
The Mechanism of Room-Temperature Ionic-Liquid-Based Electrochemical CO2 Reduction: A Review



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
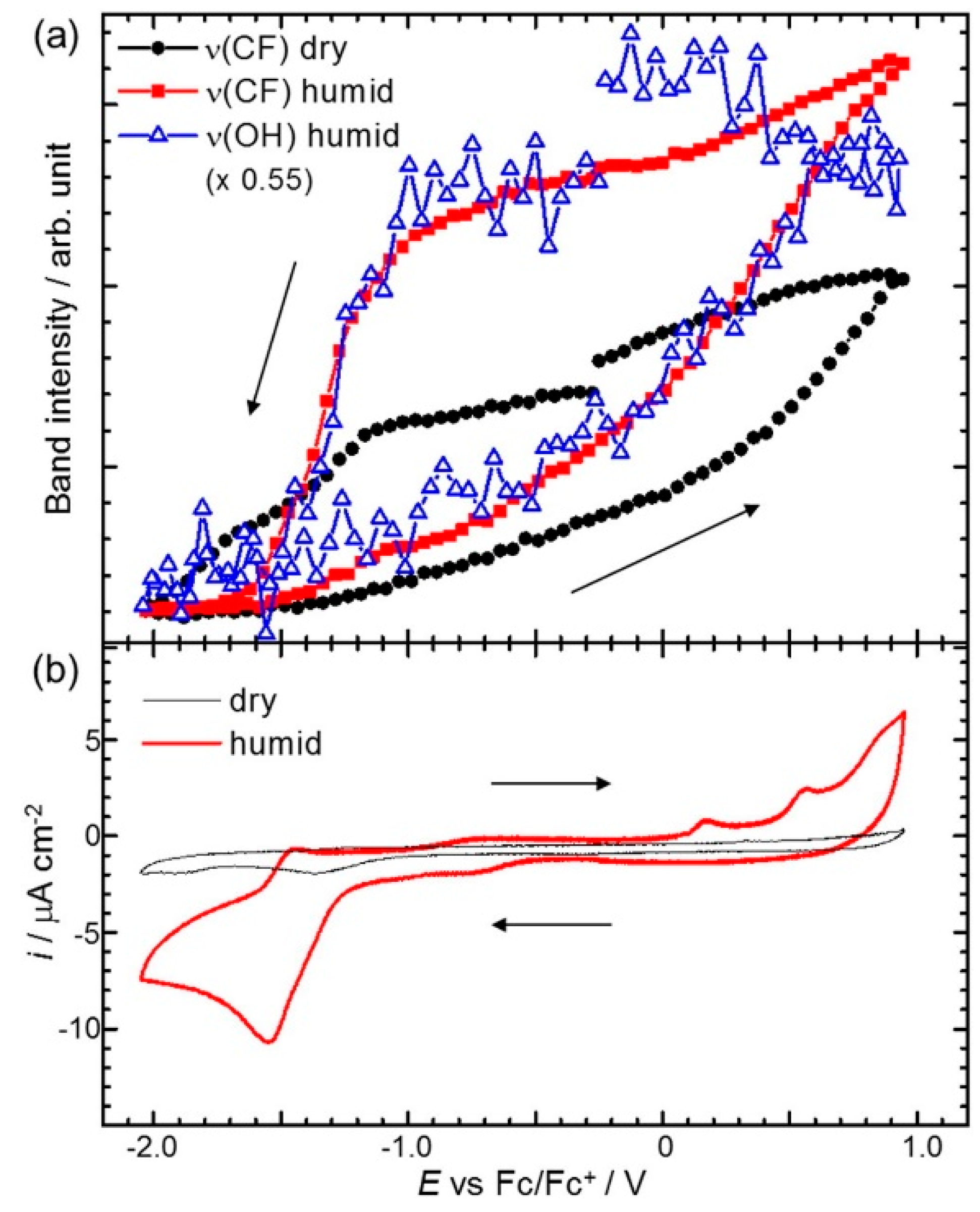{kind=link}
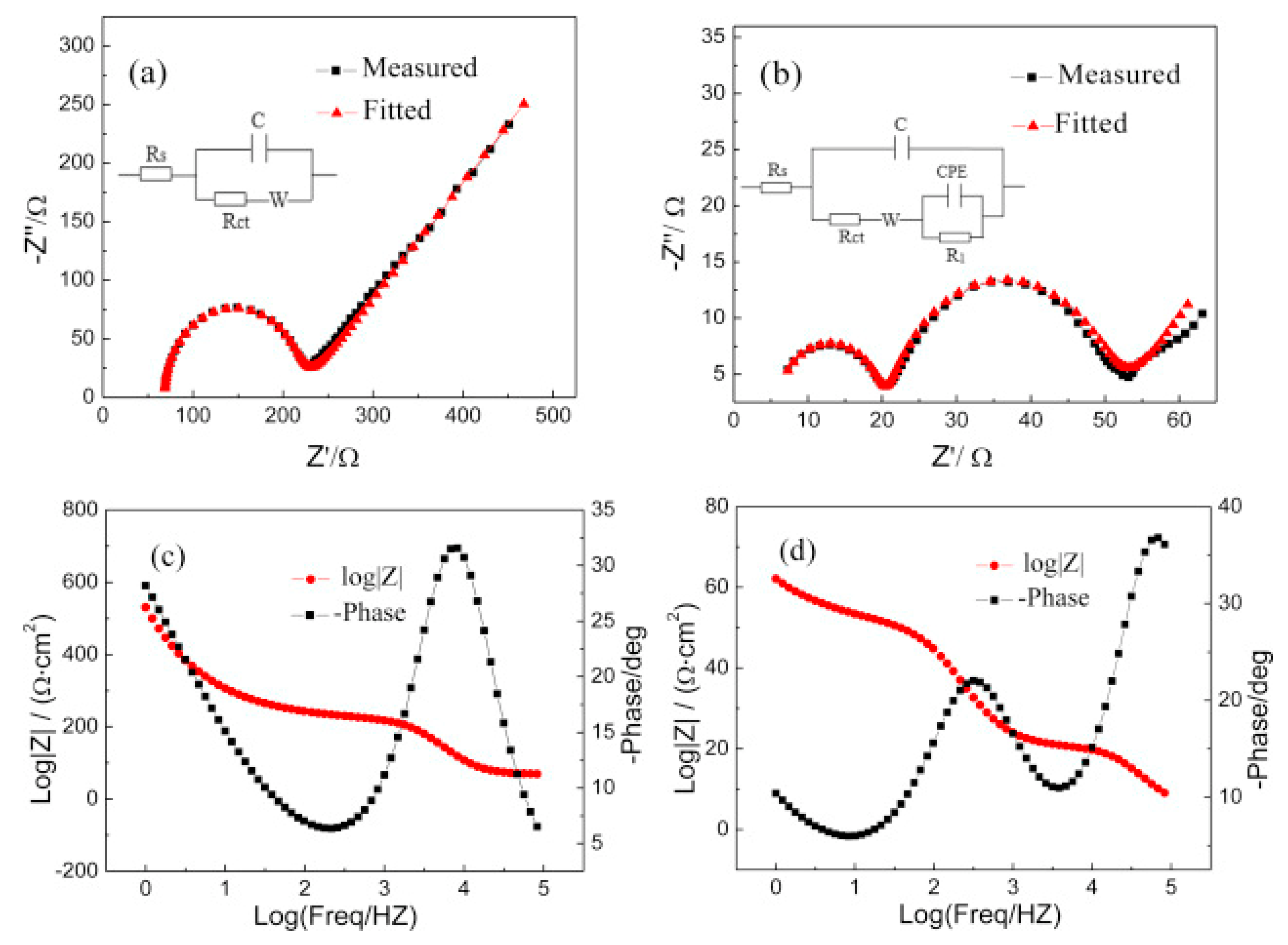{kind=link}
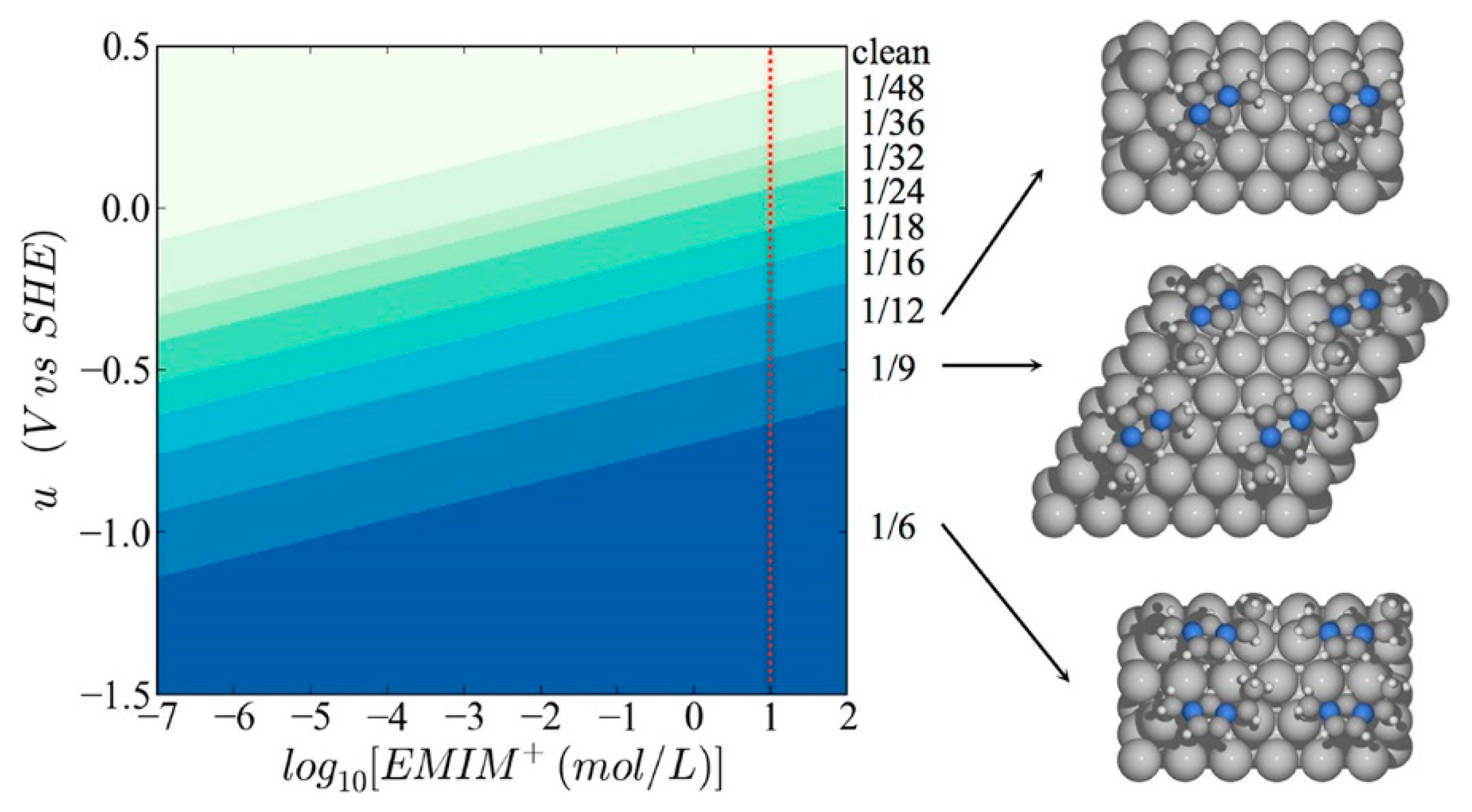{kind=link}
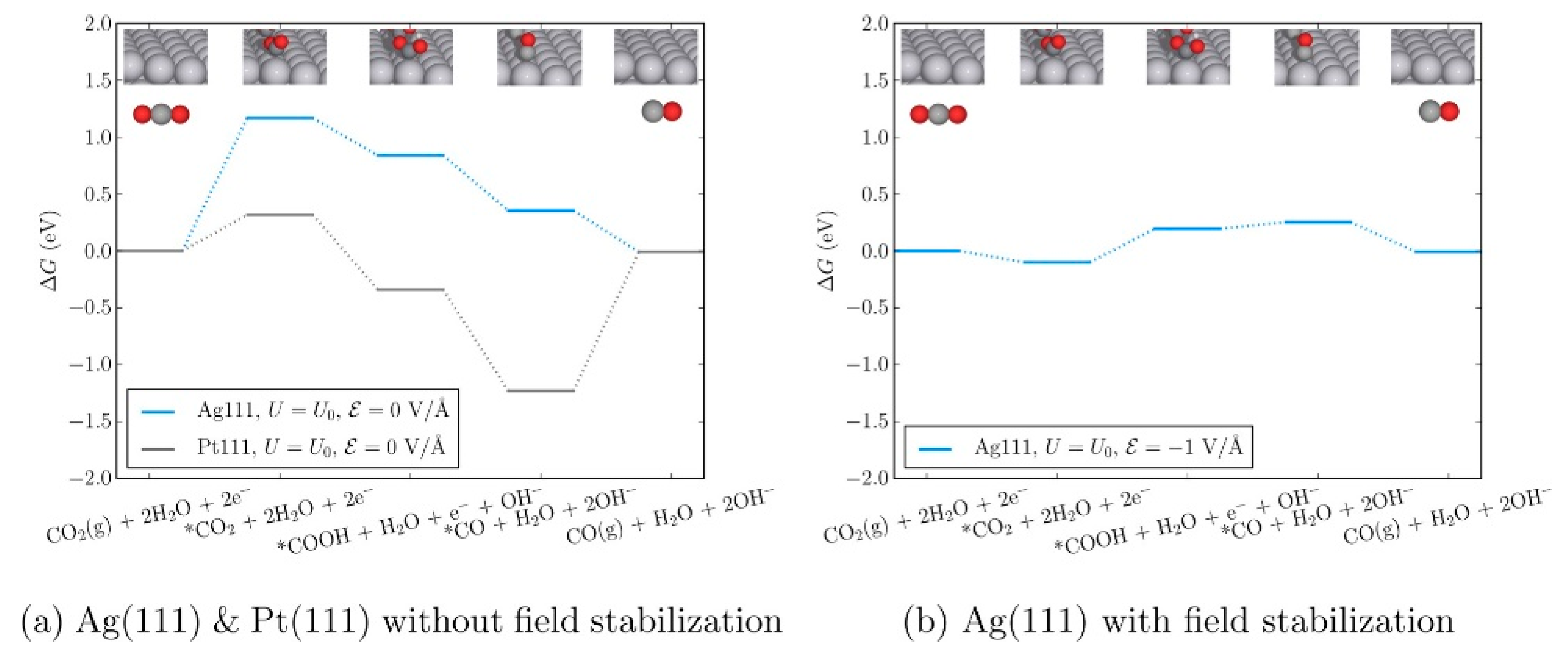{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Roles of RTILs at the Electrode–RTIL Interface
3. The Roles of RTILs in Chemical Interactions
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| [Emim] | 1-Ethyl-3-methylimidazolium |
| [Bmim] | 1-Butyl-3-methylimidazolium |
| [Pmim] | 1-Propyl-3-methylimidazolium |
| [Bmpyrr] | 1-Butyl-1-methylpyrrolidinium |
| [Bu4N] | Tetrabutylammonium |
| [BF4] | Tetrafluoroborate |
| [PF6] | Hexafluorophosphate |
| [MS] | Methylsulfate |
| [TFSA] | Bis(trifluoromethanesulfonyl)amide |
| [CF3SO3] | Trifluoromethanesulfonate |
| [NTf2] | Bis(trifluoromethylsulfonyl)imide |
| [FAP] | Tris(pentafluoroethyl)trifluorophosphate |
References
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Ansari, M.B.; Park, S.-E. Carbon dioxide utilization as a soft oxidant and promoter in catalysis. Energy Environ. Sci. 2012, 5, 9419–9437. [Google Scholar] [CrossRef]
- Xiaoding, X.; Moulijn, J.A. Mitigation of CO2 by Chemical Conversion: Plausible Chemical Reactions and Promising Products. Energy Fuels 1996, 10, 305–325. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [PubMed]
- Wood, H.G.; Werkman, C.H. The utilization of CO2 by the propionic acid bacteria. Biochem. J. 1938, 32, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Michiki, H. Biological CO2 fixation and utilization project. Energy Convers. Manag. 1995, 36, 701–705. [Google Scholar] [CrossRef]
- Wang, B.; Li, Y.; Wu, N.; Lan, C.Q. CO2 bio-mitigation using microalgae. Appl. Microbiol. Biotechnol. 2008, 79, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Lee, J.-P. Review of advances in biological CO2 mitigation technology. Biotechnol. Bioprocess Eng. 2003, 8, 354. [Google Scholar] [CrossRef]
