On the Catalytic Effect of Water in the Intramolecular Diels–Alder Reaction of Quinone Systems: A Theoretical Study



Abstract
:1. Introduction
2. Computational Details






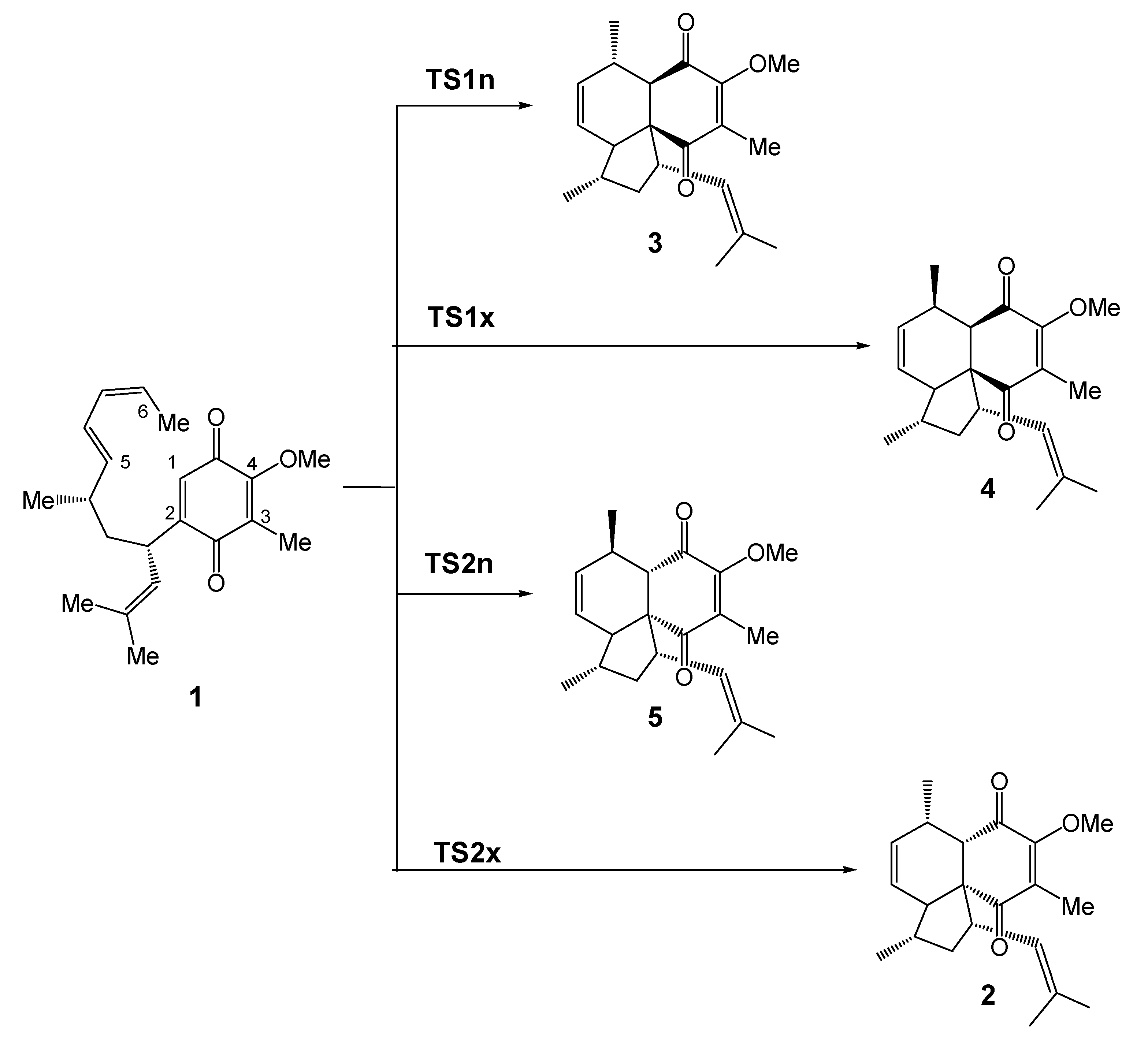
3. Results and Discussion
4. Concluding Remarks
Supplementary Materials
Acknowledgments
References
- Carruthers, W. Cycloaddition Reactions in Organic Synthesis; Pergamon Press: Oxford, UK, 1990. [Google Scholar]
- Takao, K.; Munakata, R.; Tadano, K. Recent Advances in Natural Product Synthesis by Using Intramolecular Diels−Alder Reactions. Chem. Rev. 2005, 105, 4779–4807. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.D.; González, E.; Huang, S.D. Unusual Terpenes with Novel Carbon Skeletons from the West Indian Sea Whip Pseudopterogorgia elisabethae (Octocorallia). J. Org. Chem. 1998, 63, 7083–7091. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.D.; Ramírez, C.; Rodríguez, I.I.; Barnes, C.L. Novel Terpenoids from the West Indian Sea Whip Pseudopterogorgia elisabethae (Bayer). Elisapterosins A and B: Rearranged Diterpenes Possessing an Unprecedented Cagelike Framework. J. Org. Chem. 2000, 65, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Heckrodt, T.J.; Mulzer, J. Total Synthesis of Elisabethin A: Intramolecular Diels−Alder Reaction under Biomimetic Conditions. J. Am. Chem. Soc. 2003, 125, 4680–4681. [Google Scholar] [CrossRef] [PubMed]
- Cativela, C.; Garcia, J.I.; Mayoral, J.A.; Salvatella, L. Modelling of solvent effects on the Diels–Alder reaction. Chem. Soc. Rev. 1996, 25, 209–218. [Google Scholar] [CrossRef]
- Blokzijl, W.; Blandamer, M.J.; Engberts, J.B.F.N. Diels–Alder reactions in aqueous solutions. Enforced hydrophobic interactions between diene and dienophile. J. Am. Chem. Soc. 1991, 113, 4241–4246. [Google Scholar] [CrossRef]
- Otto, S.; Blokzijl, W.; Engberts, J.B.F.N. Diels–Alder Reactions in Water. Effects of Hydrophobicity and Hydrogen Bonding. J. Org. Chem. 1994, 59, 5372–5376. [Google Scholar] [CrossRef]
- Wijnen, J.W.; Engberts, J.B.F.N. Retro-Diels−Alder Reaction in Aqueous Solution: Toward a Better Understanding of Organic Reactivity in Water. J. Org. Chem. 1997, 62, 2039–2044. [Google Scholar] [CrossRef] [PubMed]
- Blake, J.F.; Jorgensen, W.L. Solvent effects on a Diels−Alder reaction from computer simulations. J. Am. Chem. Soc. 1991, 113, 7430–7432. [Google Scholar] [CrossRef]
