
Synthesis and Spectral Properties of meso-Arylbacteriochlorins, Including Insights into Essential Motifs of their Hydrodipyrrin Precursors


Abstract
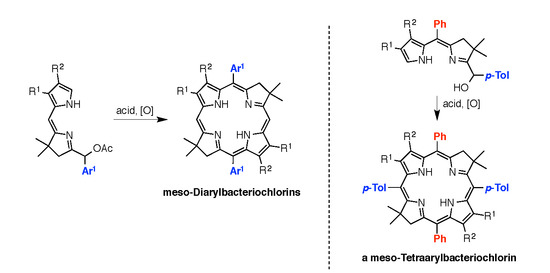
:
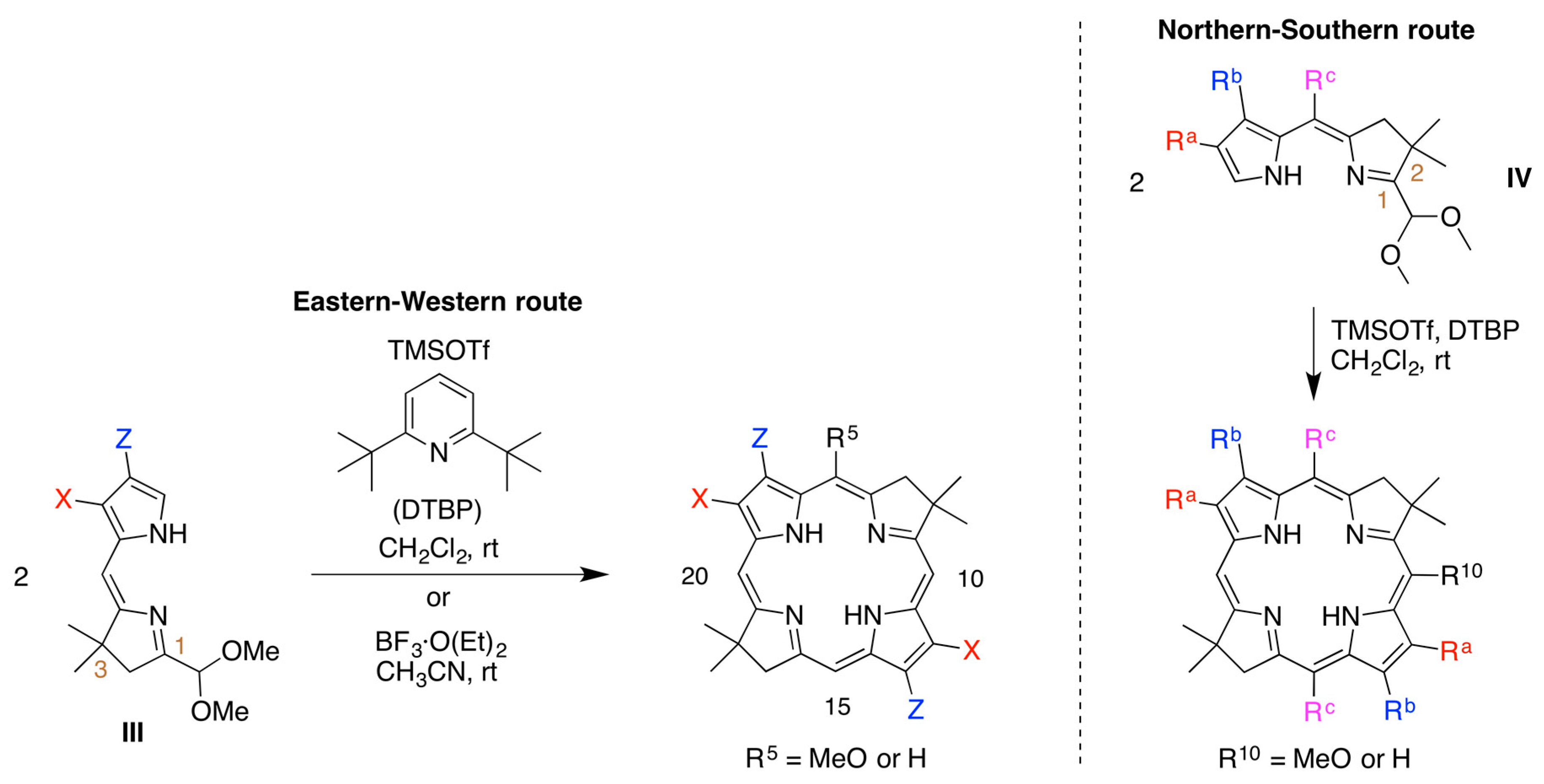
1. Introduction
2. Results
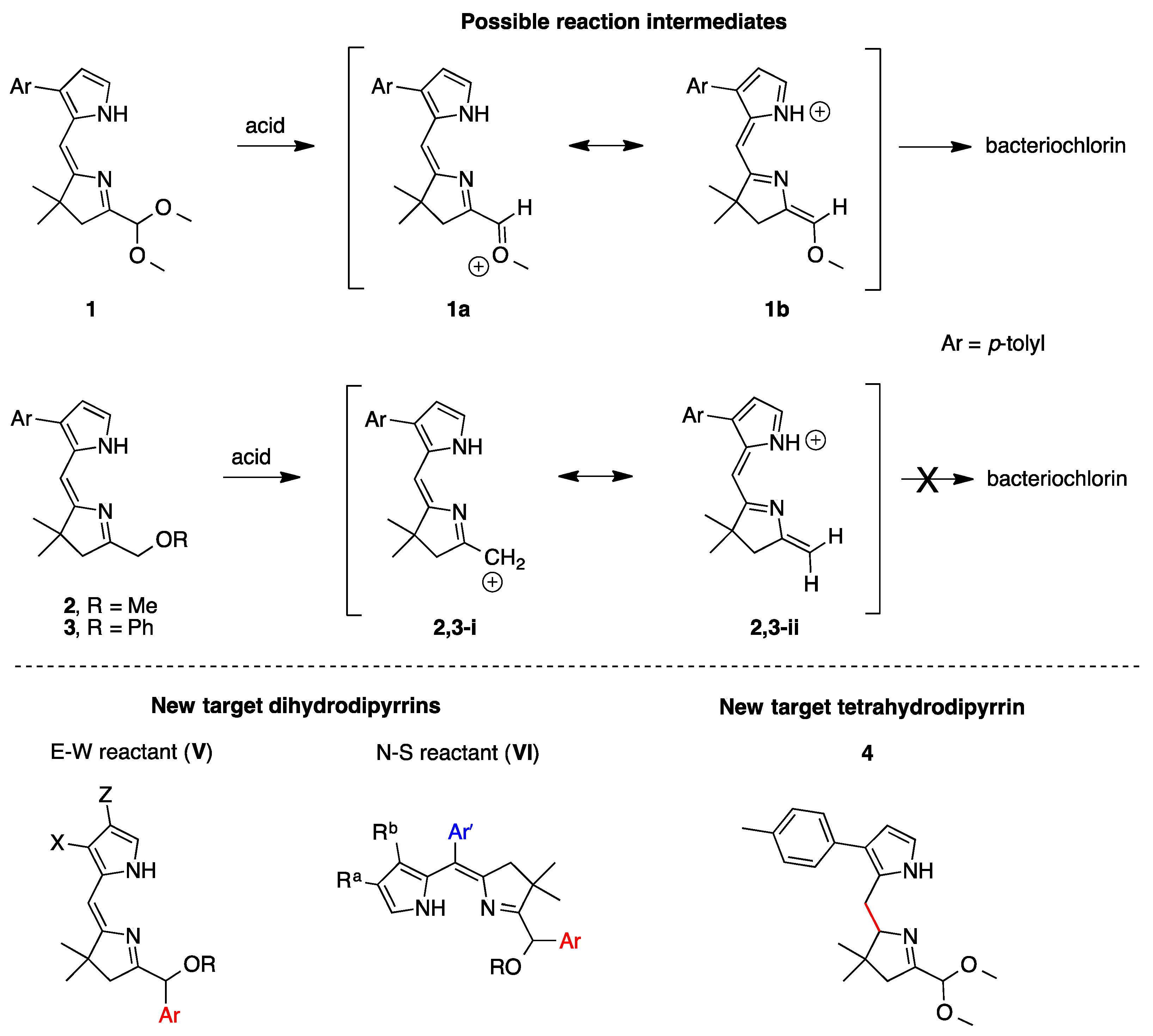
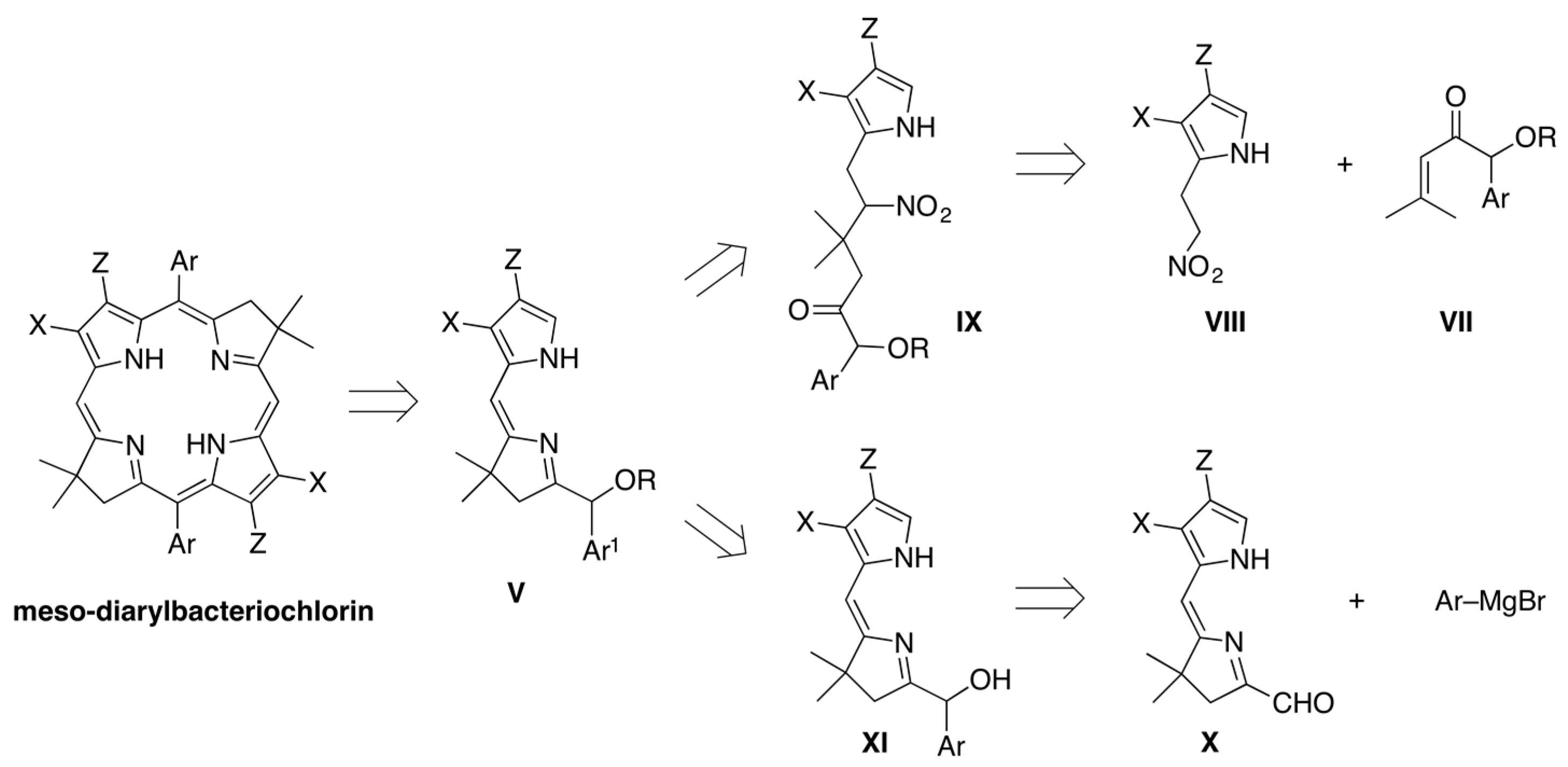