- Smestad, G.P.; Steinfeld, A. Review: Photochemical and Thermochemical Production of Solar Fuels from H2O and CO2 Using Metal Oxide Catalysts. Ind. Eng. Chem. Res. 2012, 51, 11828–11840. [Google Scholar] [CrossRef]
- Lyman, J.L.; Jensen, R.J. Chemical reactions occurring during direct solar reduction of CO2. Sci. Total Environ. 2001, 277, 7–14. [Google Scholar] [CrossRef]
- Nigara, Y.; Cales, B. Production of Carbon-Monoxide by Direct Thermal Splitting of Carbon-Dioxide at High-Temperature. Bull. Chem. Soc. Jpn. 1986, 59, 1997–2002. [Google Scholar] [CrossRef]
- Traynor, A.J.; Jensen, R.J. Direct solar reduction of CO2 to fuel: First prototype results. Ind. Eng. Chem. Res. 2002, 41, 1935–1939. [Google Scholar] [CrossRef]
- Furler, P.; Scheffe, J.; Gorbar, M.; Moes, L.; Vogt, U.; Steinfeld, A. Solar Thermochemical CO2 Splitting Utilizing a Reticulated Porous Ceria Redox System. Energy Fuels 2012, 26, 7051–7059. [Google Scholar] [CrossRef]
- Kumar, B.; Llorente, M.; Froehlich, J.; Dang, T.; Sathrum, A.; Kubiak, C.P. Photochemical and Photoelectrochemical Reduction of CO2. Annu. Rev. Phys. Chem. 2012, 63, 541–569. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Zhou, Y.; Zou, Z. Photocatalytic Conversion of CO2 into Renewable Hydrocarbon Fuels: State-of-the-Art Accomplishment, Challenges, and Prospects. Adv. Mater. 2014, 26, 4607–4626. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.C.; Varghese, O.K.; Paulose, M.; Grimes, C.A. Toward Solar Fuels: Photocatalytic Conversion of Carbon Dioxide to Hydrocarbons. ACS Nano 2010, 4, 1259–1278. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.N.; Soulis, J.; Yang, Y.J.; Biswas, P. Comparison of CO2 Photoreduction Systems: A Review. Aerosol Air Qual. Res. 2014, 14, 533–549. [Google Scholar] [CrossRef]
- Jhong, H.-R.M.; Ma, S.; Kenis, P.J.A. Electrochemical conversion of CO2 to useful chemicals: Current status, remaining challenges, and future opportunities. Curr. Opin. Chem. Eng. 2013, 2, 191–199. [Google Scholar] [CrossRef]
- Savéant, J.-M. Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Robert, M.; Saveant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Spinner, N.S.; Vega, J.A.; Mustain, W.E. Recent progress in the electrochemical conversion and utilization of CO2. Catal. Sci. Technol. 2012, 2, 19–28. [Google Scholar] [CrossRef]