- Acevedo, O.; Jorgensen, W.L. Understanding Rate Accelerations for Diels–Alder Reactions in Solution Using Enhanced QM/MM Methodology. J. Chem. Theory Comput. 2007, 3, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Evanseck, J. Density Functional Theory Study of Aqueous-Phase Rate Acceleration and Endo/Exo Selectivity of the Butadiene and Acrolein Diels−Alder Reaction. J. Am. Chem. Soc. 2000, 122, 10418–10427. [Google Scholar] [CrossRef]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Simón, L.; Goodman, J.M. How reliable are DFT transition structures? Comparison of GGA, hybrid-meta-GGA and meta-GGA functionals. Org. Biomol. Chem. 2011, 9, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Pieniazek, S.N.; Clemente, F.R.; Houk, K.N. Sources of Error in DFT Computational of C–C Bond Formation Thermochemidtries: π→σ Transformations and Error Cancellation by DFT Methods. Angew. Chem. Int. Ed. 2008, 47, 7746–7749. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.; Johansson, A.J.; Brink, T. Mechanism Insights into the Stepwise Diels–Alder Reaction of 4,6-Dinitrobenzofuroxan. Org. Lett. 2012, 14, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.; Brink, T. Stepwise Diels–Alder: More than Just an Oddity? A Computational Mechanism Study. J. Org. Chem. 2012, 77, 6563–6573. [Google Scholar] [CrossRef] [PubMed]
- Krenske, E.H.; Perry, E.W.; Jerome, S.V.; Maimone, T.J.; Baran, P.S.; Houk, K.N. Why a Proximity-Induce Diels–Alder Reaction Is So Fast. Org. Lett. 2012, 14, 3016–3019. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.V.; Martin, D.B.C.; Vanderwal, C.D.; Houk, K.N. The intramolecular Diels–Alder reaction of tryptamine-derived Zincke aldehydes is a stepwise process. Chem. Sci. 2012, 3, 1650–1655. [Google Scholar] [CrossRef] [PubMed]
- Soto-Delgado, J.; Domingo, L.R.; Contreras, R. Quantitative characterization of group electrophilicity and nucleophilicity for intramolecular Diels–Alder reactions. Org. Biomol. Chem. 2010, 8, 3678–3683. [Google Scholar] [CrossRef] [PubMed]
- Soto-Delgado, J.; Aizman, A.; Domingo, L.R.; Contreras, R. Invariance of electrophilicity of independent fragments. Application to intramolecular Diels–Alder reactions. Chem. Phys Lett. 2010, 499, 272–277. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- DeLano, W. PyMOL, version 0.99; DeLano Scientific LLC: South San Francisco, CA, USA, 2002; Available online: http://www.pymol.org (accessed on 16 November 2012).