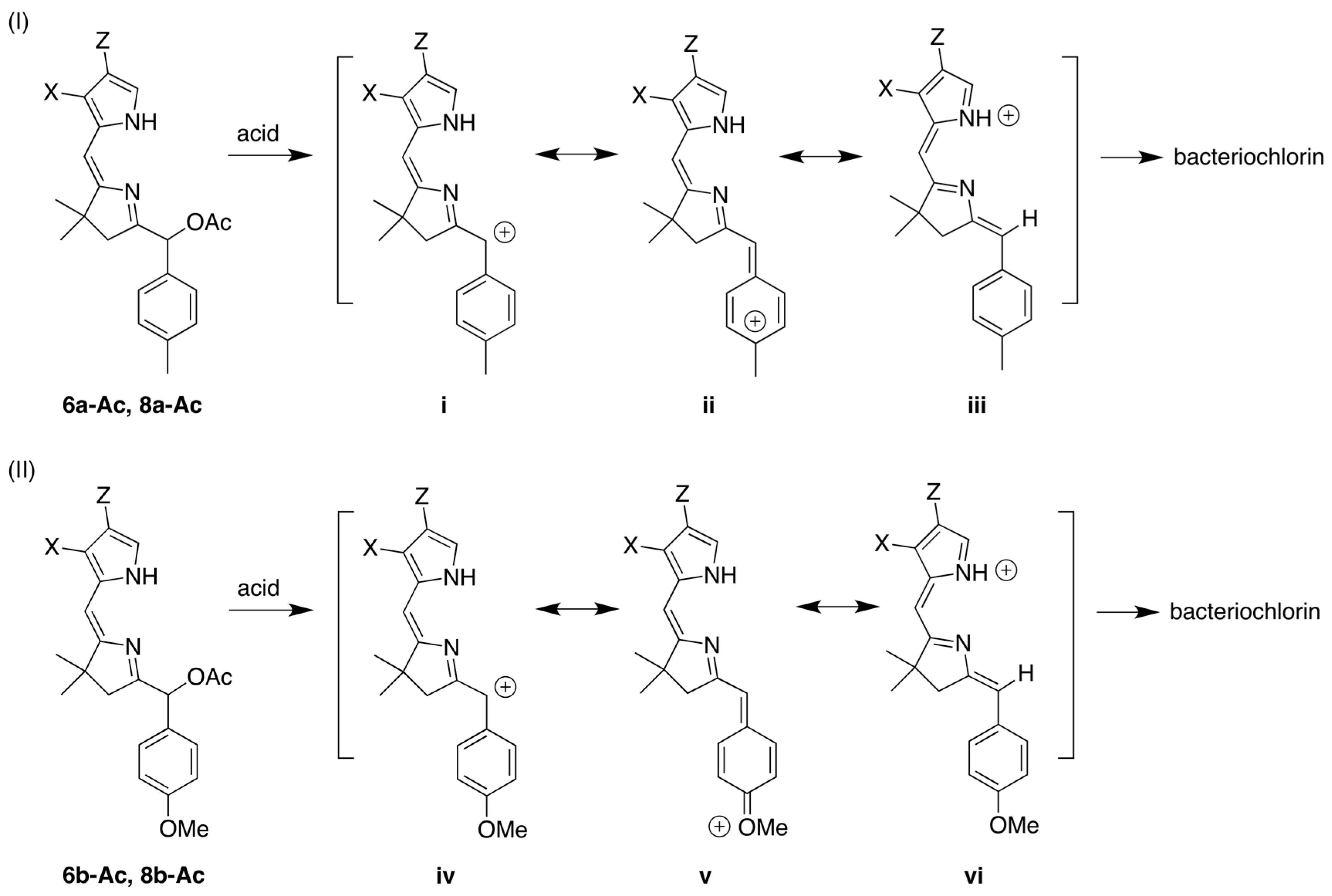
2.1. Reconnaissance
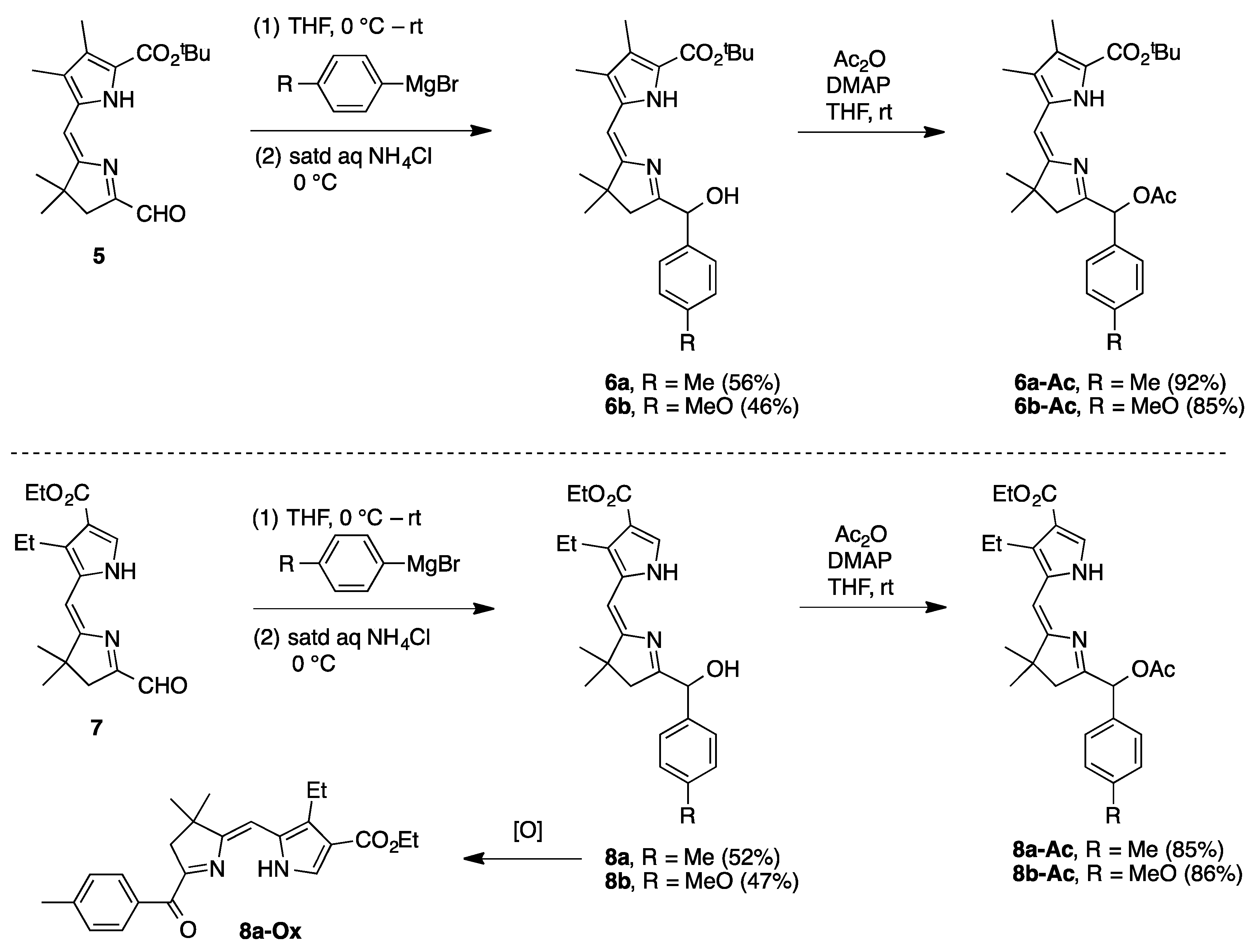
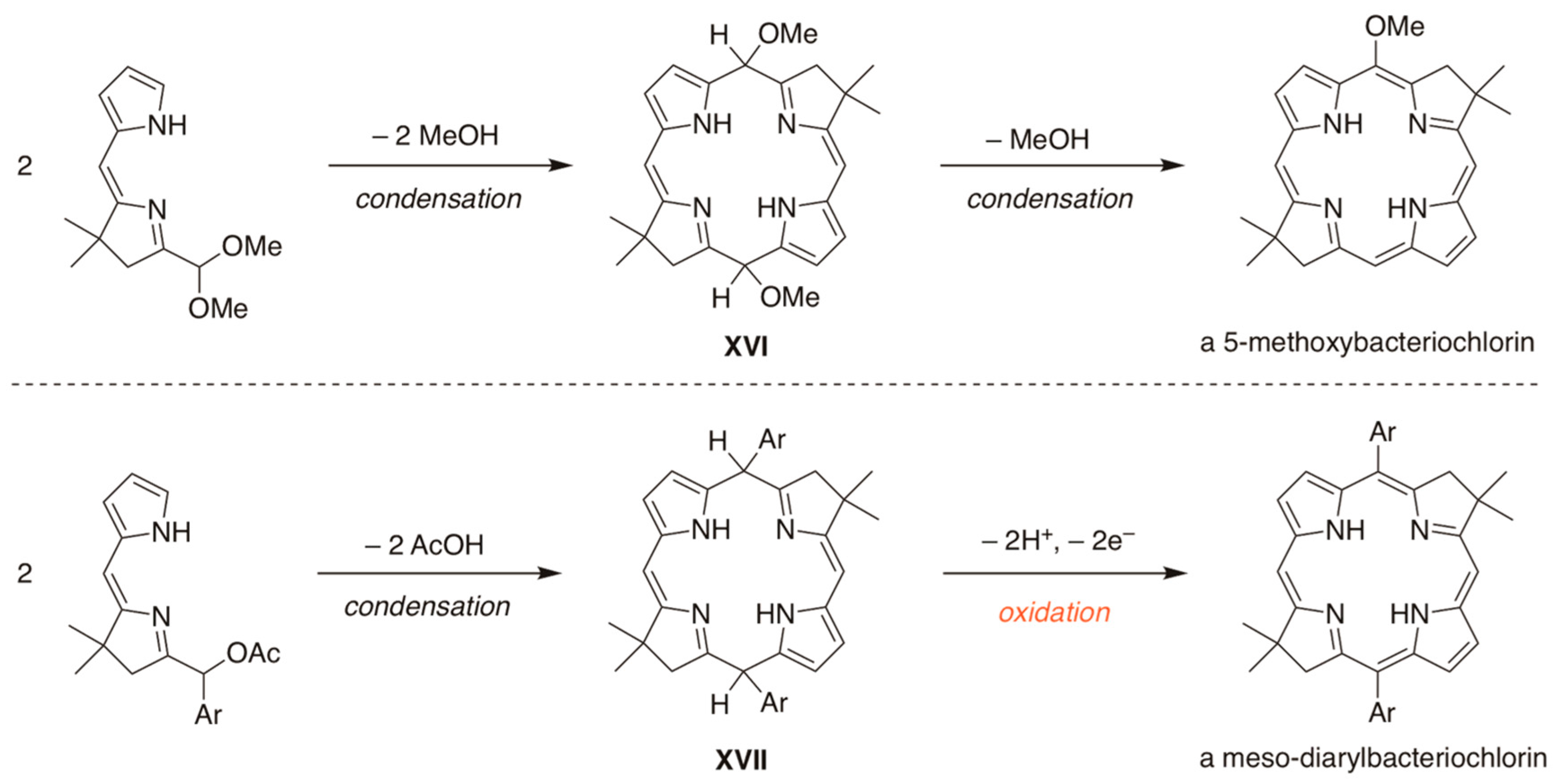
2.2. Synthesis of Dihydrodipyrrin–Carbinols and Dihydrodipyrrin–Acetates
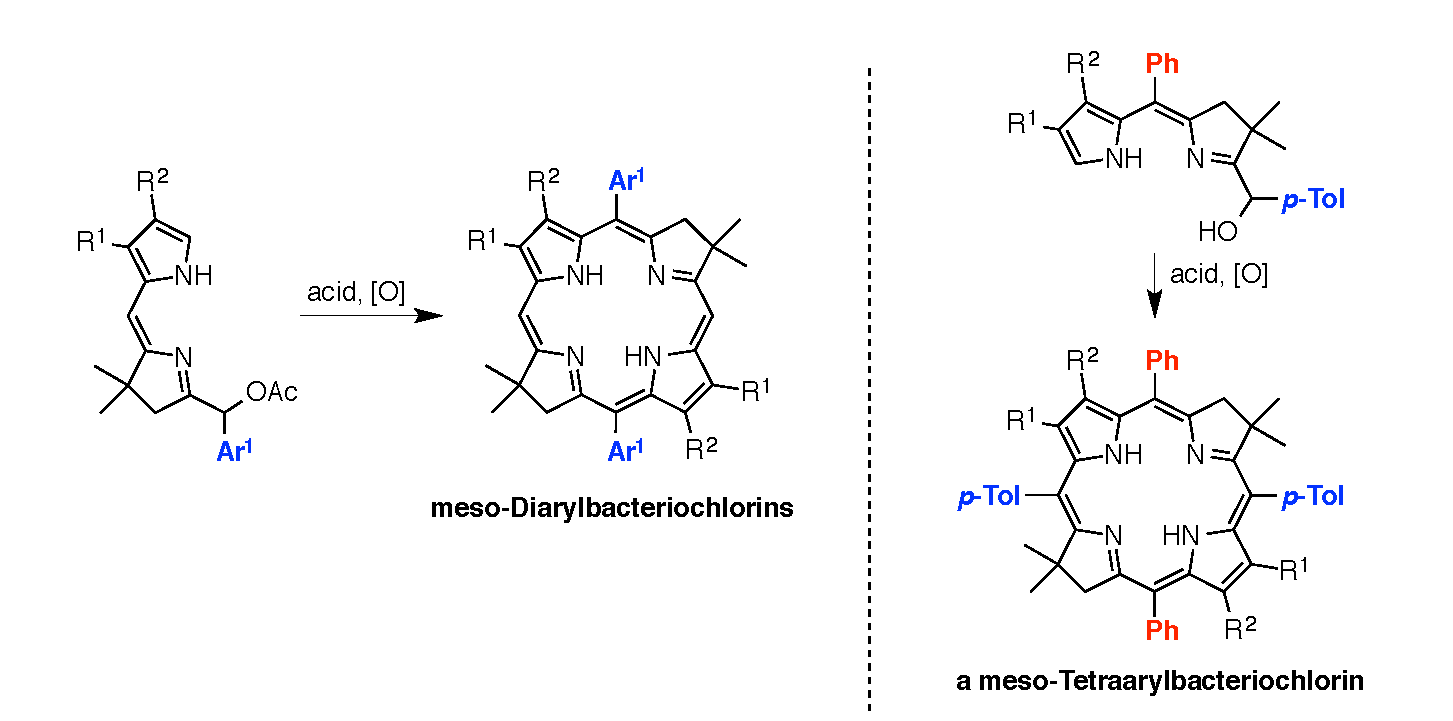
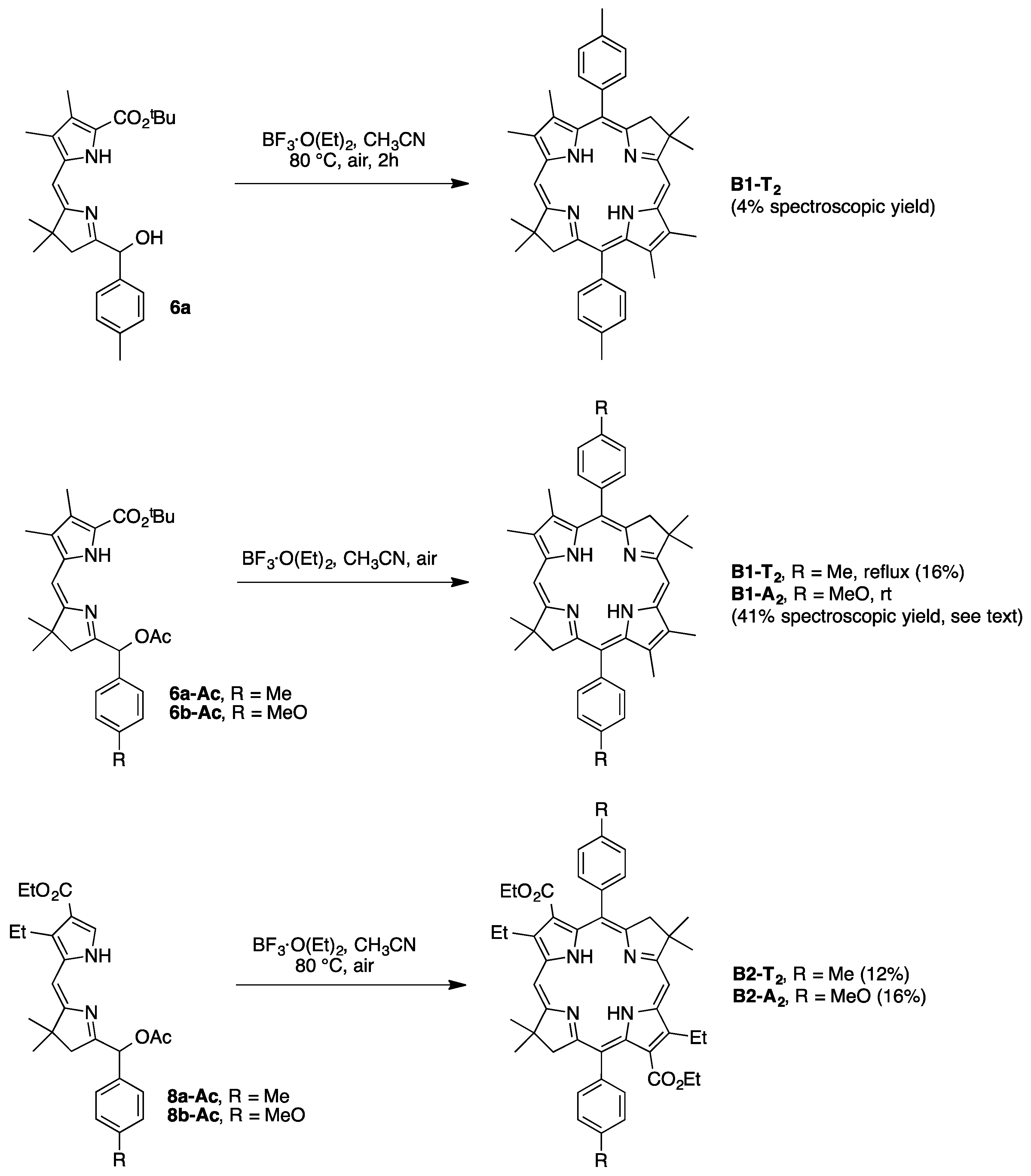