- Whipple, D.T.; Kenis, P.J.A. Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction. J. Phys. Chem. Lett. 2010, 1, 3451–3458. [Google Scholar] [CrossRef]
- Kauffman, D.R.; Thakkar, J.; Siva, R.; Matranga, C.; Ohodnicki, P.R.; Zeng, C.; Jin, R. Efficient Electrochemical CO2 Conversion Powered by Renewable Energy. ACS Appl. Mater. Interfaces 2015, 7, 15626–15632. [Google Scholar] [CrossRef] [PubMed]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Zhan, Z.; Kobsiriphat, W.; Wilson, J.R.; Pillai, M.; Kim, I.; Barnett, S.A. Syngas Production by Coelectrolysis of CO2/H2O: The Basis for a Renewable Energy Cycle. Energy Fuels 2009, 23, 3089–3096. [Google Scholar] [CrossRef]
- Hu, B.; Guild, C.; Suib, S.L. Thermal, electrochemical, and photochemical conversion of CO2 to fuels and value-added products. J. CO2 Util. 2013, 1, 18–27. [Google Scholar] [CrossRef]
- Lamy, E.; Nadjo, L.; Saveant, J.M. Standard Potential and Kinetic-Parameters of Electrochemical Reduction of Carbon-Dioxide in Dimethylformamide. J. Electroanal. Chem. 1977, 78, 403–407. [Google Scholar] [CrossRef]
- Hoshi, N.; Kato, M.; Hori, Y. Electrochemical reduction of CO2 on single crystal electrodes of silver Ag(111), Ag(100) and Ag(110). J. Electroanal. Chem. 1997, 440, 283–286. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Hatsukade, T.; Cave, E.R.; Abram, D.N.; Kibsgaard, J.; Jaramillo, T.F. Electrocatalytic Conversion of Carbon Dioxide to Methane and Methanol on Transition Metal Surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G.S.; Kimmel, Y.C.; Chen, J.G.; Jiao, F. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 2014, 5, 3242. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, J.; Hadadzadeh, H.; Tavakol, H. Photocatalytic reduction of CO2 to CO by a dinuclear carbonyl polypyridyl rhenium(I) complex. Polyhedron 2014, 78, 112–122. [Google Scholar] [CrossRef]
