [M+H]+ 225 (100) [M+Na].
(5-Bromofuran-2-yl)(2,3-dihydroxy-4-methoxyphenyl)methanone (7a). Mol. formula (MW): C12H9BrO5 (313 g/mol); light yellow solid; Yield: 70%; mp: 128–130 °C; 1H-NMR (DMSO-d6) δ 11.01 (br s, 1H, OH), 8.83 (br s, 1H, OH), 7.43 (dd, J = 8.8 Hz, J =2.4 Hz, 1H, Ph-6-H), 7.38 (d, J = 3.6 Hz, 1H, 3’-H), 6.93 (d, J = 3.6 Hz, 1H, 4’-H), 6.71 (d, J = 8.8 Hz, 1H, Ph-5-H), 3.88 (s, 3H, OCH3); 13C-NMR (DMSO-d6) δ 56.4, 104.2, 115.4, 122.4, 122.4, 123.3, 129.2, 134.5, 150.1, 153.3, 153.6, 182.0; ESI-MS (%): m/z = 313 (100) 315 (99.1) [M+H]+ 335 (100) 337 (99.1%) [M+Na]; HRMS (ESI): Calcd. for [M + H]+:312.9706; Found: 312.9708.
(5-Bromo-2-methoxyphenyl)(5-bromofuran-2-yl)methanone (7b). Mol. formula (MW): C12H8Br2O3 (360 g/mol); light yellow solid; Yield: 76%; mp: 98–100 °C; 1H-NMR (CDCl3) δ 7.59 (dd, 1H, J = 8.8 Hz, J = 2.4 Hz, Ph-4-H ), 7.53 (d, J = 2.4 Hz, 1H, Ph-6-H), 7.01 (d, J = 3.6 Hz, 1H, 3’-H ), 6.91 (d, J = 8.8 Hz, 1H, Ph-3-H ), 6.53 (d, J =3.6 Hz, 1H, 4’-H ), 3.81 (s, 3H, OCH3 ); 13C-NMR (CDCl3) δ 56.1, 112.6, 113.6, 114.5, 122.5, 128.9, 129.9, 132.0, 135.1, 154.1, 156.6, 179.8; ESI-MS (%): m/z = 359 (51) 361(100) 363 (50) [M+H]+ 381 (51) 383 (100) 385 (50) [M+Na].
(5-Bromofuran-2-yl)(3-hydroxy-4-methoxyphenyl)methanone(7c). Mol. formula (MW): C12H9BrO4 (297 g/mol); light yellow solid; Yield: 72%; mp: 128–130 °C; 1H-NMR (CDCl3) δ 7.67 (s, 1H, Ph-2-H), 7.62 (d, J = 8.4 Hz, 1H, Ph-6-H), 7.32 (d, J = 3.6 Hz, 1H, 3’-H), 7.12 (d, J = 8.4 Hz, 1H, Ph-5-H), 6.50 (d, J = 3.6 Hz, 1H, 4’-H) 3.90 (s, 3H, OCH3); 13C-NMR (CDCl3) δ 56.2, 111.1, 113.8, 118.0, 119.6, 122.1, 123.4, 127.9, 148.6, 152.6, 155.1, 181.0; ESI-MS (%): m/z = 295 (100) 297 (98.9) [M−H]−; HRMS (ESI): Calcd. for [M+H]+:296.9757; Found: 296.9768.
(4,5-Dibromofuran-2-yl)(3-hydroxy-4-methoxyphenyl)methanone (13). Mol. formula (MW): C12H8Br2O4 (376 g/mol); light yellow solid; Yield: 60%; mp: 107–109 °C; 1H-NMR (CDCl3) δ 7.66 (d, J = 8.4 Hz, 1H, Ph-5-H), 7.54 (s, 1H, 3’-H), 7.23 (s, 1H, Ph-2-H), 7.02 (d, J = 8.4 Hz, 1H, Ph-6-H), 3.98 (s, 3H, OCH3); 13C-NMR (CDCl3) δ 56.2, 104.3, 111.5, 114.1, 123.2, 124.9, 128.4, 129.0, 146.8, 150.9, 153.6, 180.0; ESI-MS (%): m/z = 373 (51) 375 (100) 377 (50) [M−H]−; HRMS (ESI): Calcd. for [M+H]+: 374.8865; Found: 374.8867.