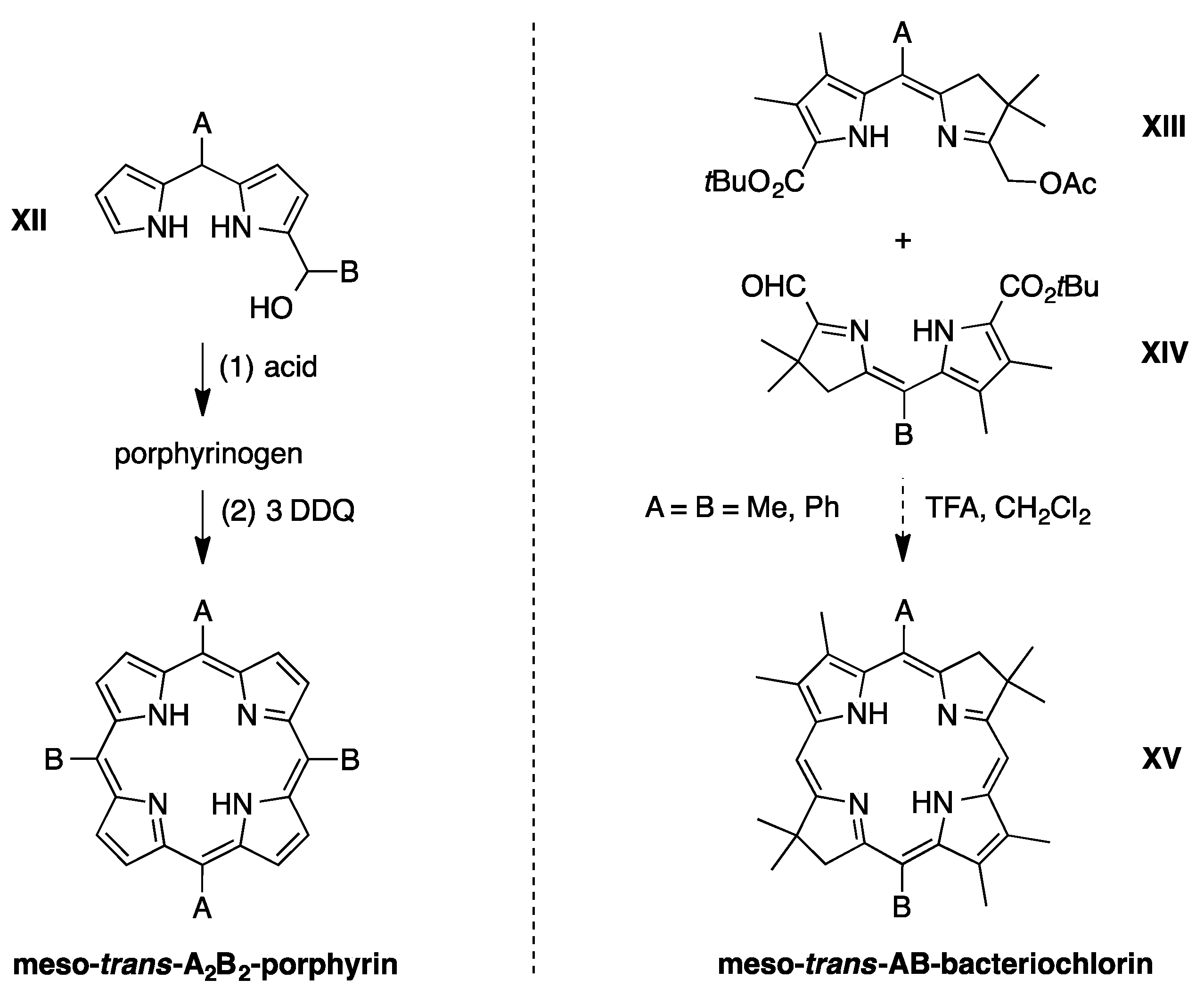
2.3. Synthesis of Meso-Diarylbacteriochlorins (via E-W Route)
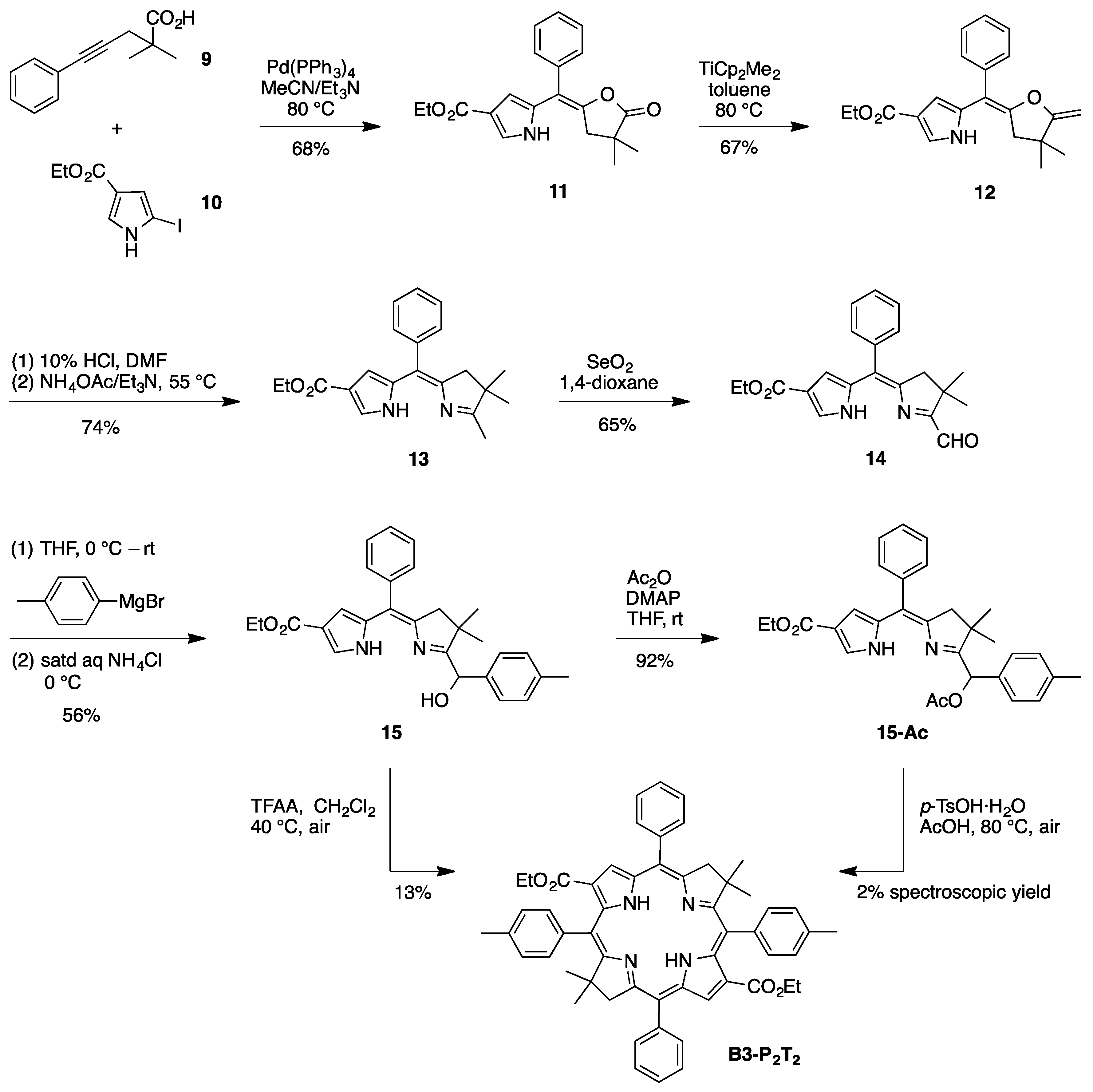
2.4. Synthesis of a Meso-Tetraarylbacteriochlorin (via the N-S Route)
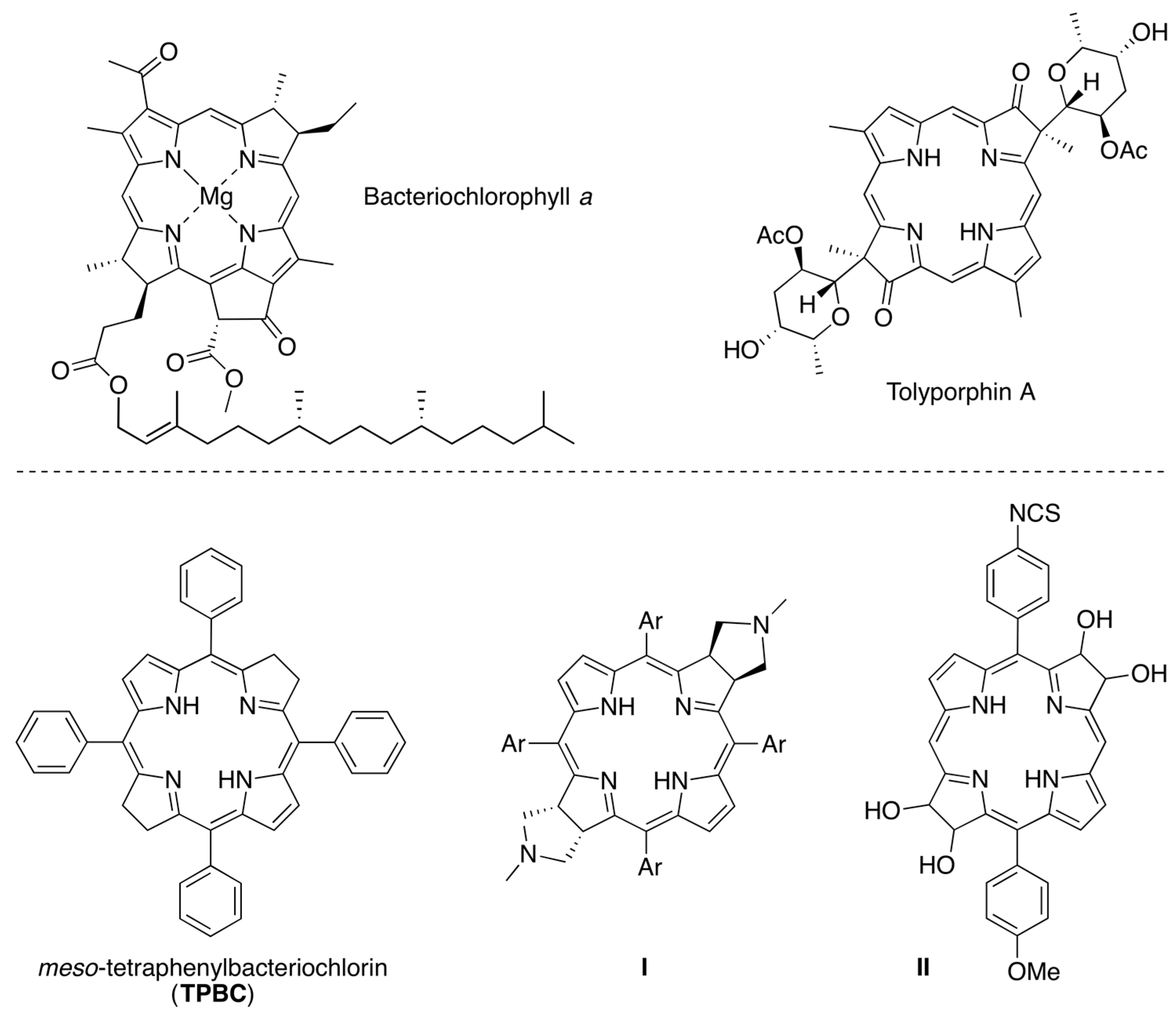
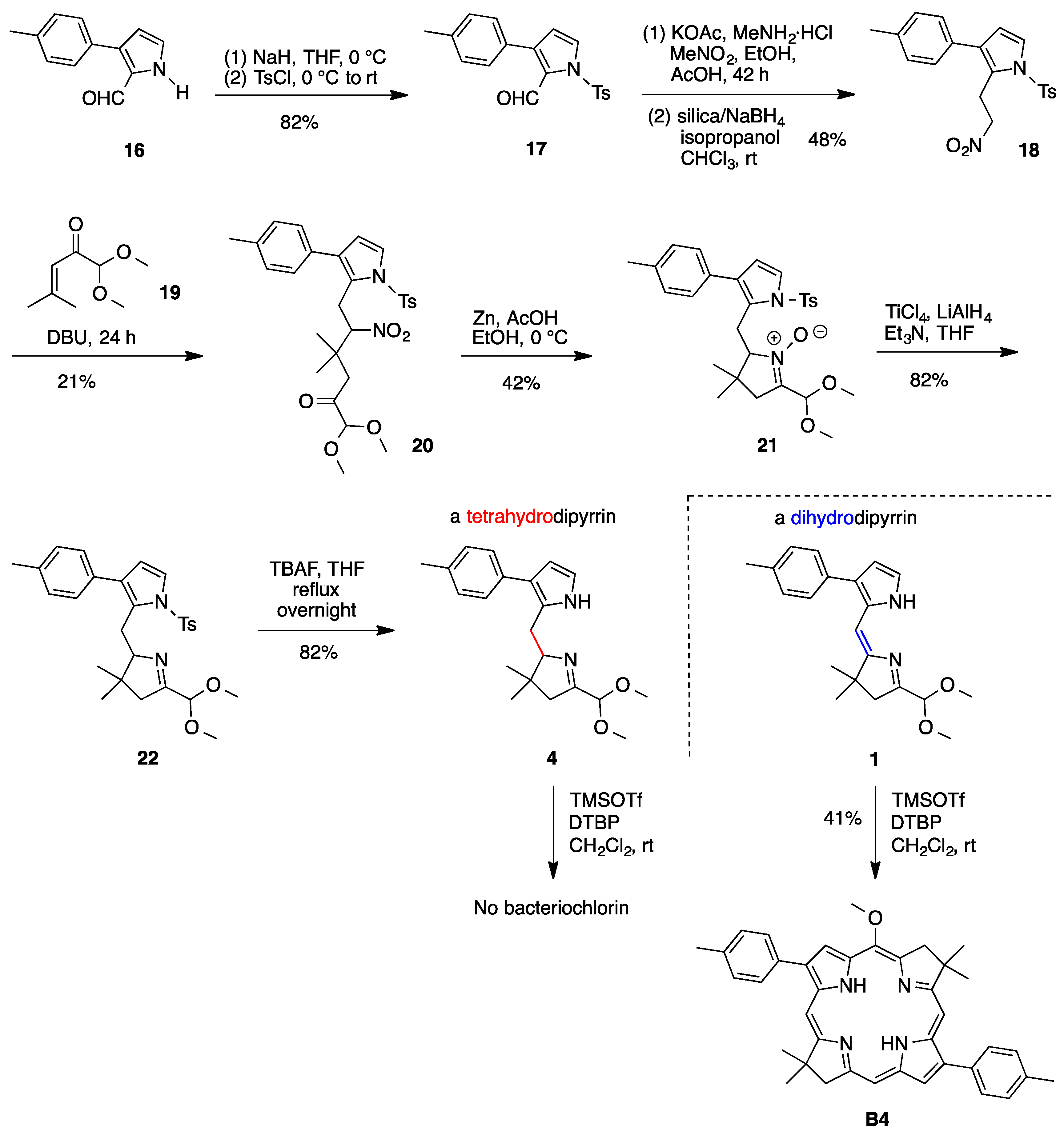
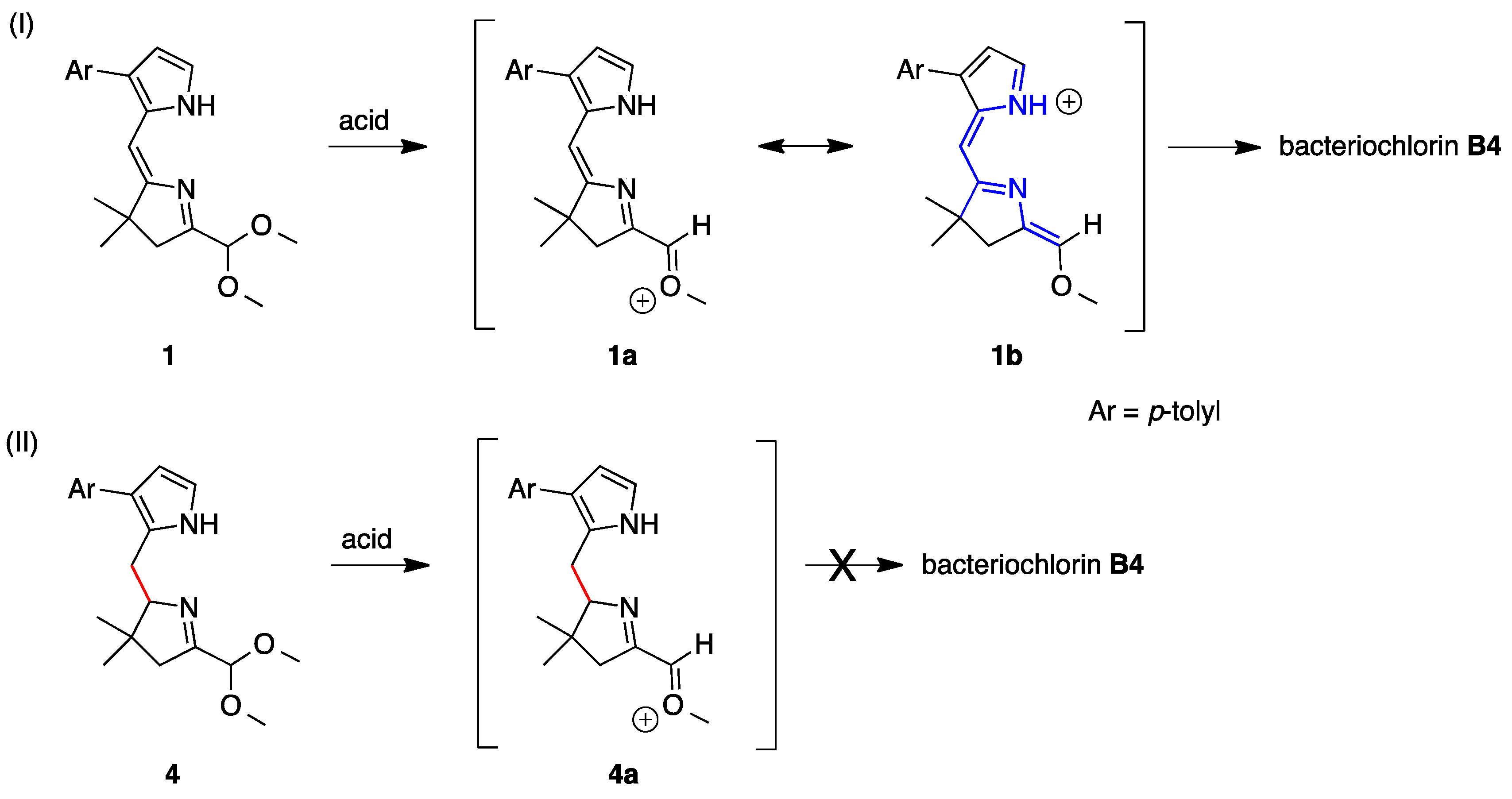