- Rosen, B.A.; Salehi-Khojin, A.; Thorson, M.R.; Zhu, W.; Whipple, D.T.; Kenis, P.J.A.; Masel, R.I. Ionic Liquid–Mediated Selective Conversion of CO2 to CO at Low Overpotentials. Science 2011, 334, 643. [Google Scholar] [CrossRef] [PubMed]
- Marsh, K.N.; Boxall, J.A.; Lichtenthaler, R. Room temperature ionic liquids and their mixtures—A review. Fluid Phase Equilib. 2004, 219, 93–98. [Google Scholar] [CrossRef]
- Aparicio, S.; Atilhan, M.; Karadas, F. Thermophysical Properties of Pure Ionic Liquids: Review of Present Situation. Ind. Eng. Chem. Res. 2010, 49, 9580–9595. [Google Scholar] [CrossRef]
- Marsh, K.N.; Deev, A.; Wu, A.C.-T.; Tran, E.; Klamt, A. Room temperature ionic liquids as replacements for conventional solvents—A review. Korean J. Chem. Eng. 2002, 19, 357–362. [Google Scholar] [CrossRef]
- Seddon, K.R. Ionic Liquids for Clean Technology. J. Chem. Technol. Biotechnol. 1997, 68, 351–356. [Google Scholar] [CrossRef]
- Armand, M.; Endres, F.; MacFarlane, D.R.; Ohno, H.; Scrosati, B. Ionic-liquid materials for the electrochemical challenges of the future. Nat. Mater. 2009, 8, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Guerra, M.; Albo, J.; Alvarez-Guerra, E.; Irabien, A. Ionic liquids in the electrochemical valorisation of CO2. Energy Environ. Sci. 2015, 8, 2574–2599. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, J.; Zhang, X.; Xin, J.; Miao, Q.; Wang, J. Ionic liquid-based green processes for energy production. Chem. Soc. Rev. 2014, 43, 7838–7869. [Google Scholar] [CrossRef] [PubMed]
- Buzzeo, M.C.; Evans, R.G.; Compton, R.G. Non-Haloaluminate Room-Temperature Ionic Liquids in Electrochemistry—A Review. ChemPhysChem 2004, 5, 1106–1120. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, M.V.; Kornyshev, A.A. Ionic Liquids at Electrified Interfaces. Chem. Rev. 2014, 114, 2978–3036. [Google Scholar] [CrossRef] [PubMed]
- Bazant, M.Z.; Storey, B.D.; Kornyshev, A.A. Double Layer in Ionic Liquids: Overscreening versus Crowding. Phys. Rev. Lett. 2011, 106, 046102. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Pereira, C.M.; Silva, F. Double layer in room temperature ionic liquids: Influence of temperature and ionic size on the differential capacitance and electrocapillary curves. Phys. Chem. Chem. Phys. 2010, 12, 11125–11132. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.M.; Alam, M.T.; Okajima, T.; Ohsaka, T. Electrical Double Layer Structure in Ionic Liquids: An Understanding of the Unusual Capacitance–Potential Curve at a Nonmetallic Electrode. J. Phys. Chem. C 2009, 113, 3386–3389. [Google Scholar] [CrossRef]
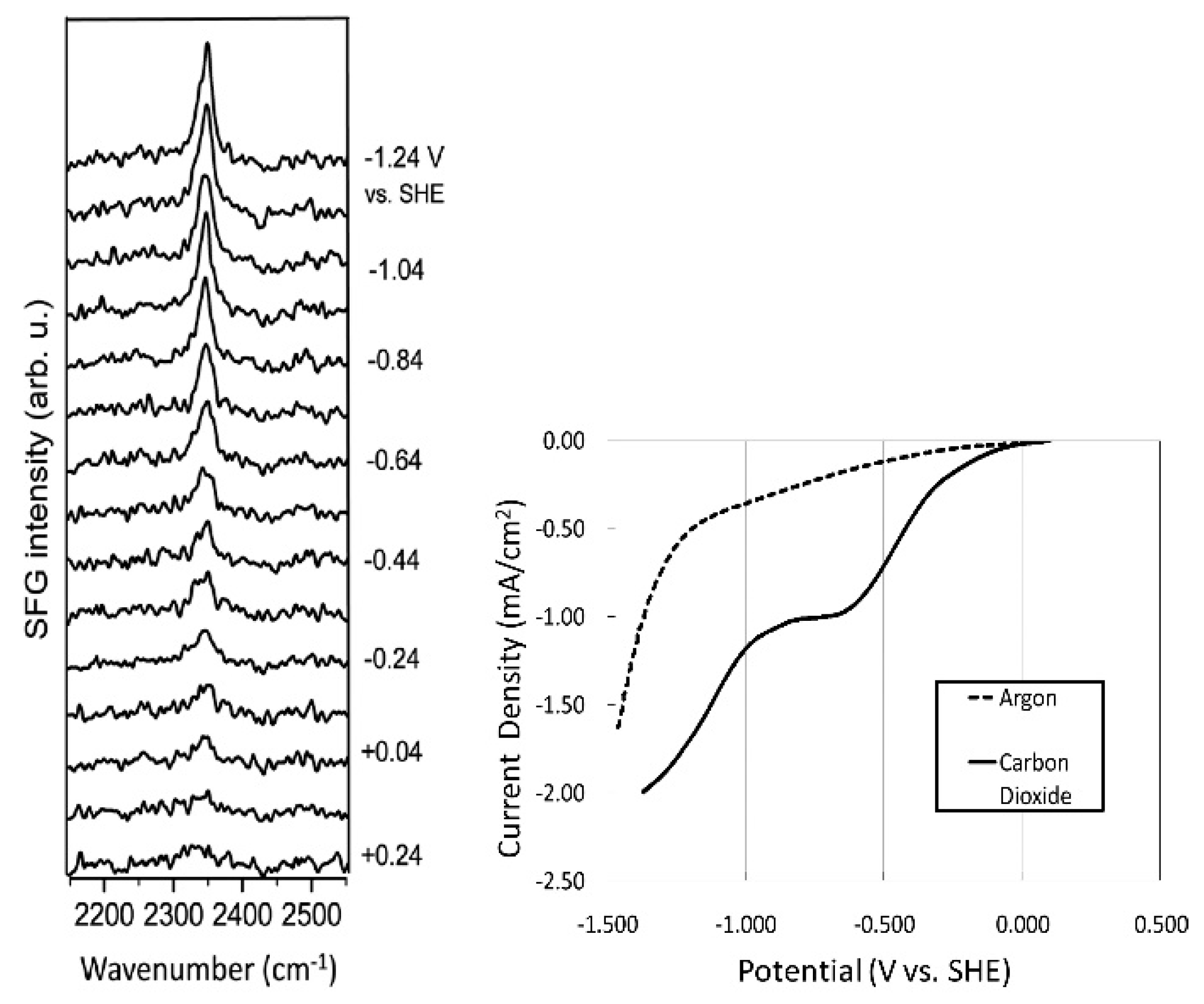
- Rosen, B.A.; Haan, J.L.; Mukherjee, P.; Braunschweig, B.; Zhu, W.; Salehi-Khojin, A.; Dlott, D.D.; Masel, R.I. In Situ Spectroscopic Examination of a Low Overpotential Pathway for Carbon Dioxide Conversion to Carbon Monoxide. J. Phys. Chem. C 2012, 116, 15307–15312. [Google Scholar] [CrossRef]
- Rosen, B.A.; Zhu, W.; Kaul, G.; Salehi-Khojin, A.; Masel, R.I. Water Enhancement of CO2 Conversion on Silver in 1-Ethyl-3-Methylimidazolium Tetrafluoroborate. J. Electrochem. Soc. 2013, 160, H138–H141. [Google Scholar] [CrossRef]
- Verma, S.; Lu, X.; Ma, S.; Masel, R.I.; Kenis, P.J.A. The effect of electrolyte composition on the electroreduction of CO2 to CO on Ag based gas diffusion electrodes. Phys. Chem. Chem. Phys. 2016, 18, 7075–7084. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, S. Probing Electric Fields at the Ionic Liquid–Electrode Interface Using Sum Frequency Generation Spectroscopy and Electrochemistry. J. Phys. Chem. B 2005, 109, 13049–13051. [Google Scholar] [CrossRef] [PubMed]
- Richmond, G.L. Molecular Bonding and Interactions at Aqueous Surfaces as Probed by Vibrational Sum Frequency Spectroscopy. Chem. Rev. 2002, 102, 2693–2724. [Google Scholar] [CrossRef] [PubMed]
- Shultz, M.J.; Schnitzer, C.; Simonelli, D.; Baldelli, S. Sum frequency generation spectroscopy of the aqueous interface: Ionic and soluble molecular solutions. Int. Rev. Phys. Chem. 2000, 19, 123–153. [Google Scholar] [CrossRef]
- Braunschweig, B.; Mukherjee, P.; Haan, J.L.; Dlott, D.D. Vibrational sum-frequency generation study of the CO2 electrochemical reduction at Pt/EMIM-BF4 solid/liquid interfaces. J. Electroanal. Chem. 2016. [Google Scholar] [CrossRef]
