
The Effect of Glycosaminoglycans (GAGs) on Amyloid Aggregation and Toxicity

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
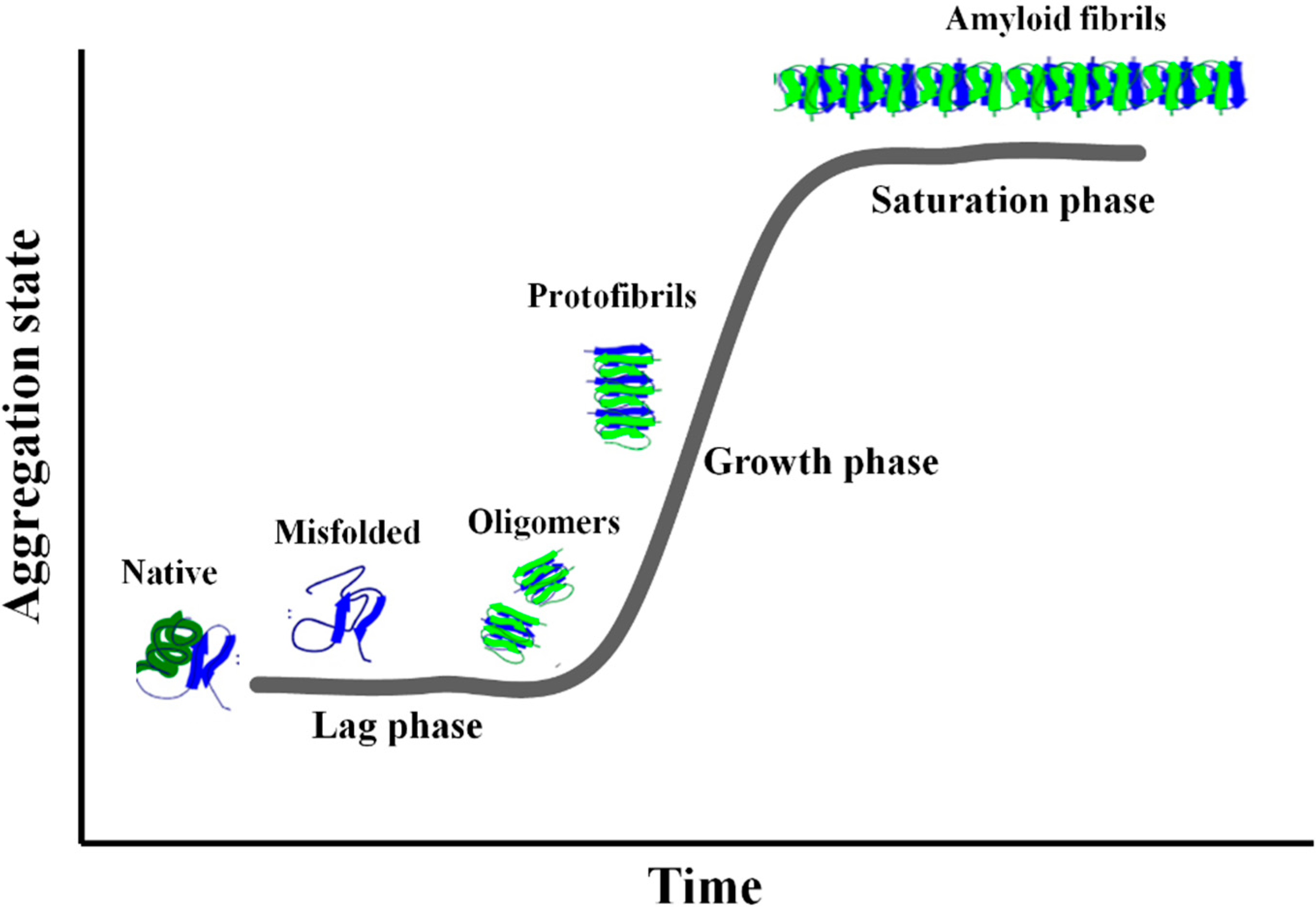
2. Amyloid Aggregation

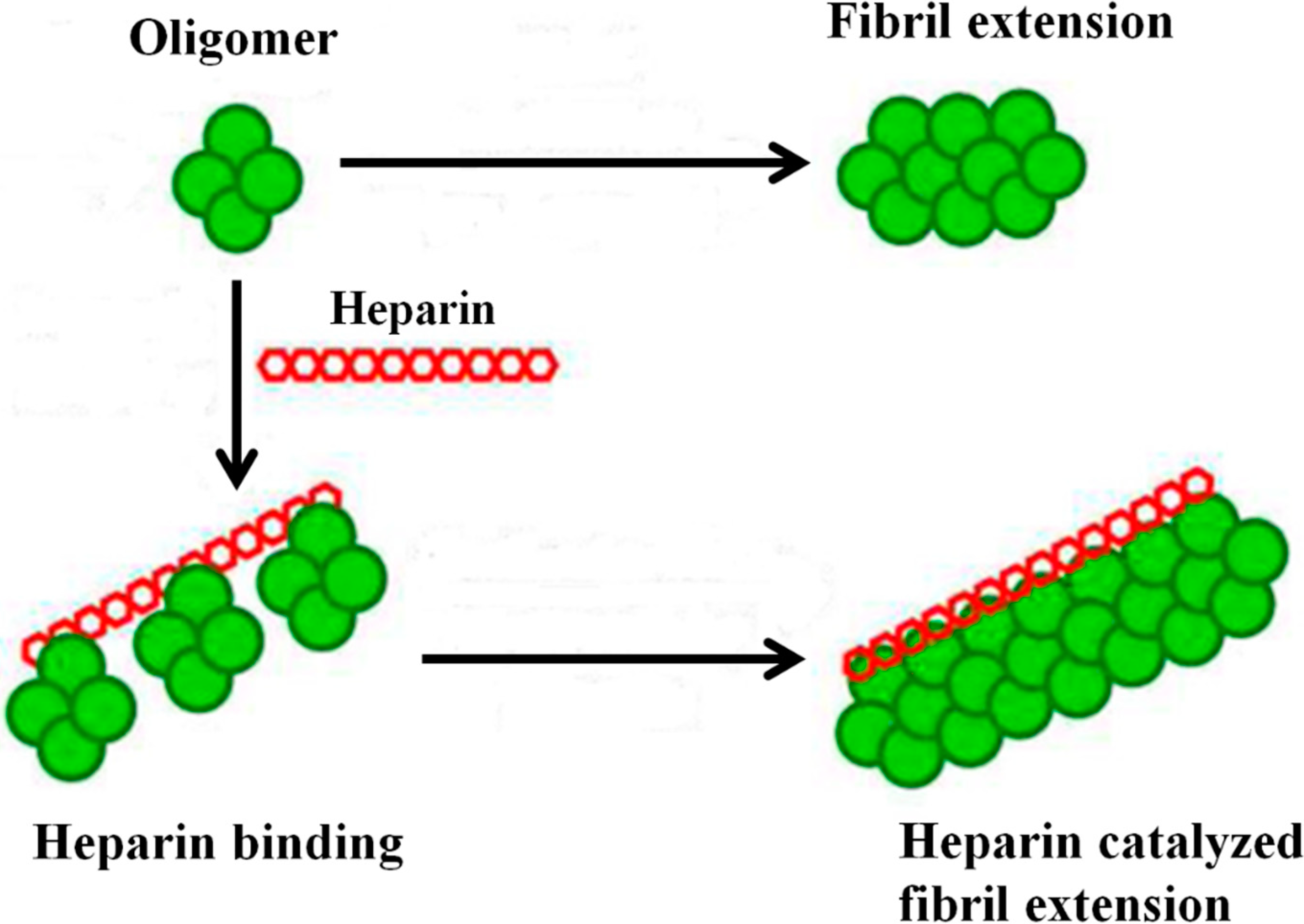
3. GAGs and Amyloid Deposition


4. Molecular Recognition of Heparin by Proteins
5. Effect of GAGs on Peptides and Proteins Having a Weak or None Propensity to Aggregate
5.1. Apomyoglobin
5.2. 23-Residue Peptide of the Phospholamban Transmembrane Protein (PLB(1–23))
5.3. Islet Amyloid Polypeptide (IAPP) Variants
5.4. N-Terminal Domain of Escherichia coli HypF (HypF-N)
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Horssen, J.; Wesseling, P.; van den Heuvel, L.P.; de Waal, R.M.; Verbeek, M.M. Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol. 2003, 2, 482–492. [Google Scholar]
- Snow, A.D.; Wight, T.N. Proteoglycans in the pathogenesis of Alzheimer’s disease and other amyloidosis. Neurobiol. Aging 1989, 10, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S. Basement membrane and beta amyloid fibrillogenesis in Alzheimer’s disease. In International Review of Cytology—A Survey of Cell Biology; Academic Press Inc.: San Diego, CA, USA, 2001; Volume 210, pp. 121–161. [Google Scholar]
- Potter-Perigo, S.; Hull, R.L.; Tsoi, C.; Braun, K.R.; Andrikopoulos, S.; Teague, J.; Verchereb, C.B.; Kahnb, S.E.; Wight, T.N. Proteoglycans synthesized and secreted by pancreatic islet beta-cells bind amylin. Arch. Biochem. Biophys. 2003, 413, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Ancsin, J.B. Amyloidogenesis: Historical and modern observations point to heparan sulfate proteoglycans as a major culprit. Amyloid 2003, 10, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Ailles, L.; Narindrasorasak, S.; Tan, R.; Kisilevsky, R. Localization of the basement membrane heparan sulfate proteoglycan in islet amyloid deposits in type II diabetes mellitus. Arch. Pathol. Lab. Med. 1992, 116, 951–954. [Google Scholar] [PubMed]
- Snow, A.D.; Wight, T.N.; Nochlin, D.; Koike, Y.; Kimata, K.; de Armond, S.J.; Prusiner, S.B. Immunolocalization of heparan sulfate proteoglycans to the prion protein amyloid plaques of Gerstmann-Straussler syndrome, Creutzfeldt-Jakob disease and scrapie. Lab. Investig. 1990, 63, 601–611. [Google Scholar] [PubMed]
- Diaz-Nido, J.; Wandossel, F.; Avila, J. Glycosaminoglycans and beta-amyloid, prion and tau peptides in neurodegenerative diseases. Peptides 2002, 23, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Gruys, E.; Ultee, A.; Upragarin, N. Glycosaminoglycans are part of amyloid fibrils: Ultrastructural evidence in avian AA amyloid stained with cuprolinic blue and labeled with immunogold. Amyloid 2006, 13, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Papy-Garcia, D.; Christophe, M.; Huynh, M.B.; Fernando, S.; Ludmilla, S.; Sepulveda-Diaz, J.E.; Raisman-Vozari, R. Glycosaminoglycans, protein aggregation and neurodegeneration. Curr. Protein Pept. Sci. 2011, 12, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Souillac, P.O.; Ionesco-Zanetti, C.; Carter, S.A.; Fink, A.L. Surface-catalyzed amyloid fibril formation. J. Biol. Chem. 2002, 277, 50914–50922. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Franklin, T.; Zhang, X.; Deng, J.; Fraser, P.E. Interactions of Alzheimer amyloid-beta peptides with glycosaminoglycans effects on fibril nucleation and growth. Eur. J. Biochem. 1999, 266, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Motamedi-Shad, N.; Monsellier, E.; Chiti, F. Amyloid formation by the model protein muscle acylphosphatase is accelerated by heparin and heparan sulphate through a scaffolding-based mechanism. J. Biochem. 2009, 146, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Hardy, J.; Fischbeek, K.H. Toxic proteins in neurodegenerative disease. Science 2005, 296, 1991–1995. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C. Protein misfolding, functional amyloid, and human diseases. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.; Merlini, G.; Saraiva, M.J.; Westermark, P. Amyloid fibril protein nomenclature: 2010 Recommendations of the nomenclature committee of the international society of amyloidosis. Amyloid 2010, 17, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarström, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005, 121, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Blake, C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Adv. Protein Chem. 1997, 50, 123–159. [Google Scholar] [PubMed]
- Makin, O.S.; Serpell, L.C. Examining the structure of the mature amyloid fibril. Biochem. Soc. Trans. 2002, 30, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.O.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, C.; Maritato, R.; Irace, G.; Sirangelo, I. Misfolding and amyloid aggregation of apomyoglobin. Int. J. Mol. Sci. 2013, 14, 14287–14300. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.W. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 1998, 8, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Serio, T.R.; Cashikar, A.G.; Kowal, A.S.; Sawicki, G.J.; Moslehi, J.J.; Serpell, L.; Arnsdorf, M.F.; Lindquist, S.L. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 2000, 289, 1317–1321. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Jiang, P.; Xu, W.; Li, H.; Zhang, H.; Yan, L.; Chan-Park, M.B.; Liu, X.W.; Tang, K.; Mu, Y.; et al. The molecular basis of distinct aggregation pathways of islet amyloid polypeptide. J. Biol. Chem. 2011, 286, 6291–6300. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Culyba, E.K.; Powers, E.T.; Kelly, J.W. Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 2012, 7, 602–609. [Google Scholar] [CrossRef]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Ann. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.J.; Murphy, R.M. Inhibition of insulin fibrillogenesis with targeted peptides. Prot. Sci. 2006, 15, 1133–1141. [Google Scholar] [CrossRef]
- Bernacki, J.P.; Murphy, R.M. Model discrimination and mechanistic interpretation of kinetic data in protein aggregation studies. Biophys. J. 2009, 96, 2871–2887. [Google Scholar] [CrossRef] [PubMed]