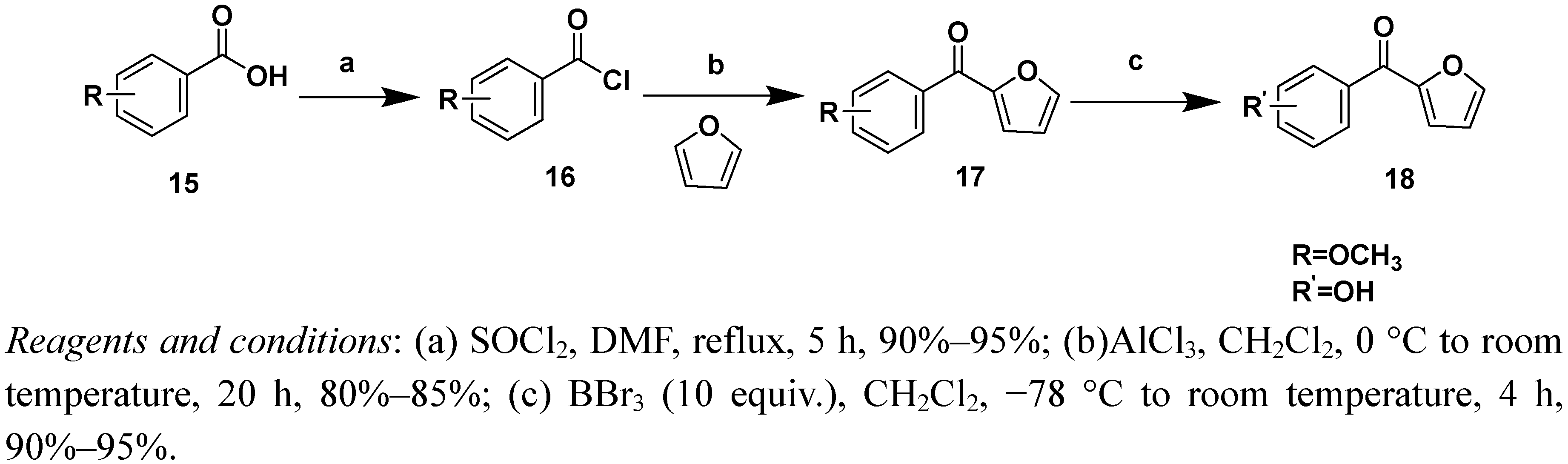
3.2.4.General procedure for the synthesis of compounds 17a and 17b
A solution of 2,3-dimethoxybenzoic acid (2.0 g) and a catalytic amount of DMF in thionyl chloride (5 mL) was stirred at 80–90 °C for 3 h. After concentration under reduced pressure, the acid chloride 16 was dissolved in dried CH2Cl2 and reacted with furan (3 mL) catalyzed by AlCl3. The reaction mixture was stirred at 0 °C for 2 h and then warmed to room temperature and stirred for additional 12 h. After the reaction was completed, the reaction was quenched by carefully pouring the mixture into iced water (100 mL) and the resultant compound was collected by filtration. Then, the filtrate was extracted with CH2Cl2 (3 × 20 mL), and dried with anhydrous Na2SO4. The solvent was removed under reduced pressure to yield a brown oil that was purified by silica gel column chromatography (petroleum ether/EtOAc 80:20, v/v) to give (2,3-dimethoxyphenyl)(furan-2-yl)methanone (17a) as light yellow oil (1.78 g, 70% yield). Molecular formula (MW): C13H12O4 (232 g/mol); 1H-NMR (CDCl3) δ 7.67 (d, 1H, J = 1.0 Hz, 5’-H), 7.15 (dd, 1H, J = 8.0 Hz, J = 7.6 Hz, Ph-5-H), 7.08 (dd, 1H, J = 8.4 Hz, J =1.6 Hz, Ph-4-H), 7.06 (d, J = 3.6 Hz, 1H, 3’-H), 7.02 (dd, 1H, J = 7.6 Hz, J = 1.6 Hz, Ph-6-H), 6.55 (dd, J = 3.6 Hz, J = 2.0 Hz, 1H, 4’-H), 3.92 (s, 3H, OCH3), 3.83 (s, 3H, OCH3); 13C-NMR (CDCl3) δ 56.0, 62.1, 112.3, 114.7, 120.5, 121.1, 123.9, 133.4, 147.1, 147.5, 152.7, 152.8, 182.8; ESI-MS (%): m/z = 233 (100) [M+H]+ 255 (100) ([M+Na].