2.5. A Tetrahydrodipyrrin for Bacteriochlorin Formation
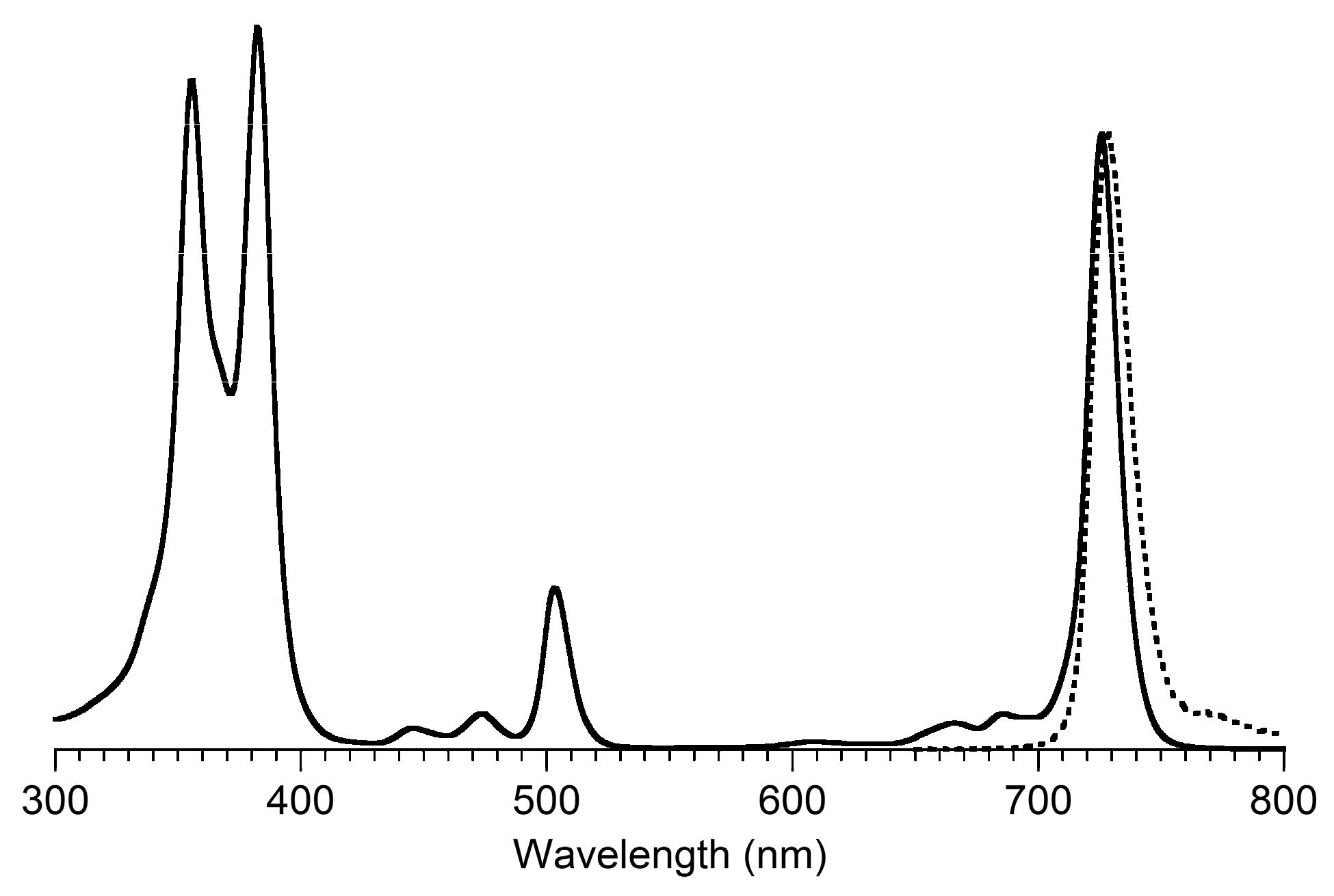
2.6. Spectroscopic Properties
3. Discussion
3.1. Essential Motifs
3.2. Comparison of Routes
3.3. Dehydrogenation
3.4. Meso-Arylbacteriochlorins
3.5. Spectroscopic Properties
- (1)
- (2)
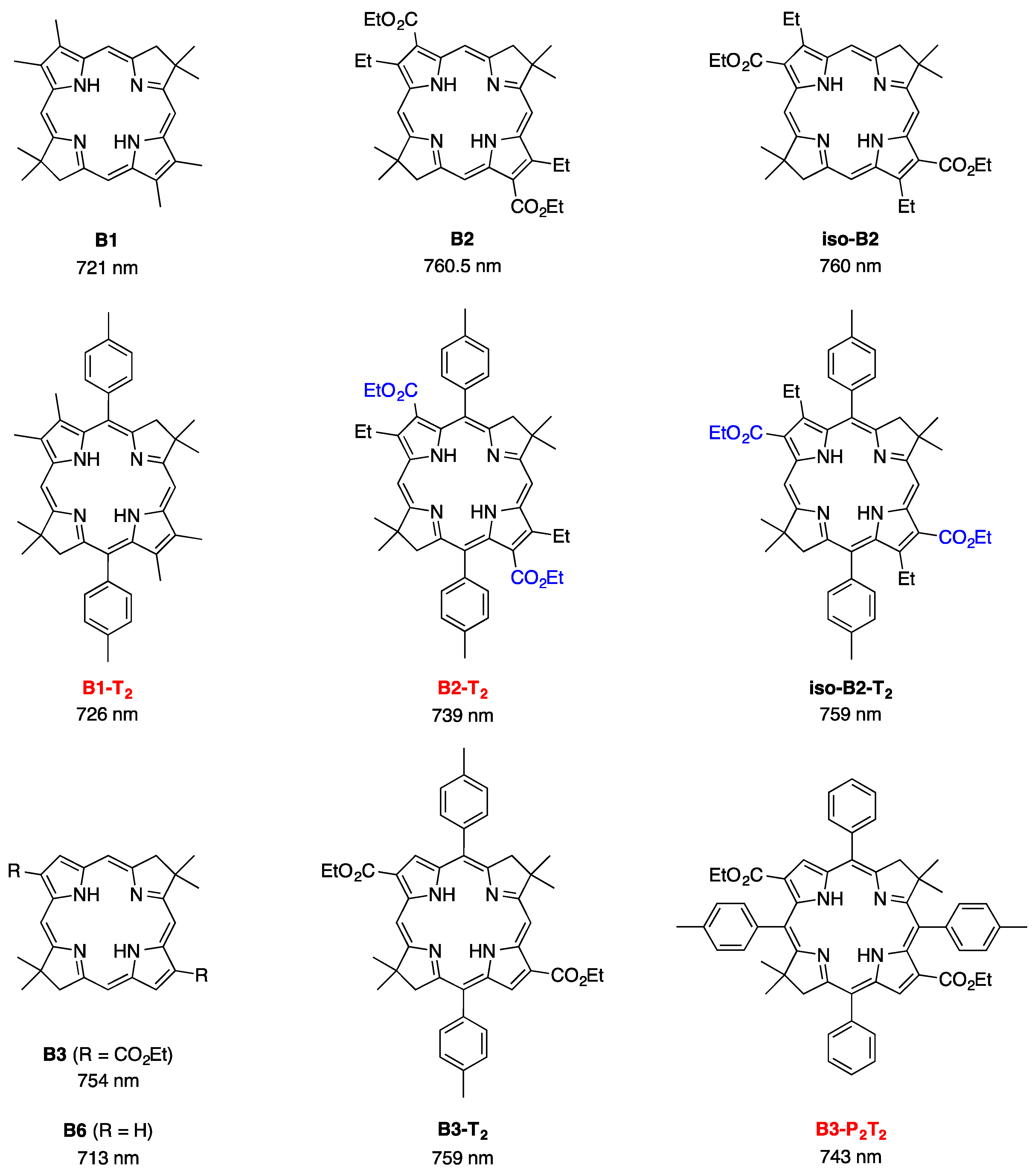