- Phelps, E.M.; Hall, C.K. Structural transitions and oligomerization along polyalanine fibril formation pathways from computer simulations. Proteins 2012, 80, 582–597. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowark, R.J.; Waltz, T.; Lansbury, P.T. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Poirier, M.A.; Li, H.; Macosko, J.; Cail, S.; Amzel, M.; Ross, C.A. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 2002, 277, 41032–41037. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 2012, 421, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Ortore, M.G.; Spinozzi, F.; Vilasi, S.; Sirangelo, I.; Irace, G.; Shukla, A.; Narayanan, T.; Sinibaldi, R.; Mariani, P. Time-resolved small-angle X-ray scattering study of the early stage of amyloid formation of an apomyoglobin mutant. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2011, 84, 061904. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, non fibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Oligomers on the brain: The emerging role of soluble protein aggregates in neurodegeneration. Protein Pept. Lett. 2000, 11, 213–228. [Google Scholar] [CrossRef]
- Reixach, N.; Deechongkit, S.; Jiang, X.; Kelly, J.W.; Buxbaum, J.N. Tissue damage in the amyloidosis: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. USA 2004, 101, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, C.; Stefani, M. The amyloid-cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity. Biophys. Chem. 2013, 182, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Malmo, C.; Vilasi, S.; Iannuzzi, C.; Tacchi, S.; Cametti, C.; Irace, G.; Sirangelo, I. Tetracycline inhibits W7FW14F apomyoglobin fibril extension and keeps the amyloid protein in a pre-fibrillar, highly cytotoxic state. FASEB J. 2006, 20, 346–347. [Google Scholar] [PubMed]
- Bucciantini, M.; Calloni, G.; Chiti, F.; Formigli, L.; Nosi, D.; Dobson, C.M.; Stefani, M. Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem. 2004, 279, 31374–31382. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thomson, J.L.; Mcintire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 2010, 277, 4602–4613. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, J.I.; Sunde, M.; Jones, J.A.; Campbell, I.D.; Dobson, C.M. Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. USA 1998, 95, 4224–4228. [Google Scholar] [CrossRef] [PubMed]
- Litvinovich, S.V.; Brew, S.A.; Aota, S.; Akiyama, S.K.; Haudenschild, C.; Ingham, K.C. Formation of amyloid-like fibrils by self-association of a partially unfolded fibronectin type III module. J. Mol. Biol. 1998, 280, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Fandrich, M.; Fletcher, M.A.; Dobson, C.M. Amyloid fibrils from muscle myoglobin. Nature 2001, 410, 165–166. [Google Scholar] [CrossRef] [PubMed]
- Infusini, G.; Iannuzzi, C.; Vilasi, S.; Birolo, L.; Pagnozzi, D.; Pucci, P.; Irace, G.; Sirangelo, I. Resolution of the effects induced by W→F substitutions on the conformation and dynamics of the amyloid-forming apomyoglobin mutant W7FW14F. Eur. Biophys. J. 2012, 41, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [PubMed]
- Snow, A.D.; Kisilevsky, R. Temporal relationship between glycosaminoglycan accumulation and amyloid deposition during experimental amyloidosis. A histochemical study. Lab. Investig. 1985, 53, 37–44. [Google Scholar] [PubMed]