(2,6-Dimethoxyphenyl)(furan-2-yl)methanone (17b). Mol. formula (MW): C13H12O4 (232 g/mol); light yellow solid; Yield: 85%; mp: 64–66 °C; 1H-NMR (CDCl3) δ 7.87 (d, 1H, J = 1.0 Hz, 5’-H) 7.42 (dd, 1H, J = 8.0 Hz, J = 8.0 Hz, Ph-4-H), 7.08 (d, 1H, J = 8.0 Hz, J = 1.6 Hz, Ph-3-H), 7.06 (d, J = 3.6 Hz, 1H, 3’-H), 7.02 (dd, 1H, J = 8.0 Hz, J = 1.2 Hz, Ph-5-H), 6.55 (dd, J = 3.6 Hz, J = 1.0 Hz, 1H, 4’-H), 3.91 (s, 3H, OCH3), 3.83 (s, 3H, OCH3); 13C-NMR (CDCl3) δ 56.0, 62.5, 112.3, 114.7, 120.5, 121.1, 123.9, 133.3, 147.1, 147.5, 152.7, 152.8, 182.8; ESI-MS (%): m/z = 233 (100) [M+H]+ 255 (100) [M+Na].
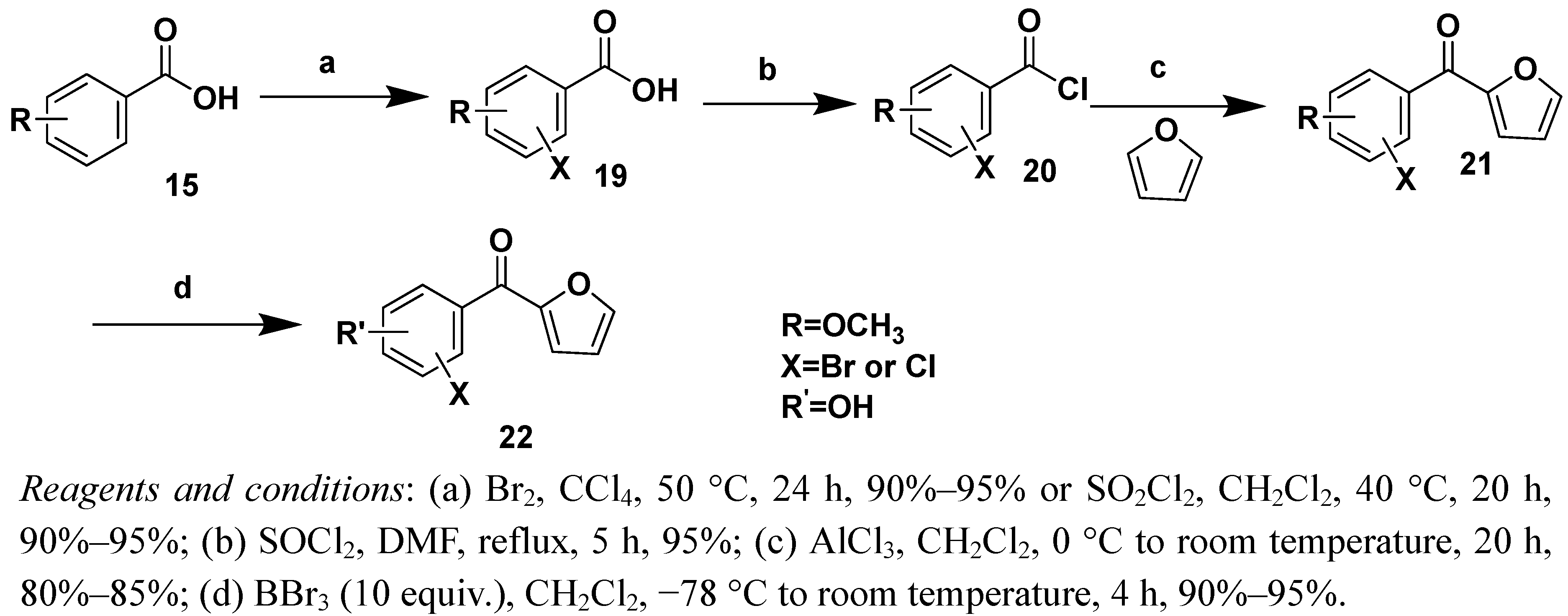
3.2.5. General procedure for the synthesis of compounds 21a–21d
Br2 (2 mL) was slowly added to a solution of 4-methoxybenzoic acid (5.0 g) in CCl4 (20 mL). The reaction mixture was stirred at 40–45 °C for 24 h. After concentration under reduced pressure, the residue was re-crystallized from boiling water to give 3-bromo-4-methoxybenzoic acid (19) as white needles. To chlorinate the methoxybenzoic acid, we used SO2Cl2. A solution of 19 (2.0 g) and a catalytic amount of DMF in thionyl chloride (5 mL) was stirred at 80–90 °C for 3 h. After concentration under reduced pressure, the acid chloride 20 was dissolved in dried CH2Cl2 and reacted with furan (3 mL) catalyzed by AlCl3. The reaction mixture was stirred at 0 °C for 2 h and then warmed to room temperature and stirred for an additional 12 h. After the reaction was completed, it was