- Bacteriochlorins B2 and iso-B2 are positional isomers (due to swapping the positions of the β-ethyl and β-carbethoxy groups).
- (3)
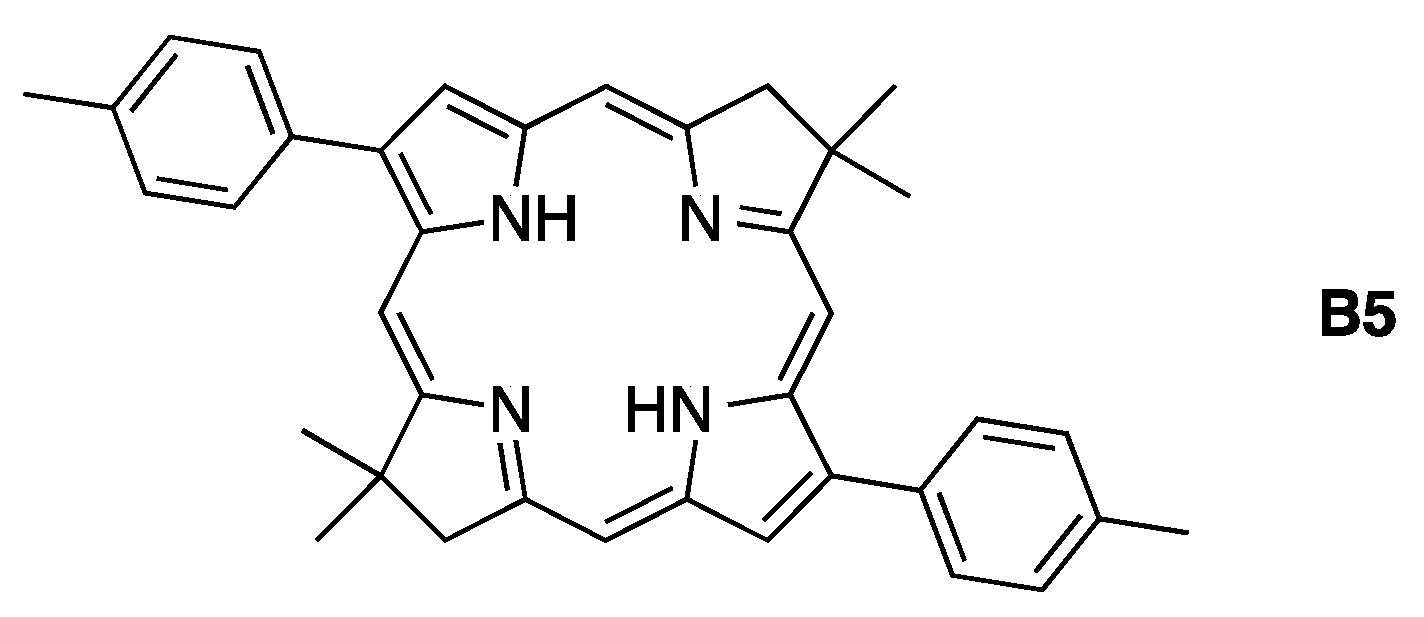
- Bacteriochlorins in the second row (B1-T2, B2-T2, and iso-B2-T2 [19]) differ from the first row (B1, B2, iso-B2) in the presence of two meso-aryl groups.