- Deng, G.-H.; Li, X.; Liu, S.; Zhang, Z.; Lu, Z.; Guo, Y. Successive Adsorption of Cations and Anions of Water–1-Butyl-3-methylimidazolium Methylsulfate Binary Mixtures at the Air–Liquid Interface Studied by Sum Frequency Generation Vibrational Spectroscopy and Surface Tension Measurements. J. Phys. Chem. C 2016, 120, 12032–12041. [Google Scholar] [CrossRef]
- Rey, N.G.; Dlott, D.D. Structural Transition in an Ionic Liquid Controls CO2 Electrochemical Reduction. J. Phys. Chem. C 2015, 119, 20892–20899. [Google Scholar]
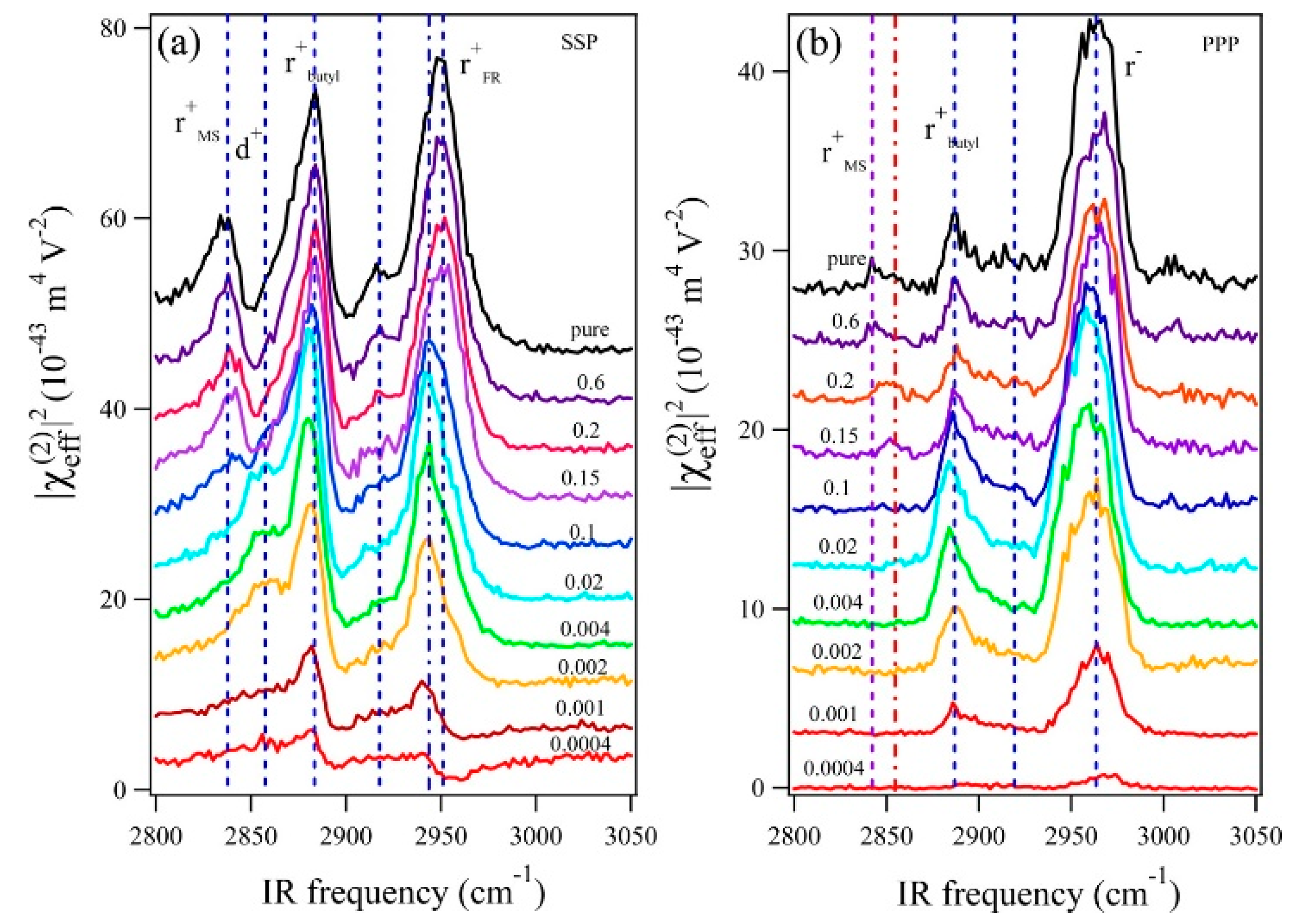
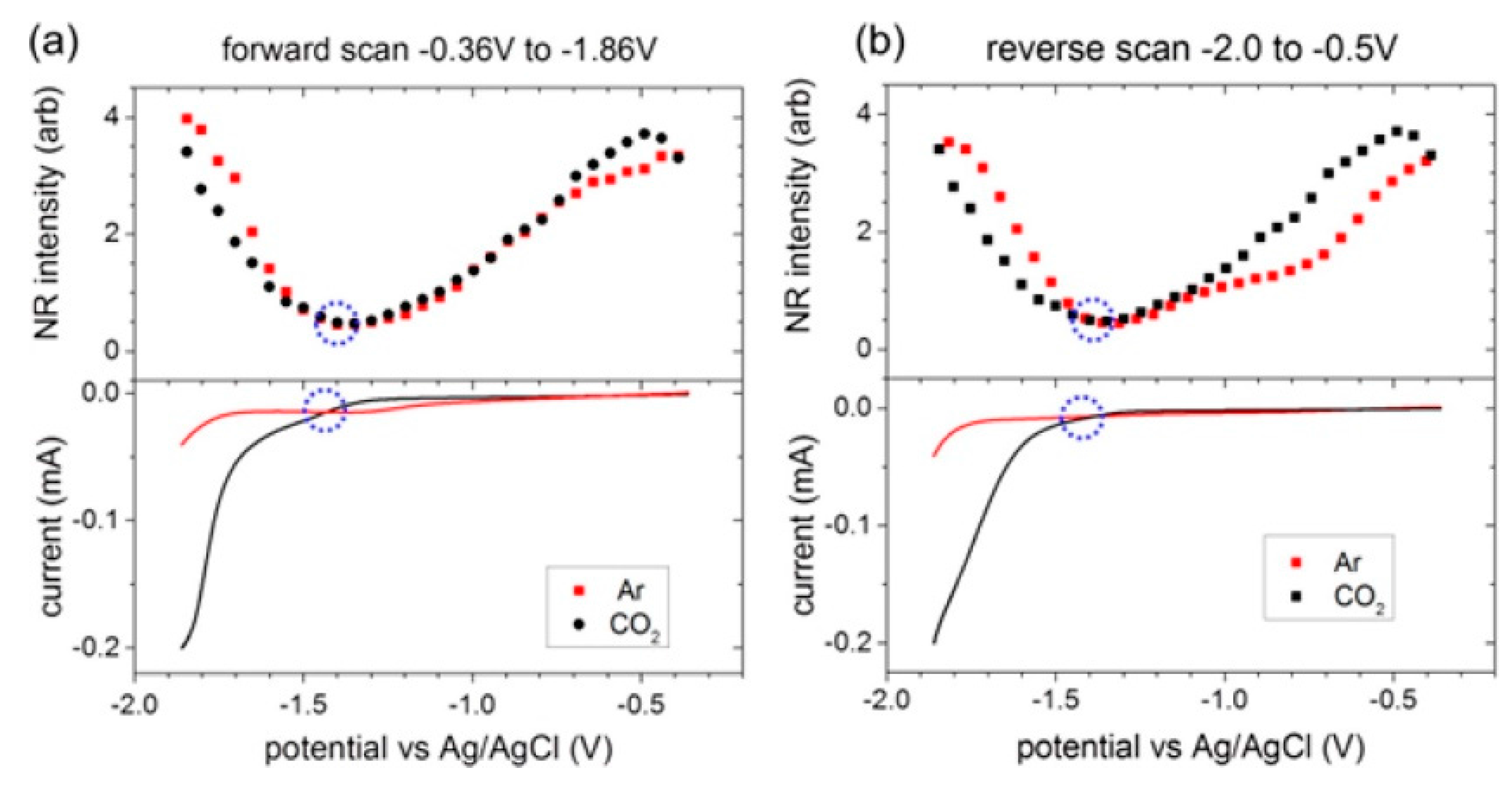
- Motobayashi, K.; Minami, K.; Nishi, N.; Sakka, T.; Osawa, M. Hysteresis of Potential-Dependent Changes in Ion Density and Structure of an Ionic Liquid on a Gold Electrode: In Situ Observation by Surface-Enhanced Infrared Absorption Spectroscopy. J. Phys. Chem. Lett. 2013, 4, 3110–3114. [Google Scholar] [CrossRef]
- Motobayashi, K.; Osawa, M. Potential-dependent condensation of Water at the Interface between ionic liquid [BMIM][TFSA] and an Au electrode. Electrochem. Commun. 2016, 65, 14–17. [Google Scholar] [CrossRef]
- Irvine, J.T.S.; Sinclair, D.C.; West, A.R. Electroceramics: Characterization by Impedance Spectroscopy. Adv. Mater. 1990, 2, 132–138. [Google Scholar] [CrossRef]
- Pajkossy, T. Impedance spectroscopy at interfaces of metals and aqueous solutions — Surface roughness, CPE and related issues. Solid State Ion. 2005, 176, 1997–2003. [Google Scholar] [CrossRef]
- Yang, D.-W.; Li, Q.-Y.; Shen, F.-X.; Wang, Q.; Li, L.; Song, N.; Dai, Y.-N.; Shi, J. Electrochemical Impedance Studies of CO2 Reduction in Ionic Liquid/Organic Solvent Electrolyte on Au Electrode. Electrochim. Acta 2016, 189, 32–37. [Google Scholar] [CrossRef]
- Chen, L.D.; Urushihara, M.; Chan, K.; Nørskov, J.K. Electric Field Effects in Electrochemical CO2 Reduction. ACS Catal. 2016, 6, 7133–7139. [Google Scholar] [CrossRef]
- Dai, C.; Yang, Y.; Fisher, A.; Liu, Z.; Cheng, D. Interaction of CO2 with metal cluster-functionalized ionic liquids. J. CO2 Util. 2016, 16, 257–263. [Google Scholar] [CrossRef]