- Parr, R.G.; von Szentpály, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamoro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [PubMed]
- Domingo, L.R.; Pérez, P. The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Duley, S.; Domingo, R.L. Understanding local electrophylicity/ Nucleophilicity activation through a single reactivity difference index. Org. Biomol. Chem. 2012, 10, 2855–2861. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the local electrophilicity of organic molecules. Understanding the regioselectivity on Diels–Alder reactions. J. Phys. Chem. A 2002, 106, 6871–6875. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Duque-Noreña, M.; Chamorro, E. A condensed-to-atom nucleophilicity index. An application to the director effects on the electrophilic aromatic substitutions. J. Mol. Struct. (Theochem) 2009, 895, 86–91. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Contreras, R.; Fuentealba, F.; Galván, M.; Pérez, P. A direct evaluation of regional Fukui functions in molecules. Chem. Phys. Lett. 1999, 304, 405–413. [Google Scholar] [CrossRef]
- Tachibana, A. Chemical potential inequality principle. Theor. Chem. Acc. 1999, 102, 188–195. [Google Scholar] [CrossRef]
- Soto-Delgado, J.; Domingo, L.R.; Araya-Maturana, R.; Contreras, R. Understanding the stereo- and regioselectivities of the polar Diels–Alder reactions between 2-acetyl-1,4-benzoquinone and methyl substituted 1,3-butadienes: A DFT study. J. Phys. Org. Chem. 2009, 22, 578–584. [Google Scholar] [CrossRef]
- Domingo, L.R.; Saez, J.A. Understanding the mechanism of polar Diels–Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Soto-Delgado, J.; Aizman, A.; Contreras, R.; Domingo, L.R. A DFT Study of the Regioselectivity in Intramolecular Diels–Alder Reactions with Formation of a Tricyclodecane Skeleton. Lett. Org. Chem. 2011, 8, 125–131. [Google Scholar] [CrossRef]
- Soto-Delgado, J.; Domingo, L.R.; Contreras, R. Understanding the influence of Lewis acids in the regioselectivity of the Diels–Alder reactions of 2-methoxy-5-methyl-1,4-benzoquinone: A DFT study. J. Mol. Struct. (Theochem) 2009, 902, 103–108. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Manoharan, M.; Peabody, S.; Weinhold, F. Electronic Basis of Improper Hydrogen Bonding: A Subtle Balance of Hyperconjugation and Rehybridization. J. Am. Chem. Soc. 2003, 125, 5973–5987. [Google Scholar] [CrossRef] [PubMed]
- Bent, H.A. An Appraisal of Valence-bond Structures and Hybridization in Compounds of the First-row elements. Chem. Rev. 1961, 61, 275–311. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Domingo, L.R.; Zaragoza, R.J.; Saéz, J.A.; Arnó, M. Understanding the Mechanism of the Intramolecular Stetter Reaction. A DFT Study. Molecules 2012, 17, 1335–1353. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







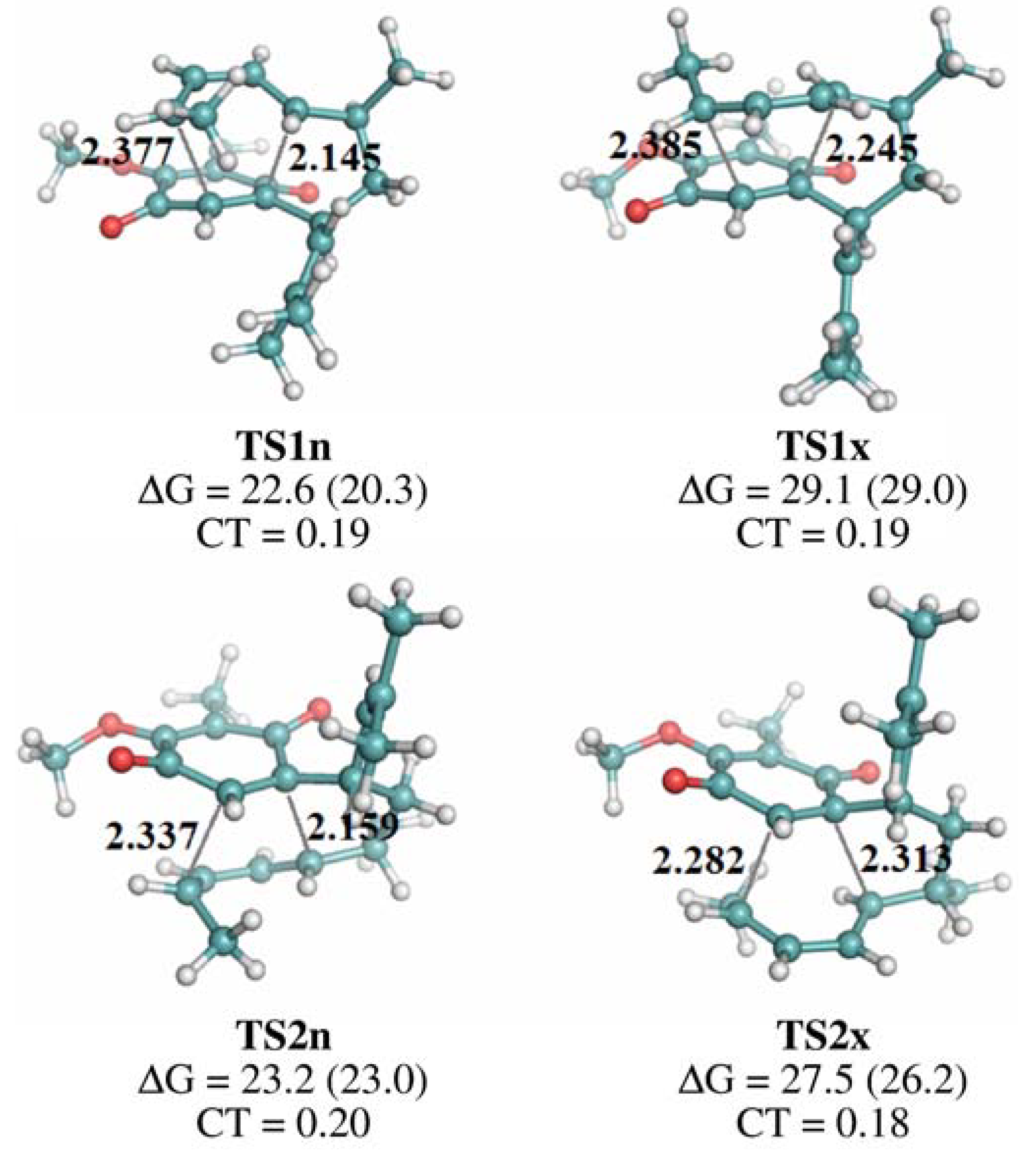{kind=link}
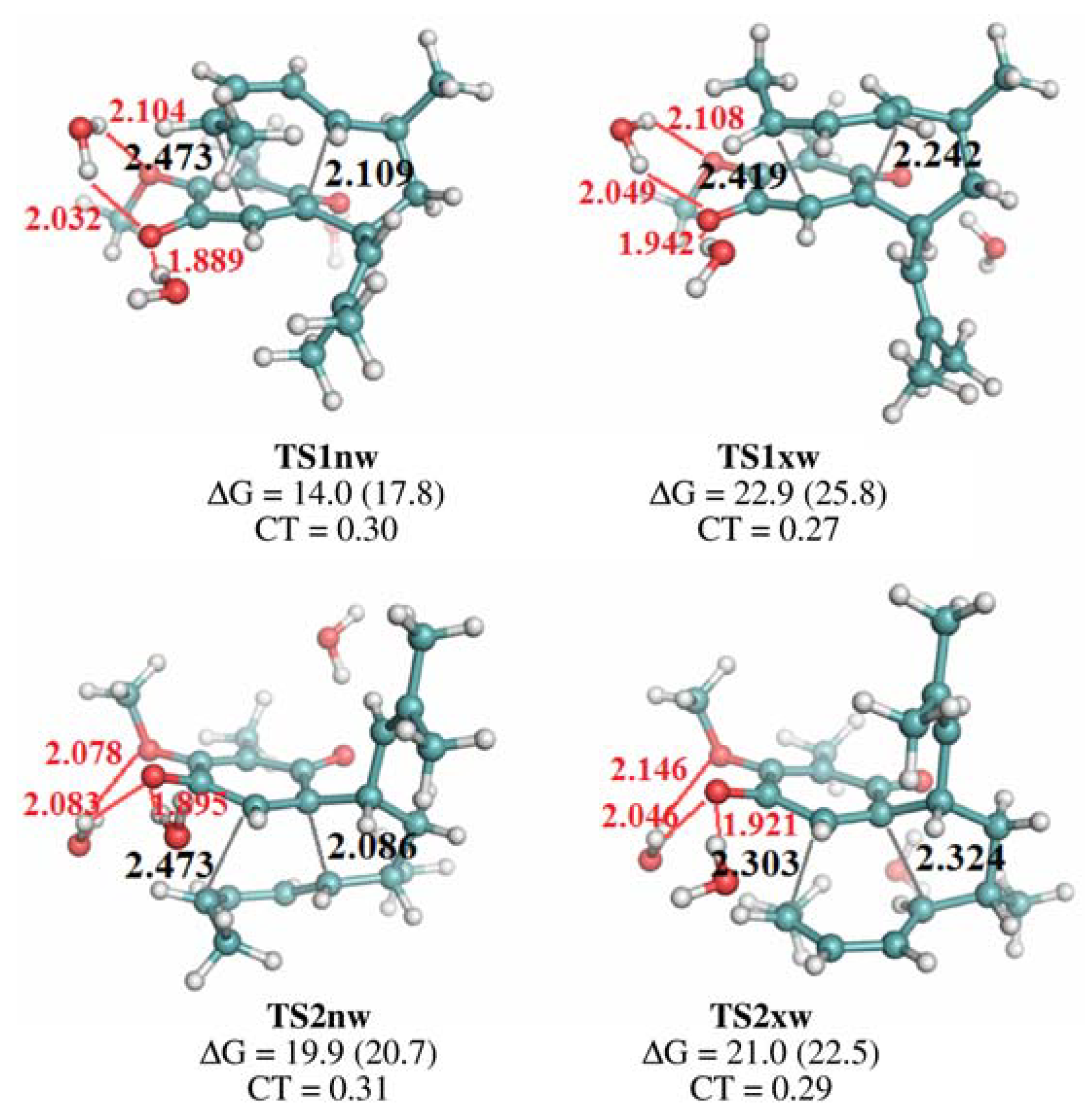{kind=link}
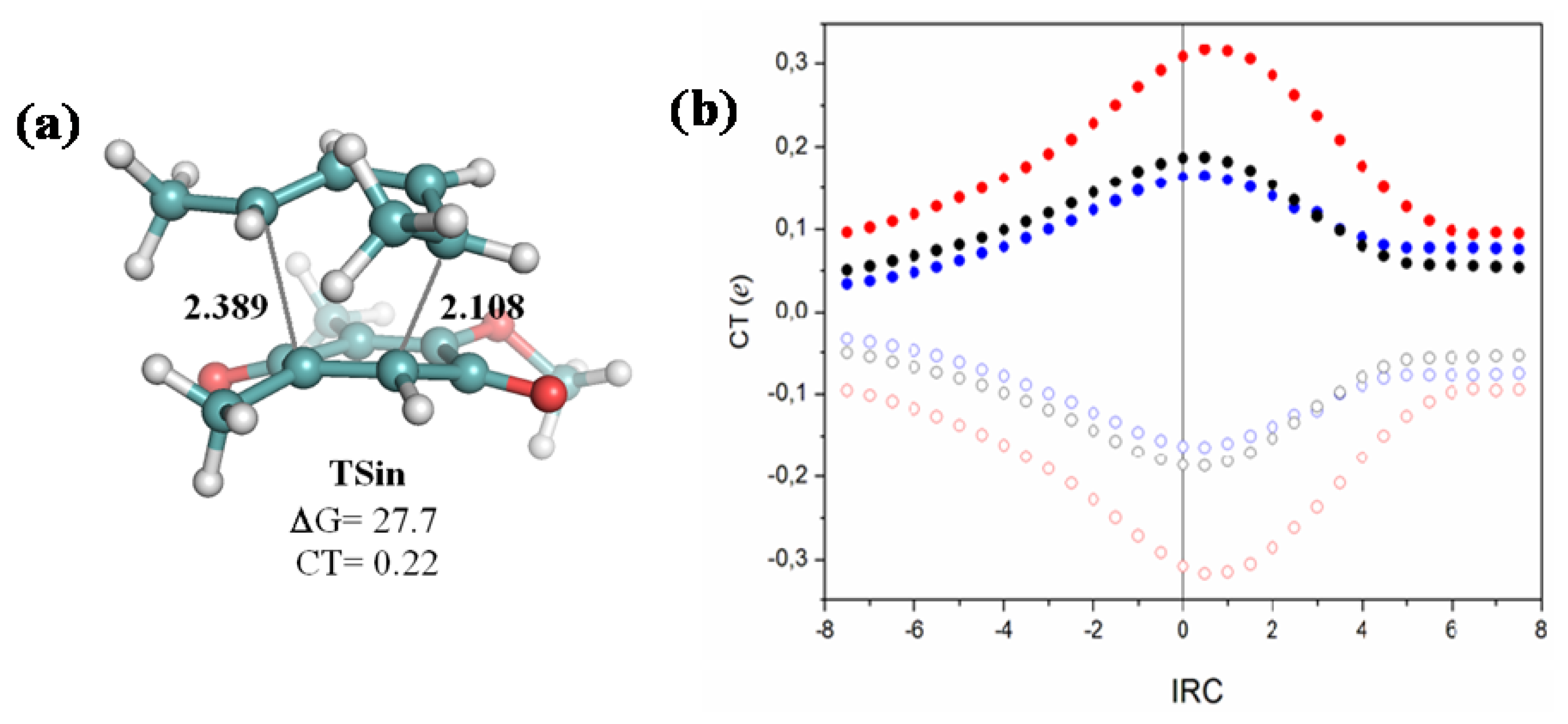{kind=link}
{kind=link}
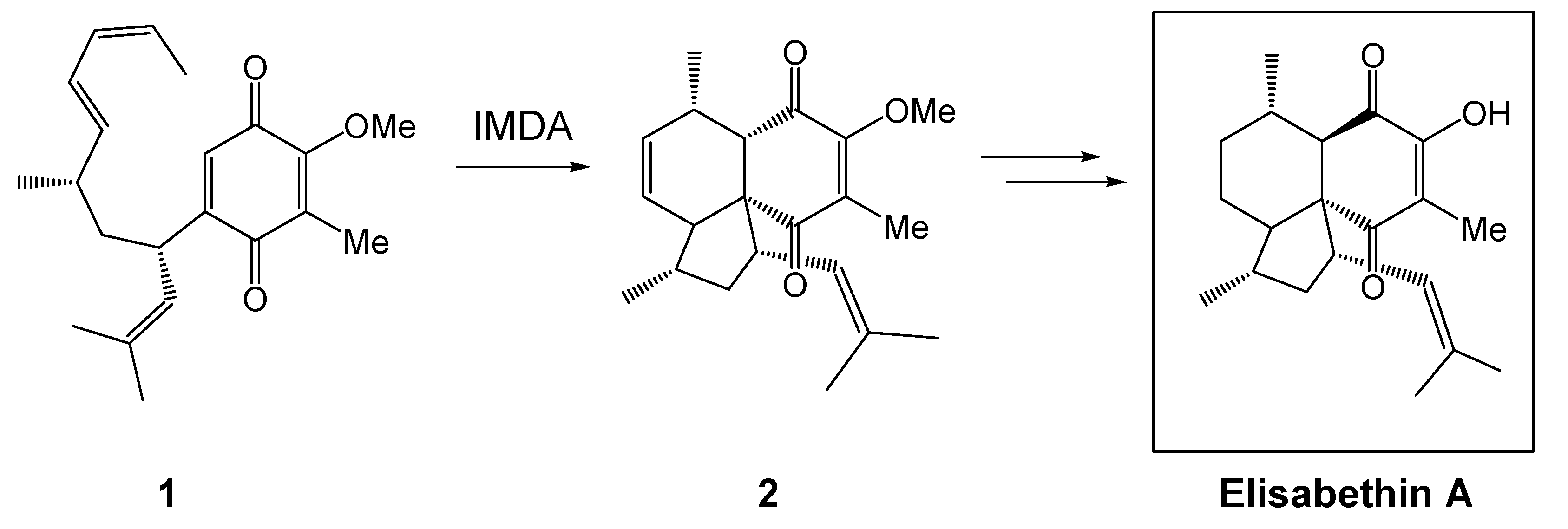{kind=link}
{kind=link}

| Distance | Charge | |||||
|---|---|---|---|---|---|---|
| 1w | TS1nw | 1 | 1w | TS1nw | ||
| d1 | 1.965 | 1.889 | 1 | −0.533 | −0.585 | −0.673 |