- (4)
- Bacteriochlorins in the third row (B3, B3-T2 [19], and B3-P2T2) each contain two β-carbethoxy groups but differ in the number (0, 2, 4) of meso-aryl groups.
- (1)
- Bacteriochlorin positional isomers B2 and iso-B2 exhibit absorption and fluorescence spectral properties that are nearly identical with each other.
- (2)
- β-Carbethoxy groups are established auxochromes in bacteriochlorins [53]. The presence of two β-carbethoxy groups in B3 (754 nm) causes a bathochromic shift of the Qy absorption band by 41 nm compared to that of the unsubstituted benchmark bacteriochlorin B6 (713 nm).
- (3)
- Aryl groups also serve as auxochromes in bacteriochlorins upon incorporation at the meso- or β-positions. Two β-aryl groups (2,12- or 3,13-diarylbacteriochlorins) cause a bathochromic shift of the Qy absorption band by ~23 nm relative to that of B6 [30], whereas a single meso-aryl group typically causes a bathochromic shift of the Qy absorption band by 3 to 6 nm [29].
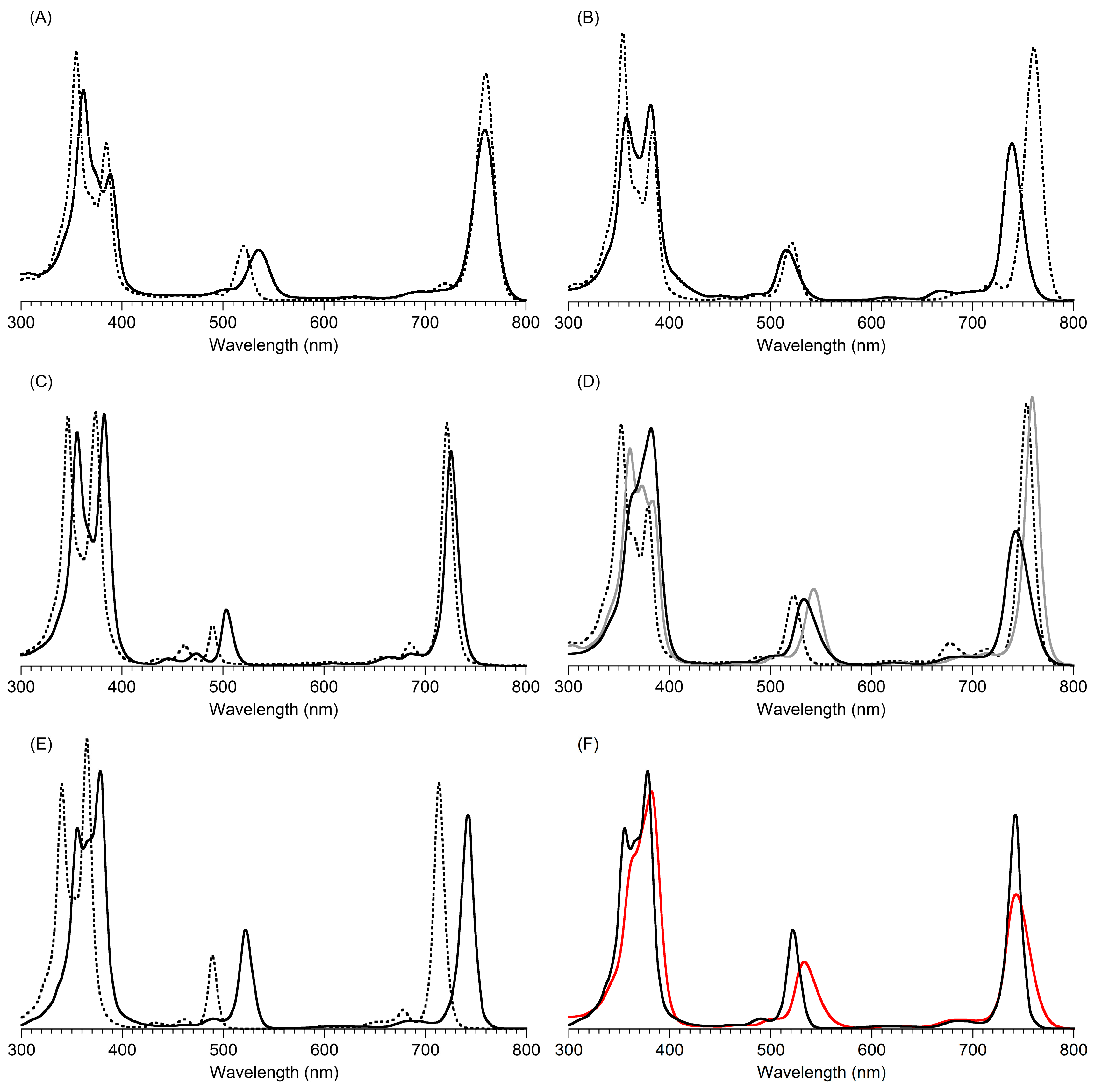
- (1)
- The position of the Qy absorption band is nearly unchanged, while a hypochromic change is observed together with an increase in the fwhm by 5 nm for iso-B2-T2.
- (2)
- The By, Bx, and Qx absorption bands exhibit bathochromic shifts (+7, +4, and +14 nm, respectively) for iso-B2-T2. At odds with this observation, the absorption spectra of B2 and B2-T2 are distinct from each other (Figure 5, Panel B). Compared to the benchmark bacteriochlorin B2, B2-T2 shows a large (21 nm) hypsochromic shift of the Qy absorption band, together with a large hypochromic change (~40% decline), while the fwhm is slightly increased. The positions of the By, Bx, and Qx absorption bands of B2 and B2-T2 are relatively unchanged.
- (3)
- The contrasting results for the meso-di-p-tolylbacteriochlorins B2-T2 and iso-B2-T2 are quite surprising, since the two are positional isomers, where the only structural difference is the swapped position of the β-ethyl and β-carbethoxy groups. Reasonable expectations are that the absorption spectra of B2-T2 and iso-B2-T2 should be nearly identical, as shown for their respective non-aryl analogues B2 and iso-B2. The origin of the hypsochromic shift of the Qy absorption band due to pairwise juxtaposition of a β-carbethoxy group and a meso-aryl substituent remains unclear.
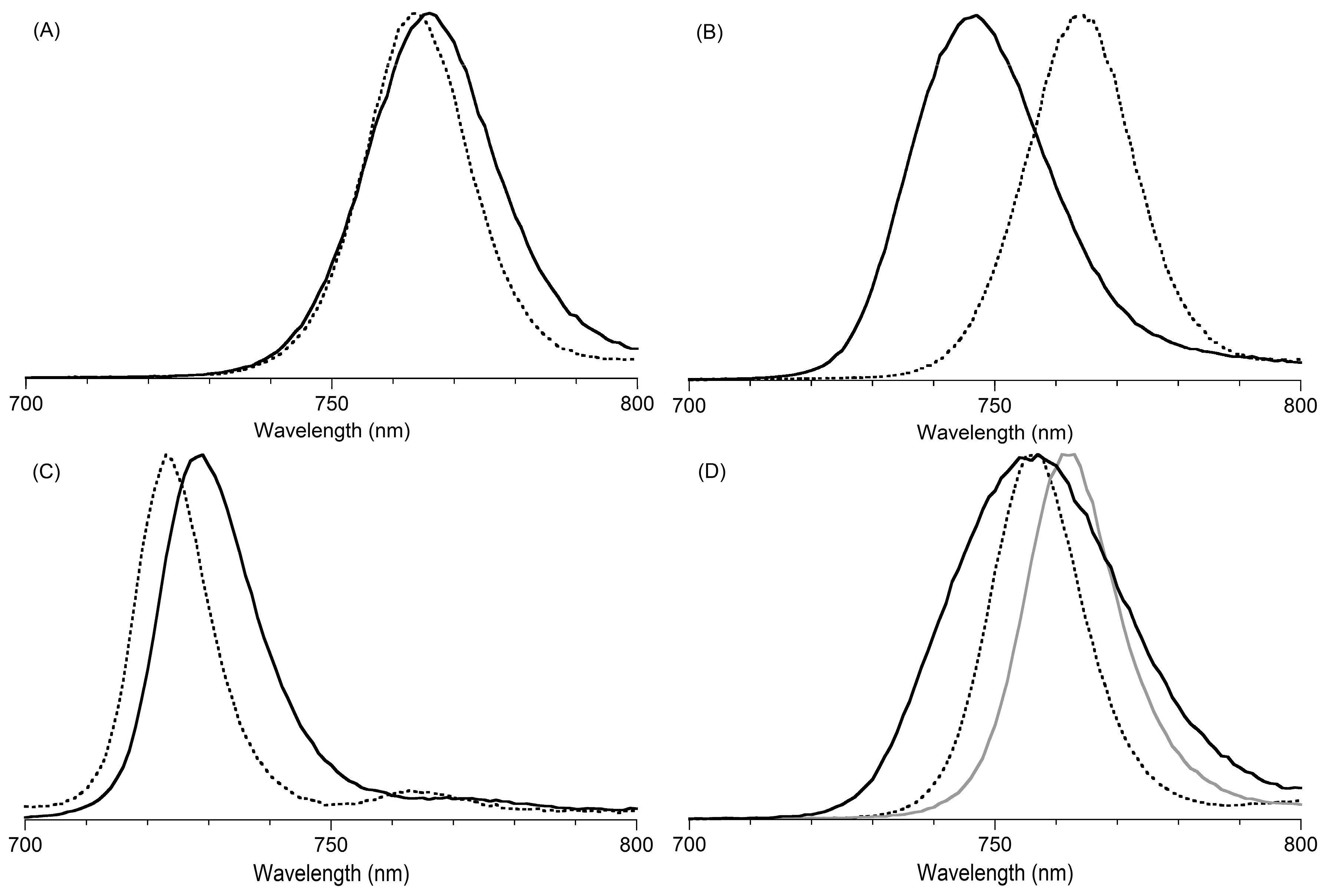
- (1)
- The difference in position of the Qy fluorescence band versus that of the benchmarks (Δ) corresponds very well to the difference of the Qy absorption band compared to that of benchmarks (Δ), except for that of B3-P2T2. The position of the Qy fluorescence band of B3-P2T2 was unchanged compared to that of benchmark B3, which results in a large Stokes shift (vide infra).
- (2)
- The Stokes shift of each benchmark bacteriochlorin lacking meso-aryl substituents is small (<70 cm−1 (<4 nm)). On the other hand, meso-arylbacteriochlorins exhibit larger Stokes shifts (>120 cm−1 (>7 nm)), and B3-P2T2 shows the largest Stokes shift (250 cm−1 (12 nm)).
- (3)
- The presence of meso-aryl substituents causes broadening of the fluorescence spectra. Such an effect is most pronounced for B3-P2T2, where the fwhm has almost doubled (34 nm, +16 nm) compared to that of benchmark bacteriochlorin B3 (18 nm).
- (4)
- The fluorescence quantum yield (Φf) of meso-arylbacteriochlorins is increased (1.2–1.6 times) compared to that of the benchmarks lacking meso-aryl substituents.