- Kisilevsky, R.; Szarek, W.A.; Ancsin, J.B.; Elimova, E.; Marone, S.; Bhat, S.; Berkin, A. Inhibition of amyloid A amyloidogenesis in vivo and in tissue culture by 4-deoxy analogues of peracetylated 2-acetamido-2-deoxy-alpha- and beta-d-glucose: Implications for the treatment of various amyloidosis. Am. J. Pathol. 2004, 164, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Elimova, E.; Kisilevsky, R.; Szarek, W.A.; Ancsin, J.B. Amyloidogenesis recapitulated in cell culture: A peptide inhibitor provides direct evidence for the role of heparan sulfate and suggests a new treatment strategy. FASEB J. 2004, 18, 1749–1751. [Google Scholar] [PubMed]
- Li, J.P.; Galvis, M.L.; Gong, F.; Zhang, X.; Zcharia, E.; Metzger, S.; Vlodavsky, I.; Kisilevsky, R.; Lindahl, U. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc. Natl. Acad. Sci. USA 2005, 102, 6473–6477. [Google Scholar] [CrossRef] [PubMed]
- Castillo, G.M.; Lukito, W.; Wight, T.N.; Snow, A.D. The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J. Neurochem. 1999, 72, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Castillo, G.M.; Ngo, C.; Cummings, J.; Wight, T.N.; Snow, A.D. Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer’s disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J. Neurochem. 1997, 69, 2452–2465. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.E.; Nguyen, J.T.; Chin, D.T.; Kirschner, D.A. Effects of sulfate ions on Alzheimer beta/A4 peptide assemblies: Implications for amyloid fibril-proteoglycan interactions. J. Neurochem. 1992, 59, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.E.; Darabie, A.A.; McLaurin, J.A. Amyloid-beta interactions with chondroitin sulfate-derived monosaccharides and disaccharides. Implications for drug development. J. Biol. Chem. 2001, 276, 6412–6419. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Fraser, P.E. Effect of amino-acid substitutions on Alzheimer’s amyloid-beta peptide-glycosaminoglycan interactions. Eur. J. Biochem. 2000, 267, 6353–6361. [Google Scholar] [CrossRef] [PubMed]
- Valle-Delgado, J.J.; Alfonso-Prieto, M.; de Groot, N.S.; Ventura, S.; Samitier, J.; Rovira, C.; Fernàndez-Busquets, X. Modulation of Abeta42 fibrillogenesis by glycosaminoglycan structure. FASEB J. 2010, 24, 4250–4261. [Google Scholar] [CrossRef] [PubMed]
- Ariga, T.; Miyatake, T.; Yu, R.K. Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer’s disease and related disorders: Amyloidogenesis and therapeutic strategies—A review. J. Neurosci. Res. 2010, 88, 2303–2315. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschini, L.; Rossi, E.; Vergani, C.; de Simoni, M.G. Alzheimer’s disease: Another target for heparin therapy. Sci. World J. 2009, 9, 891–908. [Google Scholar] [CrossRef]
- Scholefield, Z.; Yates, E.A.; Wayne, G.; Amour, A.; McDowell, W.; Turnbull, J.E. Heparan sulfate regulates amyloid precursor protein processing by BACE1, the Alzheimer’s beta-secretase. J. Cell Biol. 2003, 163, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Shankar, G.M.; Townsend, M.; Fadeeva, J.V.; Betts, V.; Podlisny, M.B.; Cleary, J.P.; Ashe, K.H.; Rowan, M.J.; et al. The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic intervention. Biochem. Soc. Trans. 