| d2 | 2.012 | 1.963 | 2 | −0.545 | −0.561 | −0.610 |
| d3 | 2.092 | 2.032 | 3 | −0.538 | −0.561 | −0.581 |
| d4 | 2.341 | 2.104 | 4 | 0.487 | 0.505 | |
| 5 | 0.495 | 0.498 | ||||
| 6 | 0.487 | 0.491 | ||||
| 7 | 0.477 | 0.486 | ||||
| % H–O s character | σ*(H–O) Occupation | E(2) | % H–O s character | σ*(H–O) Occupation | E(2) | |
|---|---|---|---|---|---|---|
| 1w | TS1nw | |||||
| d1 | 24.2 | 0.0218 | 6.7 | 23.3 | 0.0334 | 11.9 |
| d2 | 24.6 | 0.0159 | 4.9 | 24.3 | 0.0201 | 5.8 |
| d3 | 25.0 | 0.0140 | 4.0 | 24.7 | 0.0162 | 3.8 |
| d4 | 25.9 | 0.0037 | 0.2 | 25.2 | 0.0109 | 4.9 |

| μ | η | ω | N | ωDp | ωD | NDp | ND | E1 | E2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −0.1607 | 0.0940 | 3.65 | 3.46 | 3.64 | 0.00 | 0.00 | 3.44 | 7.08 | 0.00 |
| 1w | −0.1705 | 0.0619 | 6.39 | 3.64 | 6.26 | 0.00 | 0.00 | 3.29 | 9.55 | 0.00 |
| FA | FB | |||||||||
| μA | ηA | ωA | μB | ηB | ωB | ΔωΩ | ||||
| 1 | −0.1646 | 0.1189 | 3.10 | −0.1064 | 0.1901 | 0.81 | 2.39 | |||
| 1w | −0.1869 | 0.0749 | 6.34 | −0.1091 | 0.1726 | 0.94 | 5.40 | |||