quenched by carefully pouring the mixture into iced water (100 mL) and the resultant compound was collected by filtration. The filtrate was then extracted with CH2Cl2 (3 × 20 mL) and dried with anhydrous Na2SO4. The solvent was removed under reduced pressure to yield a brown oil that was purified by silica gel column chromatography (petroleum ether/EtOAc 80:20, v/v) to give compound 21a as a light yellow solid (1.85 g, 76% yield).
(3-Bromo-4-methoxyphenyl)(furan-2-yl)methanone(21a). Mol. formula (MW): C12H9BrO3 (281 g/mol); mp: 112–114 °C; 1H-NMR (CDCl3) δ 8.30 (d, 1H, J = 2.0 Hz, 5’-H), 8.06 (dd, 1H, J = 8.0 Hz, J = 2.0 Hz, Ph-6-H), 7.93 (s, 1H, Ph-2-H), 7.30 (d, 1H, J = 3.6 Hz, 3’-H), 7.01 (d, 1H, J = 8.0 Hz, Ph-5-H), 6.63 (dd, J =3.6 Hz, J = 2.0 Hz, 1H, 4’-H), 4.00 (s, 3H, OCH3); 13C-NMR (CDCl3) δ 56.5, 111.1, 112.3, 113.7, 120.1, 130.7, 131.7, 134.9, 146.9, 152.3, 159.4, 179.7; ESI-MS (%): m/z = 281(100) 283 (98.7) [M+H]+ 303 (100) 305 (98.7) [M+Na].
(2,6-Dibromo-3,4,5-trimethoxyphenyl)(furan-2-yl)methanone (21b). Mol. formula (MW): C14H12Br2O5 (420 g/mol); light yellow solid; Yield: 85%; mp: 96–98 °C; 1H-NMR (CDCl3) δ 7.71 (d, J = 1.0 Hz, 1H, 5’-H), 7.13 (d, J = 3.6 Hz, 1H, 3’-H), 6.61 (dd, J = 3.6 Hz, J = 1.6 Hz, 1H, 4’-H), 4.01 (s, 9H, OCH3); 13C-NMR (CDCl3) δ 61.2, 61.4, 61.4, 110.1, 112.9, 121.2, 136.0, 148.3, 148.6, 151.0, 151.1, 180.5; ESI-MS (%): m/z = 419 (51) 421 (100) 423 (50) [M+H]+ 441 (51) 443 (100) 445 (50) [M+Na] 457 (51) 459 (100) 461 (50) [M+K].