2005, 33, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Tsai, J.; Naslund, J.; Vincent, B.; Edgar, M.; Checler, F.; Greenfield, J.P.; Haroutunian, V.; Buxbaum, J.D.; Xu, H.; et al. Intraneuronal Abeta42 accumulation in human brain. Am. J. Pathol. 2000, 156, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Multhaup, G.; Bayer, T.A. A modified beta-amyloid hypothesis: Intraneuronal accumulation of the beta-amyloid peptide-the first step of a fatal cascade. J. Neurochem. 2004, 91, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Sandwall, E.; O’Callaghan, P.; Zhang, X.; Lindahl, U.; Lannfelt, L.; Li, J.P. Heparan sulfate mediates amyloid-beta internalization and cytotoxicity. Glycobiology 2010, 20, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Paudel, H.K.; Li, W. Heparin-induced conformational change in microtubule-associated protein Tau as detected by chemical cross-linking and phosphopeptide mapping. J. Biol. Chem. 1999, 274, 8029–8038. [Google Scholar] [CrossRef] [PubMed]
- Cohlberg, J.A.; Li, J.; Uverskky, V.N.; Fink, A.L. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha synuclein in vitro. Biochemistry 2002, 41, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.Y.; Zhang, F.; Balch, W.E.; Linhardt, R.J.; Kelly, J.F. Heparin accelerates gelsolin amyloidogenesis. Biochemistry 2006, 45, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Relini, A.; de Stefano, S.; Torrassa, S.; Cavalleri, O.; Rolandi, R.; Gliozzi, A.; Giorgetti, S.; Raimondi, S.; Marchese, L.; Verga, L.; et al. Heparin strongly enhances the formation of beta2-microglobulin amyloid fibrils in the presence of type I collagen. J. Biol. Chem. 2008, 283, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Calamai, M.; Kumita, J.R.; Mifsud, J.; Parrini, C.; Ramazzotti, M.; Ramponi, G.; Taddei, N.; Chiti, F.; Dobson, C.M. Nature and significance of the interactions between amyloid fibrils and biological polyelectrolytes. Biochemistry 2006, 45, 12806–12815. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Abedini, A.; Song, B.; Raleigh, D.P. Amyloid formation by pro-islet amyloid polypeptide processing intermediates: Examination of the role of protein heparan sulfate interactions and implications for islet amyloid formation in type 2 diabetes. Biochemistry 2007, 46, 12091–12099. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, R.W.; de Stigter, J.K.; Sikkink, L.A.; Baden, E.M.; Ramirez-Alvarado, M. The effects of sodium sulfate, glycosaminoglycans, and Congo red on the structure, stability, and amyloid formation of an immunoglobulin light-chain protein. Protein. Sci. 2006, 15, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Middleton, D.A. Comparison of aggregation enhancement and inhibition as strategies for reducing the cytotoxicity of the aortic amyloid polypeptide medin. Eur. Biophys. J. 2010, 39, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- De Carufel, C.A.; Nguyen, P.T.; Sahnouni, S.; Bourgault, S. New insights into the roles of sulfated glycosaminoglycans in islet amyloid polypeptide amyloidogenesis and cytotoxicity. Biopolymers 2013, 100, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Xiong, L.W.; Horiuchi, M.; Raymond, L.; Wehrly, K.; Chesebro, B.; Caughey, B. Sulfated glycans and elevated temperature stimulate