4. Materials and Methods
4.1. General Methods
4.2. Self-Condensation Study
4.3. Synthesis and Characterization
4.4. Plotting Absorption Spectra
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
Appendix A.1. Acid Survey for the Self-Condensation of 6a and 6a-Ac

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Reactant (mM) | Acid (mM) | Solvent | Oxidant | Conditions | Yield (%) b |
|---|---|---|---|---|---|---|
| 1 | 6a [18] | BF3·O(Et)2 [140] | CH3CN | air | r.t., 16 h | <1 |
| 2 | 6a [18] | BF3·O(Et)2 [140] | CH3CN | air | reflux, 2 h | 4.3 |
| 3 | 6a-Ac [18] | Neat TFA c | – | air | r.t., 21 h | 0.6 |
| 4 | 6a-Ac [18] | 50% (v/v) TFA c | CH2Cl2 | air | r.t., 21 h | 1.1 |
| 5 | 6a-Ac [18] | TMSOTf [72], DTBP [144] | CH2Cl2 | air | r.t., 4 h d | 0.9 |
| 6 | 6a-Ac [18] | BF3·O(Et)2 [70] | CH3CN | air | r.t., 21 h | 13 |
| 7 | 6a-Ac [18] | BF3·O(Et)2 [140] | CH3CN | air | r.t., 21 h | 17 |
| 8 | 6a-Ac [18] | BF3·O(Et)2 [280] | CH3CN | air | r.t., 21 h | 12 |
| 9 | 6a-Ac [18] | BF3·O(Et)2 [560] | CH3CN | air | r.t., 21 h | 4.4 |
| 10 | 6a-Ac [5] | BF3·O(Et)2 [50] | CH3CN | air | r.t., 21 h | 4.7 |
| 11 | 6a-Ac [18] | BF3·O(Et)2 [140] | CH3CN | air | reflux, 2 h | 22 |
| 12 | 6a-Ac [18] | BF3·O(Et)2 [140] | CH3CN | air e | r.t., 21 h | 12 |
| 13 | 6a-Ac [18] | BF3·O(Et)2 [140] | CH3CN | air e | reflux, 2 h | 21 |
| 14 | 6a-Ac [18] | BF3·O(Et)2 [140] | PhNO2 | air | reflux, 2 h | 7.1 |
| 15 | 6a-Ac [18] | BF3·O(Et)2 [140] | CH3CN | O2 | reflux, 2 h | 8.6 |
| 16 | 6a-Ac [18] | Neat TFA | – | DDQ | r.t., 1 h | 7.0 f |
Appendix A.2. Acid Survey for the Self-Condensation of 15-Ac
| Entry | Acid (mM) | Solvent | Oxidant | Conditions | Yield (%) b |
|---|---|---|---|---|---|
| 1 | BF3·O(Et)2 [140] | CH3CN | air | r.t., 24 h | – c |
| 2 | BF3·O(Et)2 [140] | CH3CN | air | 80 °C, 2 h | TDC d |
| 3 | BF3·O(Et)2 [560] | CH3CN | air | 80 °C, 10 min | TDC d |
| 4 | Neat BF3·O(Et)2 | – | air | r.t., 19 h | TDC d |
| 5 | Neat TFA | – | air | r.t., 19 h | – e |
| 6 | TMSOTf [72], DTBP [144] | CH2Cl2 | air | r.t., 19 h | – e |
| 7 | TMSOTf [72], DTBP [144] | DCE | air | 80 °C, 19 h | – c |
| 8 | InCl3 [140] | Toluene | air | 80 °C, 2 h | – c |
| 9 | Bi(OTf)3 [36] | CH2Cl2 | air | r.t., 16 h | 0.8 |
| 10 | FeCl3 [9], AgOTf [18] | DCE | air | r.t., 16 h | 1.2 |
| 11 | p-TsOH·H2O [90] | AcOH | air | 80 °C, 2 h | 1.9 |
| 12 | p-TsOH·H2O [360] | DCE | air | 50 °C, 1 h | 1.2 |
Appendix A.3. Acid Survey for the Self-Condensation of 15
| Entry | Reactant (mM) | Acid (mM) | Solvent | Oxidant | Conditions | Yield (%) b |
|---|---|---|---|---|---|---|
| 1 | 15 [18] | Neat TFA | – | air | 50 °C, 24 h | -- c |
| 2 | 15 [18] | TFAA [45] | CH2Cl2 | air | r.t., 16 h | 1.8 |
| 3 | 15 [18] | TFAA [90] | CH2Cl2 | air | r.t., 24 h | 4.0 |
| 4 | 15 [18] | TFAA [180] | CH2Cl2 | air | r.t., 24 h | 4.2 |
| 5 | 15 [18] | TFAA [45] | CH2Cl2 | air | 40 °C, 24 h | 4.5 |
| 6 | 15 [18] | TFAA [90] | CH2Cl2 | air | 40 °C, 24 h | 10 d |
| 7 | 15 [18] | TFAA [180] | CH2Cl2 | air | 40 °C, 24 h | 5.8 |
| 8 | 15 [50] | TFAA [90] | CH2Cl2 | air | 40 °C, 20 h | 4.8 |
| 9 | 15 [18] | TFAA [90] | CHCl3 | air | 40 °C, 24 h | 2.4 |
| 10 | 15 [2] | TFAA [90] | CH2Cl2 | air | 40 °C, 24 h | 10 |
| 11 | 15 [2] | TFAA [180] | CH2Cl2 | air | 40 °C, 24 h | 17 e |
| 12 | 15 [18] | Tf2O [90] | CH2Cl2 | air | r.t., 16 h | -- c |
| 13 | 15 [18] | Tf2O [90] | CH2Cl2 | air | 40 °C, 16 h | -- c |
| 14 | 15 [18] | Tf2O [90], PPh3O [180] | CH2Cl2 | air | 40 °C, 16 h | 5.2 |
References
- Scheer, H. An Overview of Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications. In Chlorophylls and Bacteriochlorophylls. Biochemistry, Biophysics, Functions and Applications; Grimm, B., Porra, R.J., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; Volume 25, pp. 1–26. [Google Scholar]
- Kobayashi, M.; Akiyama, M.; Kano, H.; Kise, H. Spectroscopy and Structure Determination. In Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications; Grimm, B., Porra, R.J., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 79–94. [Google Scholar]
- Galezowski, M.; Gryko, D.T. Recent Advances in the Synthesis of Hydroporphyrins. Curr. Org. Chem. 2007, 11, 1310–1338. [Google Scholar] [CrossRef]
- Brückner, C.; Samankumara, L.; Ogikubo, J. Syntheses of Bacteriochlorins and Isobacteriochlorins. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2012; Volume 17, pp. 1–112. [Google Scholar]
- Whitlock, H.W., Jr.; Hanauer, R.; Oester, M.Y.; Bower, B.K. Diimide Reduction of Porphyrins. J. Am. Chem. Soc. 1969, 91, 7485–7489. [Google Scholar] [CrossRef]
- Keegan, J.D.; Stolzenberg, A.M.; Lu, Y.-C.; Linder, R.E.; Barth, G.; Moscowitz, A.; Bunnenberg, E.; Djerassi, C. Magnetic Circular Dichroism Studies. 60. Substituent-Induced Sign Variation in the Magnetic Circular Dichroism Spectra of Reduced Porphyrins. 1. Spectra and Band Assignments. J. Am. Chem. Soc. 1982, 104, 4305–4317. [Google Scholar] [CrossRef]
- Brückner, C. The Breaking and Mending of meso-Tetraarylporphyrins: Transmuting the Pyrrolic Building Blocks. Acc. Chem. Res. 2016, 49, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Lindsey, J.S. Synthetic Chlorins, Possible Surrogates for Chlorophylls, Prepared by Derivatization of Porphyrins. Chem. Rev. 2017, 117, 344–535. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, G.; Pandey, R.K. Synthesis of Bacteriochlorins and Their Potential Utility in Photodynamic Therapy. Curr. Org. Chem. 2004, 8, 1105–1134. [Google Scholar] [CrossRef]
- Grin, M.A.; Mironov, A.F.; Shtil, A.A. Bacteriochlorophyll a and its Derivatives; Chemistry and Perspectives for Cancer Therapy. Anti-Cancer Agents Med. Chem. 2008, 8, 683–697. [Google Scholar] [CrossRef]
- Montforts, F.-P.; Gerlach, B.; Höper, F. Discovery and Synthesis of Less Common Natural Hydroporphyrins. Chem. Rev. 1994, 94, 327–347. [Google Scholar] [CrossRef]
- Montforts, F.-P.; Glasenapp-Breiling, M. The Synthesis of Chlorins, Bacteriochlorins, Isobacteriochlorins and Higher Reduced Porphyrins. Prog. Heterocycl. Chem. 1998, 10, 1–24. [Google Scholar]
- Prinsep, M.R.; Caplan, F.R.; Moore, R.E.; Patterson, G.M.L.; Smith, C.D. Tolyporphin, a Novel Multidrug Resistance Reversing Agent from the Blue-Green Alga Tolypothrix nodosa. J. Am. Chem. Soc. 1992, 114, 385–387. [Google Scholar] [CrossRef]
- Prinsep, M.R.; Puddick, J. Laser Desorption Ionisation–Time of Flight Mass Spectrometry of the Tolyporphins, Bioactive Metabolites from the Cyanobacterium Tolypothrix nodosa. Phytochem. Anal. 2011, 22, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Minehan, T.G.; Kishi, Y. Extension of the Eschenmoser Sulfide Contraction/Iminoester Cyclization Method to the Synthesis of Tolyporphin Chromophore. Tetrahedron Lett. 1997, 38, 6811–6814. [Google Scholar] [CrossRef]
- Minehan, T.G.; Kishi, Y. Total Synthesis of the Proposed Structure of (+)-Tolyporphin A O,O-Diacetate. Angew. Chem. Int. Ed. 1999, 38, 923–925. [Google Scholar] [CrossRef]
- Wang, W.; Kishi, Y. Synthesis and Structure of Tolyporphin A O,O-Diacetate. Org. Lett. 1999, 1, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Lindsey, J.S. De Novo Synthesis of Stable Tetrahydroporphyrinic Macrocycles: Bacteriochlorins and a Tetradehydrocorrin. J. Org. Chem. 2005, 70, 5475–5486. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lindsey, J.S. Northern–Southern Route to Synthetic Bacteriochlorins. J. Org. Chem. 2016, 81, 11882–11897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lindsey, J.S. Construction of the Bacteriochlorin Macrocycle with Concomitant Nazarov Cyclization to form the Annulated Isocyclic Ring: Analogues of Bacteriochlorophyll a. J. Org. Chem. 2017, 82, 2489–2504. [Google Scholar] [CrossRef] [PubMed]
- Alexander, V.M.; Sano, K.; Yu, Z.; Nakajima, T.; Choyke, P.L.; Ptaszek, M.; Kobayashi, H. Galactosyl Human Serum Albumin-NMP1 Conjugate: A Near infrared (NIR)-Activatable Fluorescence Imaging Agent to Detect Peritoneal Ovarian Cancer Metastases. Bioconjug. Chem. 2012, 23, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Ptaszek, M. Multifunctional Bacteriochlorins from Selective Palladium-Coupling Reactions. Org. Lett. 2012, 14, 3708–3711. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Ptaszek, M. Near-IR Emissive Chlorin–Bacteriochlorin Energy-Transfer Dyads with a Common Donor and Acceptors with Tunable Emission Wavelength. J. Org. Chem. 2013, 78, 10678–10691. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Sano, K.; Sato, K.; Watanabe, R.; Yu, Z.; Hanaoka, H.; Nakajima, T.; Choyke, P.L.; Ptaszek, M.; Kabayashi, H. Activatable Organic Near-Infrared Fluorescent Probes Based on a Bacteriochlorin Platform: Synthesis and Multicolor in Vivo Imaging with a Single Excitation. Bioconjug. Chem. 2014, 25, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pancholi, C.; Bhagavathy, G.V.; Kang, H.S.; Nguyen, J.K.; Ptaszek, M. Strongly Conjugated Hydroporphyrin Dyads: Extensive Modification of Hydroporphyrins’ Properties by Expanding the Conjugated System. J. Org. Chem. 2014, 79, 7910–7925. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Esemoto, N.N.; Diers, J.R.; Niedzwiedzki, D.M.; Greco, J.A.; Akhigbe, J.; Yu, Z.; Pancholi, C.; Bhagavathy, G.V.; Nguyen, J.K.; et al. Effects of Strong Electronic Coupling in Chlorin and Bacteriochlorin Dyads. J. Phys. Chem. A 2016, 120, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Esemoto, N.N.; Yu, Z.; Wiratan, L.; Satraitis, A.; Ptaszek, M. Bacteriochlorin Dyads as Solvent Polarity Dependent Near-Infrared Fluorophores and Reactive Oxygen Species Photosensitizers. Org. Lett. 2016, 18, 4590–4593. [Google Scholar] [CrossRef] [PubMed]