(2-Chloro-3,4,5-trimethoxyphenyl)(furan-2-yl)methanone(21c). Mol. formula (MW): C14H13ClO5 (297 g/mol); light yellow solid; Yield: 82%; mp: 80–82 °C; 1H-NMR (CDCl3) δ 7.74 (s, 1H, 5’-H), 7.12 (d, J = 3.6 Hz, 1H, 3’-H), 6.80 (s, 1H, Ph-6-H), 6.60 (dd, J = 3.6 Hz, J = 2.0 Hz, 1H, 4’-H), 3.97 (s, 9H, OCH3); 13C-NMR (CDCl3) δ 56.3, 61.3, 61.3, 107.0, 107.8, 112.7, 121.4, 132.9, 145.2, 147.9, 150.3, 152.1, 153.0, 181.5; ESI-MS (%): m/z = 297 (100) 299 (34) [M+H]+ 319 (100) 321 (34) [M+Na] 335 (100) 337 (34) [M+K]; HRMS (ESI): Calcd. for [M+Na]: 319.0344; Found: 319.0343.
(4,5-Dibromo-2-hydroxy-3-methoxyphenyl)(furan-2-yl)methanone (21d). Mol. formula (MW): C12H8Br2O4 (376 g/mol); light yellow solid; Yield: 80%; mp: 102–104 °C; 1H-NMR (DMSO-d6) δ 9.86 (br s, 1H, OH), 8.07 (d, 1H, J = 1.2 Hz, 5’-H), 7.44 (s, 1H, Ph-6-H), 7.20 (d, J = 3.6 Hz, 1H, 3’-H), 6.74 (dd, J = 3.6 Hz, J = 2.0 Hz, 1H, 4’-H), 3.89 (s, 3H, OCH3); 13C-NMR (DMSO-d6) δ 57.0, 111.4, 113.6, 113.6, 117.3, 121.6, 129.6, 144.8, 148.6, 149.5, 151.6, 180.6; ESI-MS (%): m/z = 375 (51) 377 (100) 379 (50) [M+H]+ 397 (51) 399 (100) 401 (50) [M+Na]; HRMS (ESI): Calcd. for [M+Na]: 396.8682; Found: 396.8683.
3.2.6. General procedure for the synthesis of compounds 4a–4d, 8a–8c, 14, 18a–18b and 22a–22d
10% (equiv.) BBr3 (2 mL) was added to a solution of compound 3a (0.5 g) in CH2Cl2 (20 mL). The reaction mixture was stirred at −78 °C for 30 min and then warmed to room temperature and stirred for additional 3.5 h. After the reaction was completed, it was quenched by carefully pouring the mixture into iced water (100 mL), extraction of the aqueous layer three times with EtOAc, washing with 5% NaHSO3 (40 mL) and water (100 mL), and drying with anhydrous Na2SO4. The solvent was removed under reduced pressure to yield a light red solid compound 4a (0.403 g, 91.7% yield).
Synthesis of (3,4-Dihydroxyphenyl)(furan-2-yl)methanone (4a).Mol. formula (MW): C11H8O4 (204 g/mol); mp: 132–134 °C; 1H-NMR (CDCl3) δ 7.58 (d, J = 8.0 Hz, 1H, Ph-5-H), 7.53 (s, 1H, 5’-H), 7.40 (s, 1H, Ph-2-H), 7.10 (d, J = 3.2 Hz, 1H, 3’-H), 6.81 (d, J = 8.0 Hz, 1H, Ph-6-H), 6.46 (s, 1H, 4’-H); 13C-NMR (CDCl3) δ 111.8, 114.9, 116.5, 119.7, 124.0, 129.8, 146.6, 148.9, 152.3, 153.0, 180.1; ESI-MS (%): m/z = 205 (100) [M+H]+; HRMS (ESI): Calcd. for [M+Na]: 227.0315; Found: 227.0310.
Furan-2-yl(2,3,4-trihydroxyphenyl)methanone (4b). Mol. formula (MW): C11H8O5 (220 g/mol); light red solid; Yield: 95%; mp: 168–170 °C; 1H-NMR (DMSO-d6) δ 7.73 (d, J = 1.6 Hz, 1H, 5’-H), 7.64 (d, J = 9.2 Hz, 1H, Ph-6-H), 7.44 (d, J = 3.6 Hz, 1H, 3’-H), 6.78 (dd, J = 3.6 Hz, J = 1.6 Hz, 1H, 4’-H), 6.53 (d, J = 9.2 Hz, 1H, Ph-5-H); 13C-NMR (DMSO-d6) δ 108.6, 113.1, 113.2, 120.9, 123.7, 148.5, 148.7, 151.8, 152.9, 153.3, 183.5; ESI-MS (%): m/z = 219 (100) [M−H]−; HRMS (ESI): Calcd. for [M+Na]: 243.0264; Found: 243.0261.