PrP(Sc)-dependent cell-free formation of protease-resistant prion protein. EMBO J. 2001, 20, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Vieira, T.C.; Cordeiro, Y.; Caughey, B.; Silva, J.L. Heparin binding confers prion stability and impairs its aggregation. FASEB J. 2014, 28, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Narindrasorasak, S.; Lowery, D.; Gonzalez-DeWhitt, P.; Poorman, R.A.; Greenberg, B.; Kisilevsky, R. High affinity interactions between the Alzheimer’s beta-amyloid precursor proteins and the basement membrane form of heparan sulfate proteoglycan. J. Biol. Chem. 1991, 266, 12878–12883. [Google Scholar] [PubMed]
- Brunden, K.R.; Richter-Cook, N.J.; Chaturvedi, N.; Frederickson, R.C. pH-Dependent binding of synthetic beta-amyloid peptides to glycosaminoglycans. J. Neurochem. 1993, 61, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Brown, K.; Raymond, G.J.; Katzenstein, G.E.; Thresher, W. Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and Congo red. J. Virol. 1994, 68, 2135–2141. [Google Scholar] [PubMed]
- Warner, R.G.; Hundt, C.; Weiss, S.; Turnbull, J.E. Identification of the heparan sulfate binding sites in the cellular prion protein. J. Biol. Chem. 2002, 277, 18421–18430. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Verchere, C.B. Identification of a heparin binding domain in the N-terminal cleavage site of pro-islet amyloid polypeptide. Implications for islet amyloid formation. J. Biol. Chem. 2001, 276, 16611–16616. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Kisilevsky, R.; Yanagishita, M. Affinity binding of glycosaminoglycans with beta(2)-microglobulin. Nephron 2002, 90, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Solomon, J.P.; Reixach, N.; Kelly, J.W. Sulfated glycosaminoglycans accelerate transthyretin amyloidogenesis by quaternary structural conversion. Biochemistry 2011, 50, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.P.; Bourgault, S.; Powers, E.T.; Kelly, J.W. Heparin binds 8 kDa gelsolin cross-β-sheet oligomers and accelerates amyloidogenesis by hastening fibril extension. Biochemistry 2011, 50, 2486–2498. [Google Scholar] [CrossRef] [PubMed]
- Sasisekharan, R.; Raman, R.; Prabhakar, V. Glycomis approach to structure-function relationships of glycosaminoglycans. Annu. Rev. Biomed. Eng. 2006, 8, 181–231. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Ahmad, S. Sequence and structural features of carbohydrate binding in proteins and assessment of predictability using a neural network. BMC Struct. Biol. 2007, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Shionyu-Mitsuyama, C.; Shirai, T.; Ishida, H.; Yamane, T. An empirical approach for structure-based prediction of carbohydrate-binding sites on proteins. Protein. Eng. Des. Sel. 2003, 16, 467–478. [Google Scholar] [CrossRef]
- Taroni, C.; Jones, S.; Thornton, J.M. Analysis and prediction of carbohydrate binding sites. Protein. Eng. Des. Sel. 2000, 13, 89–98. [Google Scholar] [CrossRef]