© 2012 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Soto-Delgado, J.; Aizman, A.; Contreras, R.; Domingo, L.R. On the Catalytic Effect of Water in the Intramolecular Diels–Alder Reaction of Quinone Systems: A Theoretical Study. Molecules 2012, 17, 13687-13703. https://doi.org/10.3390/molecules171113687
Soto-Delgado J, Aizman A, Contreras R, Domingo LR. On the Catalytic Effect of Water in the Intramolecular Diels–Alder Reaction of Quinone Systems: A Theoretical Study. Molecules. 2012; 17(11):13687-13703. https://doi.org/10.3390/molecules171113687
Chicago/Turabian StyleSoto-Delgado, Jorge, Arie Aizman, Renato Contreras, and Luis R. Domingo. 2012. "On the Catalytic Effect of Water in the Intramolecular Diels–Alder Reaction of Quinone Systems: A Theoretical Study" Molecules 17, no. 11: 13687-13703. https://doi.org/10.3390/molecules171113687
APA StyleSoto-Delgado, J., Aizman, A., Contreras, R., & Domingo, L. R. (2012). On the Catalytic Effect of Water in the Intramolecular Diels–Alder Reaction of Quinone Systems: A Theoretical Study. Molecules, 17(11), 13687-13703. https://doi.org/10.3390/molecules171113687