(5-Bromo-2-hydroxyphenyl)(furan-2-yl)methanone (4c). Mol. formula (MW): C11H7BrO3 (267 g/mol); light red solid; Yield: 94%; mp: 88–90 °C; 1H-NMR (DMSO-d6) δ 10.50 (br s, 1H, OH), 8.08 (d, J = 1.0 Hz, 1H, 5’-H), 7.58 (d, J = 2.4 Hz, 1H, Ph-6-H), 7.56 (d, J = 8.4 Hz, 1H, Ph-4-H), 7.26 (d, J = 3.6 Hz, 1H, 3’-H), 6.95 (d, J = 8.4 Hz, 1H, Ph-3-H), 6.76 (dd, J = 3.6 Hz, J = 2.0 Hz, 1H, 4’-H); 13C-NMR (CDCl3) δ 110.3, 113.4, 119.4, 121.7, 127.5, 131.8, 135.5, 149.1, 152.2, 156.0, 181.7; ESI-MS (%): m/z = 265 (100) 267 (98.6) [M−H]−; HRMS (ESI): Calcd. for [M+Na]: 288.9471; Found: 288.9473.
Furan-2-yl(4-hydroxyphenyl)methanone (4d). Mol. formula (MW): C11H8O3 (188 g/mol); light red solid; Yield: 95%; mp: 168–170 °C; 1H-NMR (DMSO-d6) δ 10.44 (br s, 1H, OH), 8.07 (d, J = 1 Hz, 1H, 5’-H), 7.89 (s, 1H, Ph-2-H), 7.87 (s, 1H, Ph-6-H), 7.33 (d, J = 3.6 Hz, 1H, 3’-H), 6.93 (s, 1H, Ph-5-H), 6.91 (s, 1H, Ph-3-H), 6.76 (dd, J = 3.6 Hz, J = 2.4 Hz, 1H, 4’-H); 13C-NMR (DMSO-d6) δ 112.9, 115.8, 115.8, 120.3, 128.3, 132.1, 132.1, 148.1, 152.2, 162.4, 180.4; ESI-MS (%): m/z = 187 (100) [M−H]−; HRMS (ESI): Calcd. for [M+Na]: 211.0366; Found: 211.0368.
(5-Bromofuran-2-yl)(2,3,4-trihydroxyphenyl)methanone (8a). Mol. formula (MW): C11H7BrO5 (299 g/mol); light red solid; Yield: 95%; mp: 132–134 °C; 1H-NMR (DMSO-d6) δ 7.48 (d, J = 8.8 Hz, 1H, Ph-6-H), 7.43 (d, J = 3.6 Hz, 1H, 3’-H), 6.94 (d, J = 3.6 Hz, 1H, 4’-H), 6.50 (d, J = 8.8 Hz, 1H, Ph-5-H); 13C-NMR (DMSO-d6) δ 182.0, 158.7, 153.3, 153.1, 147.3, 127.2, 123.3, 120.5, 114.8, 112.6, 108.7; ESI-MS (%): m/z = 297 (100) 299 (99) [M−H]−; HRMS (ESI): Calcd. for [M+Na]: 320.9369; Found: 320.9374.
(5-Bromo-2-hydroxyphenyl)(5-bromofuran-2-yl)methanone (8b). Mol. formula (MW): C11H6Br2O3 (346 g/mol); light red solid; Yield: 95%; mp: 82–84 °C; 1H-NMR (DMSO-d6) δ 10.46 (br s, 1H, OH), 7.57 (d, J = 8.0 Hz, 1H, Ph-4-H), 7.54 (s, J = 8.0 Hz, 1H, Ph-6-H), 7.25 (d, J = 3.6 Hz, 1H, 3’-H), 6.95 (d, J = 8.0 Hz, 1H, Ph-3-H), 6.90 (d, J = 3.6 Hz, 1H, 4’-H); 13C-NMR (DMSO-d6) δ 110.3, 115.6, 119.4, 123.8, 127.1, 130.0, 131.9, 135.6, 153.4, 155.7, 180.3; ESI-MS (%): m/z = 343 (51) 345 (100) 347 (50) [M−H]−; HRMS (ESI): Calcd. for [M+H]+: 344.8736; Found: 344.8742.