- Fromm, J.R.; Hileman, R.E.; Caldwell, E.E.O.; Weiler, J.M.; Linhardt, R.J. Pattern and spacing of basic amino acids in heparin binding sites. Arch. Biochem. Biophys. 1997, 343, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Hileman, R.E.; Fromm, J.R.; Weiler, J.M.; Linhardt, R.J. Glycosaminoglycan-protein interactions: Definition of consensus sites in glycosaminoglycan binding proteins. BioEssays 1998, 20, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Cardin, A.D.; Randall, C.J.; Hirose, N.; Jackson, R.L. Physical-chemical interaction of heparin and human plasma low-density lipoproteins. Biochemistry 1987, 26, 5513–5518. [Google Scholar] [CrossRef] [PubMed]
- Hirose, N.; Blankenship, D.T.; Krivanek, M.A.; Jackson, R.L.; Cardin, A.D. Isolation and characterization of four heparin-binding cyanogen bromide peptides of human plasma apolipoprotein B. Biochemistry 1987, 26, 5505–5512. [Google Scholar] [CrossRef]
- Gigli, M.; Consonni, A.; Ghiselli, G.; Rizzo, V.; Naggi, A.; Torri, G. Heparin binding to human plasma low-density lipoproteins: Dependence on heparin sulfation degree and chain length. Biochemistry 1992, 31, 5996–6003. [Google Scholar] [CrossRef] [PubMed]
- Weisgraber, K.H.; Rail, S.C., Jr. Human apolipoprotein B-100 heparin-binding sites. J. Biol. Chem. 1987, 262, 11097–11103. [Google Scholar] [PubMed]
- Cardin, A.D.; Hirose, N.; Blankenship, D.T.; Jackson, R.L.; Harmony, J.A.; Sparrow, D.A.; Sparrow, J.T. Binding of a high reactive heparin to human apolipoprotein E: Identification of two heparin-binding domains. Biochem. Biophys. Res. Commun. 1986, 134, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Weisgraber, K.H.; Rail, S.C., Jr.; Mahley, R.W.; Milne, R.W.; Marcel, Y.L.; Sparrow, J.T. Human apolipoprotein E. Determination of the heparin binding sites of apolipoprotein E3. J. Biol. Chem. 1986, 261, 2068–2076. [Google Scholar] [PubMed]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Baird, A.; Schubert, D.; Ling, N.; Guillemin, R. Three-dimensional structure of human basic fibroblast growth factor. Proc. Natl. Acad. Sci. USA 1988, 85, 2324–2328. [Google Scholar] [CrossRef] [PubMed]
- Margalit, H.; Fischer, N.; Ben-Sasson, S.A. Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J. Biol. Chem. 1993, 268, 19228–19231. [Google Scholar] [PubMed]
- Torrent, M.; Nogués, M.V.; Andreu, D.; Boix, E. The “CPC Clip Motif”: A conserved structural signature for heparin-binding proteins. PLoS One 2012, 7, e42692. [Google Scholar] [CrossRef] [PubMed]
- Vilasi, S.; Sarcina, R.; Maritato, R.; de Simone, A.; Irace, G.; Sirangelo, I. Heparin induces harmless fibril formation in amyloidogenic W7FW14F apomyoglobin and amyloid aggregation in wild-type protein in vitro. PLoS One 2011, 6, e22076. [Google Scholar] [CrossRef] [PubMed]
- Campioni, S.; Mannini, B.; Pensalfini, A.; Zampagni, M.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Bismuto, E.; Colonna, G.; Irace, G. Unfolding pathway of myoglobin. Evidence for a multistate process. Biochemistry 1983, 22, 4165–4170. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Davies, H.A.; Hughes, E.; Middleton, D.A. Heparin promotes the rapid fibrillization of a peptide with low intrinsic amyloidogenicity. Biochemistry 2013, 52, 8984–8992. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cao, P.; Raleigh, D.P. Amyloid formation in heterogeneous environments: Islet amyloid polypeptide glycosaminoglycan interactions. J. Mol. Biol. 2013, 425, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Meng, F.L.; Raleigh, D.P. A single-point mutation converts the highly amyloidogenic human islet amyloid polypeptide into a potent fibrillization inhibitor. J. Am. Chem. Soc. 2007, 129, 11300–11301. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.M.; Tatarek-Nossol, M.; Velkova, A.; Kazantzis, A.; Kapurniotu, A. Design of a mimic of non amyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity inhibitor of IAPP cytotoxic fibrillogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Engstrom, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.H.; Gupta, R.; Ling, Y.L.; Strasfeld, D.B.; Raleigh, D.P.; Zanni, M.T. Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 6614–6619. [Google Scholar] [CrossRef] [PubMed]
- Rosano, C.; Zuccotti, S.; Bucciantini, M.; Stefani, M.; Ramponi, G.; Bolognesi, M. Crystal structure and anion binding in the prokaryotic hydrogenase maturation factor HypF acylphosphatase-like domain. J. Mol. Biol. 2002, 321, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Bucciantini, M.; Capanni, C.; Taddei, N.; Dobson, C.M.; Stefani, M. Solution conditions can promote formation of either amyloid protofilaments or mature fibrils from the HypF N-terminal domain. Protein Sci. 2001, 10, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Saridaki, T.; Zampagni, M.; Mannini, B.; Evangelisti, E.; Taddei, N.; Cecchi, C.; Chiti, F. Glycosaminoglycans (GAGs) suppress the toxicity of HypF-N prefibrillar aggregates. J. Mol. Biol. 2012, 421, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Sirangelo, I.; Irace, G. Inhibition of aggregate formation as therapeutic target in protein misfolding diseases: Effect of tetracycline and trehalose. Expert Opin. Ther. Targets 2010, 14, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, C.; Maritato, R.; Irace, G.; Sirangelo, I. Glycation accelerates fibrillization of the amyloidogenic W7FW14F apomyoglobin. PLoS One 2013, 8, e80768. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Hawkins, P.N. Amyloidosis: New strategies for treatment. Int. J. Biochem. Cell Biol. 2003, 35, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.P. Heparan sulfate proteoglycans in amyloidosis. Prog. Mol. Biol. Transl. Sci. 2010, 93, 309–334. [Google Scholar] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannuzzi, C.; Irace, G.; Sirangelo, I. The Effect of Glycosaminoglycans (GAGs) on Amyloid Aggregation and Toxicity. Molecules 2015, 20, 2510-2528. https://doi.org/10.3390/molecules20022510
Iannuzzi C, Irace G, Sirangelo I. The Effect of Glycosaminoglycans (GAGs) on Amyloid Aggregation and Toxicity. Molecules. 2015; 20(2):2510-2528. https://doi.org/10.3390/molecules20022510
Chicago/Turabian StyleIannuzzi, Clara, Gaetano Irace, and Ivana Sirangelo. 2015. "The Effect of Glycosaminoglycans (GAGs) on Amyloid Aggregation and Toxicity" Molecules 20, no. 2: 2510-2528. https://doi.org/10.3390/molecules20022510
APA StyleIannuzzi, C., Irace, G., & Sirangelo, I. (2015). The Effect of Glycosaminoglycans (GAGs) on Amyloid Aggregation and Toxicity. Molecules, 20(2), 2510-2528. https://doi.org/10.3390/molecules20022510