- Marjolin, A.; Keith, J.A. Thermodynamic Descriptors for Molecules That Catalyze Efficient CO2 Electroreductions. ACS Catal. 2015, 5, 1123–1130. [Google Scholar] [CrossRef]
- Matsubara, Y.; Grills, D.C.; Kuwahara, Y. Thermodynamic Aspects of Electrocatalytic CO2 Reduction in Acetonitrile and with an Ionic Liquid as Solvent or Electrolyte. ACS Catal. 2015, 5, 6440–6452. [Google Scholar] [CrossRef]
- Niu, D.; Wang, H.; Li, H.; Wu, Z.; Zhang, X. Roles of ion pairing on electroreduction of carbon dioxide based on imidazolium-based salts. Electrochim. Acta 2015, 158, 138–142. [Google Scholar] [CrossRef]
- Thorson, M.R.; Siil, K.I.; Kenis, P.J.A. Effect of Cations on the Electrochemical Conversion of CO2 to CO. J. Electrochem. Soc. 2013, 160, F69–F74. [Google Scholar] [CrossRef]
- Urushihara, M.; Chan, K.; Shi, C.; Nørskov, J.K. Theoretical Study of EMIM+ Adsorption on Silver Electrode Surfaces. J. Phys. Chem. C 2015, 119, 20023–20029. [Google Scholar] [CrossRef]
- Wang, Y.; Hatakeyama, M.; Ogata, K.; Wakabayashi, M.; Jin, F.; Nakamura, S. Activation of CO2 by ionic liquid EMIM–BF4 in the electrochemical system: A theoretical study. Phys. Chem. Chem. Phys. 2015, 17, 23521–23531. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Xing, H.; Bao, Z.; Su, B.; Zhang, Z.; Yang, Y.; Dai, S.; Ren, Q. One of the Distinctive Properties of Ionic Liquids over Molecular Solvents and Inorganic Salts: Enhanced Basicity Stemming from the Electrostatic Environment and “Free” Microstructure. J. Phys. Chem. B 2014, 118, 3682–3688. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pang, Y.; Zhang, B.; de Luna, P.; Voznyy, O.; Xu, J.; Zheng, X.; Dinh, C.T.; Fan, F.; Cao, C.; et al. Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature 2016, 537, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Cabaço, M.I.; Besnard, M.; Danten, Y.; Coutinho, J.A.P. Carbon Dioxide in 1-Butyl-3-methylimidazolium Acetate. I. Unusual Solubility Investigated by Raman Spectroscopy and DFT Calculations. J. Phys. Chem. A 2012, 116, 1605–1620. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ramesha, G.K.; Kamat, P.V.; Brennecke, J.F. Switching the Reaction Course of Electrochemical CO2 Reduction with Ionic Liquids. Langmuir 2014, 30, 6302–6308. [Google Scholar] [CrossRef] [PubMed]