(5-Bromofuran-2-yl)(3,4-dihydroxyphenyl)methanone (8c). Mol. formula (MW): C11H7BrO4 (283 g/mol); light red solid; Yield: 95%; mp: 138–140 °C; 1H-NMR (CDCl3) δ 7.54 (d, J = 2.0 Hz, 1H, Ph-2-H), 7.48 (dd, J = 8.4 Hz, J = 1.6 Hz, 1H, Ph-6-H), 7.12 (d, J = 3.6 Hz, 1H, 3’-H), 6.94 (d, J = 8.4 Hz, 1H, Ph-5-H), 6.49 (d, J = 3.6 Hz, 1H, 4’-H); 13C-NMR (CDCl3) δ 118.8, 120.0, 121.3, 126.2, 127.6, 132.7, 133.0, 150.0, 155.4, 158.9, 184.4; ESI-MS (%): m/z = 283 (100) 285 (98.8) [M+H]+; HRMS (ESI): Calcd. for [M+Na]: 304.9420; Found: 304.9420.
(4,5-Dibromofuran-2-yl)(3,4-dihydroxyphenyl)methanone (14). Mol. formula (MW): C11H6Br2O4 (362 g/mol); light red solid; Yield: 91%; mp: 184–186 °C; 1H-NMR (CDCl3) δ 7.53 (s, 1H, Ph-2-H), 7.48 (d, J = 8.0 Hz, 1H, Ph-6-H), 7.18 (s, 1H, 3’-H), 6.95 (d, J = 8.0 Hz, 1H, Ph-5-H); 13C-NMR (CDCl3) δ 104.0, 115.2, 116.5, 121.3, 122.8, 127.7, 128.6, 145.1, 150.9, 158.4, 179.0; ESI-MS (%): m/z = 359 (51) 361 (100) 363 (50) [M−H]−; HRMS (ESI): Calcd. for [M+Na]: 382.8531; Found: 382.8533.
(2,3-Dihydroxyphenyl)(furan-2-yl)methanone (18a). Mol. formula (MW): C11H8O4 (204 g/mol); light red solid; Yield: 95%; mp: 64–66 °C; 1H-NMR (DMSO-d6) δ 8.06 (d, 1H, J = 1.0 Hz, 5’-H), 7.30 (d, J = 3.6 Hz, 1H, 3’-H), 7.10 (dd, 1H, J = 8.0 Hz, J = 1.6 Hz, Ph-6-H), 7.06 (dd, 1H, J = 8.4 Hz, J = 2.0 Hz, Ph-4-H), 6.91 (dd, 1H, J = 8.4 Hz, J = 8.0 Hz, Ph-5-H), 6.72 (dd, J = 3.6 Hz, J = 2.0 Hz, 1H, 4’-H); 13C-NMR (DMSO-d6) δ 109.4, 113.1, 113.6, 120.1, 121.5, 130.0, 144.3, 146.8, 149.4, 151.8, 180.9; ESI-MS (%): m/z = 203 (100) [M−H]−; HRMS (ESI): Calcd. for [M+Na]: 227.0315; Found: 227.0313.
(2,6-Dihydroxyphenyl)(furan-2-yl)methanone (18b). Mol. formula (MW): C11H8O4 (204 g/mol); light red solid; Yield: 95%; mp: 66–68 °C; 1H-NMR (DMSO-d6) δ 8.08 (d, 1H, J = 1.0 Hz, 5’-H), 7.30 (d, J = 3.6 Hz, 1H, 3’-H), 7.17 (dd, 1H, J = 8.0 Hz, J = 1.6 Hz, Ph-3-H), 7.04 (dd, 1H, J = 8.4 Hz, J = 2.0 Hz, Ph-5-H), 6.81 (dd, 1H, J = 8.4 Hz, J = 8.0 Hz, Ph-4-H), 6.76 (dd, J = 3.6 Hz, J = 2.0 Hz, 1H, 4’-H); 13C-NMR (DMSO-d6) δ 113.2, 119.4, 119.6, 120.5, 121.6, 123.9, 146.5, 147.4, 148.9, 152.2, 184.0; ESI-MS (%): m/z = 203 (100) [M−H]−; HRMS (ESI): Calcd. for [M+Na]: 227.0315; Found: 227.0310.