- De Assis, F.F.; Ferreira, M.A.B.; Brocksom, T.J.; de Oliveira, K.T. NIR Bacteriochlorin Chromophores Accessed by Heck and Sonogashira Cross-coupling Reactions on a Tetrabromobacteriochlorin Derivative. Org. Biomol. Chem. 2016, 14, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Taniguchi, M.; Lindsey, J.S. Regioselective 15-Bromination and Functionalization of a Stable Synthetic Bacteriochlorin. J. Org. Chem. 2007, 72, 5350–5357. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Cramer, D.L.; Bhise, A.D.; Kee, H.L.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Accessing the Near-Infrared Spectral Region with Stable, Synthetic, Wavelength-Tunable Bacteriochlorins. New J. Chem. 2008, 32, 947–958. [Google Scholar] [CrossRef]
- Krayer, M.; Ptaszek, M.; Kim, H.-J.; Meneely, K.R.; Fan, D.; Secor, K.; Lindsey, J.S. Expanded Scope of Synthetic Bacteriochlorins via Improved Acid Catalysis Conditions and Diverse Dihydrodipyrrin-Acetals. J. Org. Chem. 2010, 75, 1016–1039. [Google Scholar] [CrossRef] [PubMed]
- Krayer, M.; Yang, E.; Diers, J.R.; Bocian, D.F.; Holten, D.; Lindsey, J.S. De Novo Synthesis and Photophysical Characterization of Annulated Bacteriochlorins. Mimicking and Extending the Properties of Bacteriochlorophylls. New J. Chem. 2011, 35, 587–601. [Google Scholar] [CrossRef]
- Mass, O.; Lindsey, J.S. A trans-AB-Bacteriochlorin Building Block. J. Org. Chem. 2011, 76, 9478–9487. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; Lubian, E.; Pavan, M.P.; Kim, H.-J.; Yang, E.; Holten, D.; Lindsey, J.S. Synthetic Bacteriochlorins with Integral Spiro-piperidine Motifs. New J. Chem. 2013, 37, 1157–1173. [Google Scholar] [CrossRef]
- Jiang, J.; Yang, E.; Reddy, K.R.; Niedzwiedzki, D.M.; Kirmaier, C.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Synthetic Bacteriochlorins Bearing Polar Motifs (Carboxylate, Phosphonate, Ammonium and a Short PEG). Water-Solubilization, Bioconjugation, and Photophysical Properties. New J. Chem. 2015, 39, 5694–5714. [Google Scholar] [CrossRef]
- Zhang, S.; Kim, H.-J.; Tang, Q.; Yang, E.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Synthesis and Photophysical Characteristics of 2,3,12,13-Tetraalkylbacteriochlorins. New J. Chem. 2016, 40, 5942–5956. [Google Scholar] [CrossRef]
- Krayer, M.; Yang, E.; Kim, H.-J.; Kee, H.L.; Deans, R.M.; Sluder, C.E.; Diers, J.R.; Kirmaier, C.; Bocian, D.F.; Holten, D.; et al. Synthesis and Photochemical Characterization of Stable Indium Bacteriochlorins. Inorg. Chem. 2011, 50, 4607–4618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Reddy, K.R.; Jiang, J.; Taniguchi, M.; Sommer, R.D.; Lindsey, J.S. Elaboration of an Unexplored Substitution Site in Synthetic Bacteriochlorins. J. Porphyr. Phthalocyanines 2015, 19, 887–902. [Google Scholar] [CrossRef]
- Srinivasan, N.; Haney, C.A.; Lindsey, J.S.; Zhang, W.; Chait, B.T. Investigation of MALDI-TOF Mass Spectrometry of Diverse Synthetic Metalloporphyrins, Phthalocyanines, and Multiporphyrin Arrays. J. Porphyr. Phthalocyanines 1999, 3, 283–291. [Google Scholar] [CrossRef]
- Komeyama, K.; Takahashi, K.; Takaki, K. Bismuth-Catalyzed Intramolecular Carbo-oxycarbonylation of 3-Alkynyl Esters. Org. Lett. 2008, 10, 5119–5122. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, J.S. De Novo Synthesis of Gem-Dialkyl Chlorophyll Analogues for Probing and Emulating our Green World. Chem. Rev. 2015, 115, 6534–6620. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Dogutan, D.K.; Ptaszek, M.; Lindsey, J.S. Synthesis of Hydrodipyrrins Tailored for Reactivity at the 1- and 9-Positions. Tetrahedron 2007, 63, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Krayer, M.; Balasubramanian, T.; Ruzié, C.; Ptaszek, M.; Cramer, D.L.; Taniguchi, M.; Lindsey, J.S. Refined Syntheses of Hydrodipyrrin Precursors to Chlorin and Bacteriochlorin Building Blocks. J. Porphyr. Phthalocyanines 2009, 13, 1098–1110. [Google Scholar] [CrossRef]
- Vairaprakash, P.; Yang, E.; Sahin, T.; Taniguchi, M.; Krayer, M.; Diers, J.R.; Wang, A.; Niedzwiedzki, D.M.; Kirmaier, C.; Lindsey, J.S.; et al. Extending the Short and Long Wavelength Limits of Bacteriochlorin Near-Infrared Absorption via Dioxo- and Bisimide-Functionalization. J. Phys. Chem. B 2015, 119, 4382–4395. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.D.; Littler, B.J.; Geier, G.R., III; Lindsey, J.S. Efficient Synthesis of Monoacyl Dipyrromethanes and Their Use in the Preparation of Sterically Unhindered trans-Porphyrins. J. Org. Chem. 2000, 65, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, J.S. Synthetic Routes to meso-Patterned Porphyrins. Acc. Chem. Res. 2010, 43, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Filatov, M.A.; Lebedev, A.Y.; Vinogradov, S.A.; Cheprakov, A.V. Synthesis of 5,15-Diaryltetrabenzoporphyrins. J. Org. Chem. 2008, 73, 4175–4183. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, W.G. New Synthetic Approaches to Chlorins and Bacteriochlorins. Ph.D. Thesis, Dartmouth College, Hanover, NH, USA, 2007. See compounds 153a–c. [Google Scholar]
- Eschenmoser, A. Robert Robinson Lecture. Post-B12 Problems in Corrin Synthesis. Chem. Soc. Rev. 1976, 5, 377–410. [Google Scholar] [CrossRef]
- Sutton, J.M.; Clarke, O.J.; Fernandez, N.; Boyle, R.W. Porphyrin, Chlorin, and Bacteriochlorin Isothiocyanates: Useful Reagents for the Synthesis of Photoactive Bioconjugates. Bioconjug. Chem. 2002, 13, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Senge, M.O. Highly Substituted Porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 1, pp. 239–347. [Google Scholar]
- Röder, B.; Büchner, M.; Rückmann, I.; Senge, M.O. Correlation of Photophysical Parameters with Macrocycle Distortion in Porphyrins with Graded Degree of Saddle Distortion. Photochem. Photobiol. Sci. 2010, 9, 1152–1158. [Google Scholar]
- Yang, E.; Kirmaier, C.; Krayer, M.; Taniguchi, M.; Kim, H.-J.; Diers, J.R.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Photophysical Properties and Electronic Structure of Stable, Tunable Synthetic Bacteriochlorins: Extending the Features of Native Photosynthetic Pigments. J. Phys. Chem. B 2011, 115, 10801–10816. [Google Scholar] [CrossRef] [PubMed]
- Dorough, G.D.; Miller, J.R. An Attempted Preparation of a Simple Tetrahydroporphine. J. Am. Chem. Soc. 1952, 74, 6106–6108. [Google Scholar] [CrossRef]
- Strachan, J.-P.; O’Shea, D.F.; Balasubramanian, T.; Lindsey, J.S. Rational Synthesis of Meso-Substituted Chlorin Building Blocks. J. Org. Chem. 2000, 65, 3160–3172. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, M.L.S.; Gago, D.J.P.; Rocha Gonsalves, A.M.d’A.; Johnstone, R.A.W.; McCarron, M.; Varejão, J.M.T.B. Investigations into the Mechanism of Action of Nitrobenzene as a Mild Dehydrogenating Agent under Acid-Catalysed Conditions. Org. Biomol. Chem. 2003, 1, 565–574. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: The small-scale syntheses, repetitive studies of reaction conditions, and photophysical studies preclude availability of most of the compounds from the authors. |

















| Compound | λBy abs (nm) | λBx abs (nm) | λQx abs (nm) | λQy abs (nm) | λQy ems (nm) | Φf b |
|---|---|---|---|---|---|---|
| B1-T2 | 356 | 382 | 503 | 726 | 729 | 0.12 |
| B2-T2 | 357 | 381 | 516 | 739 | 747 | 0.15 |
| B3-P2T2 | 362 c | 382 | 533 | 743 | 757 | 0.15 |
| Compound | λBy (Δ b) (nm) | λBx (Δ b) (nm) | λQx (Δ b) (nm) | λQy (Δ b) (nm) | Qy fwhm (Δ b) (nm) | ΣQy/ΣT c |
|---|---|---|---|---|---|---|
| iso-B2 | 355 | 384 | 521 | 760 | 20 | 0.126 |
| iso-B2-T2 | 362 (+7) | 388 (+4) | 535 (+14) | 759 (−1) | 25 (+5) | 0.117 |
| B2 d | 354 | 383 | 521 | 760 | 20 | 0.136 |
| B2-T2 | 357 (+3) | 381 (−2) | 516 (−5) | 739 (−21) | 22 (+2) | 0.106 |
| B1 e | 346 | 374 | 490 | 721 | 12 | 0.107 |
| B1-T2 | 356 (+10) | 382 (+8) | 503 (+13) | 726 (+5) | 14 (+2) | 0.101 |
| B3 | 352 | 379 | 523 | 754 | 16 | 0.133 |
| B3-T2 | 361 f | 383 | 543 (+20) | 759 (+5) | 16 (0) | 0.125 |
| B3-P2T2 | 362 g | 382 | 533 (+10) | 743 (−11) | 27 (+11) | 0.100 |
| B6 d | 340 | 365 | 489 | 713 | 12 | 0.091 |
| TPBC h | 355 (+15) | 378 (+13) | 522 (+33) | 742 (+29) | 15 (+3) | 0.091 |
| Compound | λQy (Δ b) (nm) | fwhm (Δ b) (nm) | Δν (cm−1) c | Φf d |
|---|---|---|---|---|
| iso-B2 | 764 | 20 | 70 | 0.11 |
| iso-B2-T2 | 766 (+2) | 25 (+5) | 120 | 0.15 |
| B2 e | 764 | 21 | 60 | 0.14 |
| B2-T2 | 747 (−17) | 27 (+6) | 140 | 0.15 |
| B1 f | 723 | 14 | 40 | 0.10 |
| B1-T2 | 729 (+6) | 18 (+4) | 60 | 0.12 |
| B3 | 757 | 18 | 50 | 0.12 |
| B3-T2 | 761 (+3) | 18 (0) | 35 | 0.16 |
| B3-P2T2 | 757 (0) | 34 (+16) | 250 | 0.15 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, M.N.; Zhang, S.; Kim, H.-J.; Mass, O.; Taniguchi, M.; Lindsey, J.S. Synthesis and Spectral Properties of meso-Arylbacteriochlorins, Including Insights into Essential Motifs of their Hydrodipyrrin Precursors. Molecules 2017, 22, 634. https://doi.org/10.3390/molecules22040634
Reddy MN, Zhang S, Kim H-J, Mass O, Taniguchi M, Lindsey JS. Synthesis and Spectral Properties of meso-Arylbacteriochlorins, Including Insights into Essential Motifs of their Hydrodipyrrin Precursors. Molecules. 2017; 22(4):634. https://doi.org/10.3390/molecules22040634
Chicago/Turabian StyleReddy, Muthyala Nagarjuna, Shaofei Zhang, Han-Je Kim, Olga Mass, Masahiko Taniguchi, and Jonathan S. Lindsey. 2017. "Synthesis and Spectral Properties of meso-Arylbacteriochlorins, Including Insights into Essential Motifs of their Hydrodipyrrin Precursors" Molecules 22, no. 4: 634. https://doi.org/10.3390/molecules22040634
APA StyleReddy, M. N., Zhang, S., Kim, H. -J., Mass, O., Taniguchi, M., & Lindsey, J. S. (2017). Synthesis and Spectral Properties of meso-Arylbacteriochlorins, Including Insights into Essential Motifs of their Hydrodipyrrin Precursors. Molecules, 22(4), 634. https://doi.org/10.3390/molecules22040634