- Tanner, E.E.L.; Batchelor-McAuley, C.; Compton, R.G. Carbon Dioxide Reduction in Room-Temperature Ionic Liquids: The Effect of the Choice of Electrode Material, Cation, and Anion. J. Phys. Chem. C 2016, 120, 26442–26447. [Google Scholar] [CrossRef]
- Cadena, C.; Anthony, J.L.; Shah, J.K.; Morrow, T.I.; Brennecke, J.F.; Maginn, E.J. Why Is CO2 So Soluble in Imidazolium-Based Ionic Liquids? J. Am. Chem. Soc. 2004, 126, 5300–5308. [Google Scholar] [CrossRef] [PubMed]
- Muldoon, M.J.; Aki, S.N.V.K.; Anderson, J.L.; Dixon, J.K.; Brennecke, J.F. Improving Carbon Dioxide Solubility in Ionic Liquids. J. Phys. Chem. B 2007, 111, 9001–9009. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.-F.; Horne, M.; Bond, A.M.; Zhang, J. Is the Imidazolium Cation a Unique Promoter for Electrocatalytic Reduction of Carbon Dioxide? J. Phys. Chem. C 2016, 120, 23989–24001. [Google Scholar] [CrossRef]
- Lau, G.P.S.; Schreier, M.; Vasilyev, D.; Scopelliti, R.; Grätzel, M.; Dyson, P.J. New Insights Into the Role of Imidazolium-Based Promoters for the Electroreduction of CO2 on a Silver Electrode. J. Am. Chem. Soc. 2016, 138, 7820–7823. [Google Scholar] [CrossRef]












© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, H.-K.; Kim, H. The Mechanism of Room-Temperature Ionic-Liquid-Based Electrochemical CO2 Reduction: A Review. Molecules 2017, 22, 536. https://doi.org/10.3390/molecules22040536
Lim H-K, Kim H. The Mechanism of Room-Temperature Ionic-Liquid-Based Electrochemical CO2 Reduction: A Review. Molecules. 2017; 22(4):536. https://doi.org/10.3390/molecules22040536
Chicago/Turabian StyleLim, Hyung-Kyu, and Hyungjun Kim. 2017. "The Mechanism of Room-Temperature Ionic-Liquid-Based Electrochemical CO2 Reduction: A Review" Molecules 22, no. 4: 536. https://doi.org/10.3390/molecules22040536
APA StyleLim, H. -K., & Kim, H. (2017). The Mechanism of Room-Temperature Ionic-Liquid-Based Electrochemical CO2 Reduction: A Review. Molecules, 22(4), 536. https://doi.org/10.3390/molecules22040536