(3-Bromo-4-hydroxyphenyl)(furan-2-yl)methanone (22a). Mol. formula (MW): C11H7BrO3 (267 g/mol); light red solid; Yield: 90%; mp: 100–102 °C; 1H-NMR (DMSO-d6) δ 11.32 (br s, 1H, OH), 8.10 (d, 1H, J = 2.8 Hz, Ph-2-H), 8.09 (d, 1H, J = 2.0 Hz, 5’-H), 7.90 (dd, 1H, J = 8.8 Hz, J = 2.4 Hz, Ph-6-H), 7.40 (d, J = 3.6 Hz, 1H, 3’-H), 7.12 (d, 1H, J = 8.8 Hz, Ph-5-H), 6.79 (dd, J = 3.6 Hz, J = 2.0 Hz, 1H, 4’-H); 13C-NMR (DMSO-d6) δ 110.0, 113.1, 116.5, 120.9, 129.6, 131.1, 134.7, 148.6, 151.9, 158.9, 179.2; ESI-MS (%): m/z = 265 (100) 267 (98.6) [M−H]−; HRMS(ESI): Calcd. for [M+Na]: 288.9472; Found: 288.9470.
(2,6-Dibromo-3,4,5-trihydroxyphenyl)(furan-2-yl)methanone (22b). Mol. formula (MW): C11H6Br2O5 (378 g/mol); light red solid; Yield: 93%; mp: 148–150 °C; 1H-NMR (DMSO-d6) δ 9.82 (br s, 2H, OH), 9.58 (br s, 1H, OH), 8.07 (d, J = 1.6 Hz, 1H, 5’-H), 7.13 (d, J = 3.6 Hz, 1H, 3’-H), 6.74 (dd, J = 3.6 Hz, J = 1.6 Hz, 1H, 4’-H); 13C-NMR (DMSO-d6) δ 99.1, 113.6, 113.7, 121.9, 130.9, 136.9, 143.8, 143.8, 149.7, 151.3, 181.4; ESI-MS (%): m/z = 375 (51) 377 (100) 379 (50) [M−H]−; HRMS (ESI): Calcd. for [M+H]+: 378.8635; Found: 378.8637.
(2-Chloro-3,4,5-trihydroxyphenyl)(furan-2-yl)methanone (22c). Mol. formula (MW): C11H7ClO5 (255 g/mol); light red solid; Yield: 94%; mp: 150–152 °C; 1H-NMR (DMSO-d6) δ 9.65 (br s, 3H, OH), 9.42 (br s, 3H, OH), 8.07 (s, 1H, 5’-H), 7.14 (d, J = 3.6 Hz, 1H, 3’-H), 6.74 (dd, J = 3.6 Hz, J = 1.6 Hz, 1H, 4’-H), 6.25 (s, 1H, Ph-6-H); 13C-NMR (DMSO-d6) δ 108.1, 113.3, 121.8, 127.8, 137.4, 143.5, 144.5, 146.1, 149.1, 152.2, 181.4; ESI-MS (%): m/z = 253 (100) 255 (33.7) [M−H]−; HRMS (ESI): Calcd. for [M+Na]: 276.9874; Found: 276.9876.
(4,5-Dibromo-2,3-dihydroxyphenyl)(furan-2-yl)methanone (22d). Mol. formula (MW): C11H6Br2O4 (362 g/mol); light red solid; Yield: 95%; mp: 138–140 °C; 1H-NMR (DMSO-d6) δ 9.66 (br s, 1H, OH), 9.37 (br s, 1H, OH), 8.07 (s, 1H, 5’-H), 7.40 (s, 1H, Ph-6-H), 7.14 (d, J = 3.6 Hz, 1H, 3’-H), 6.74 (dd, J = 3.6 Hz, J = 2.0 Hz, 1H, 4’-H); 13C-NMR (DMSO-d6) δ 108.1, 109.3, 113.3, 120.0, 121.8, 127.8, 143.5, 144.5, 149.1, 152.2, 181.4; ESI-MS (%): m/z = 359 (51) 361 (100) 363 (50) [M−H]−; HRMS (ESI): Calcd. for [M+Na]: 382.8526; Found: 382.8528.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}