
Electrophilic Selenium Catalysis with Electrophilic N-F Reagents as the Oxidants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. ESC with N-Fluorobenzenesulfonimide (NFSI) as the Oxidant
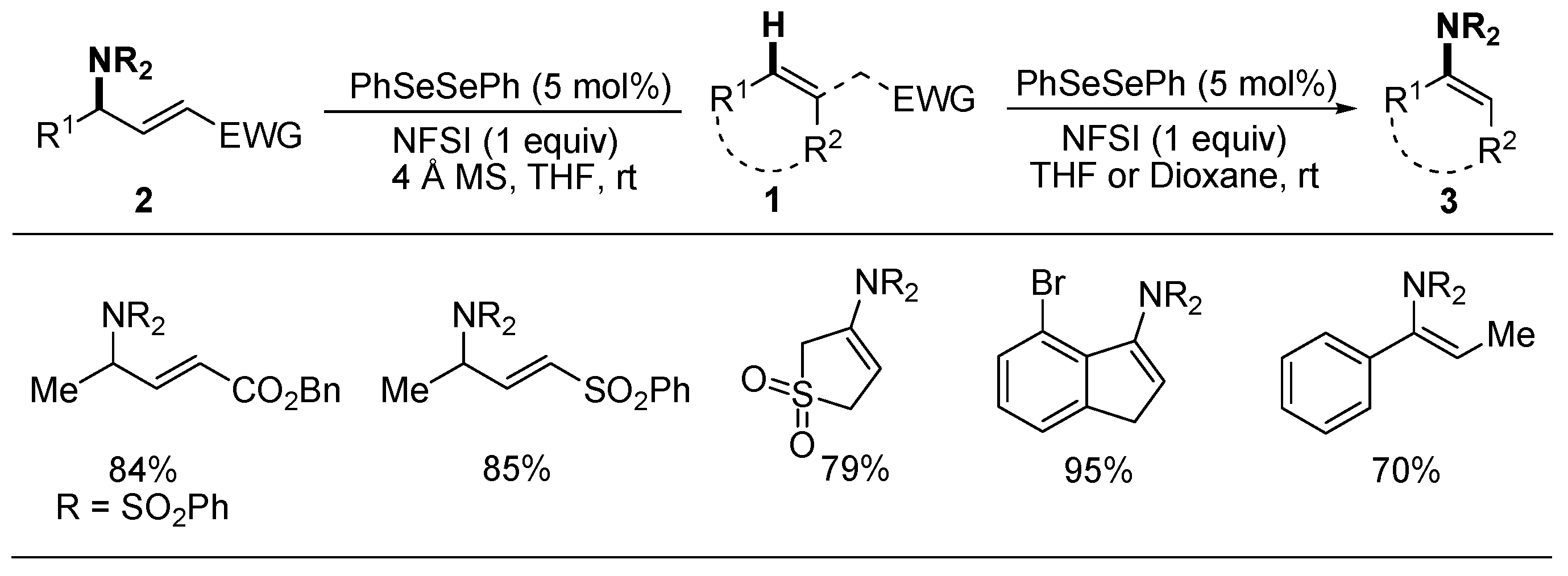
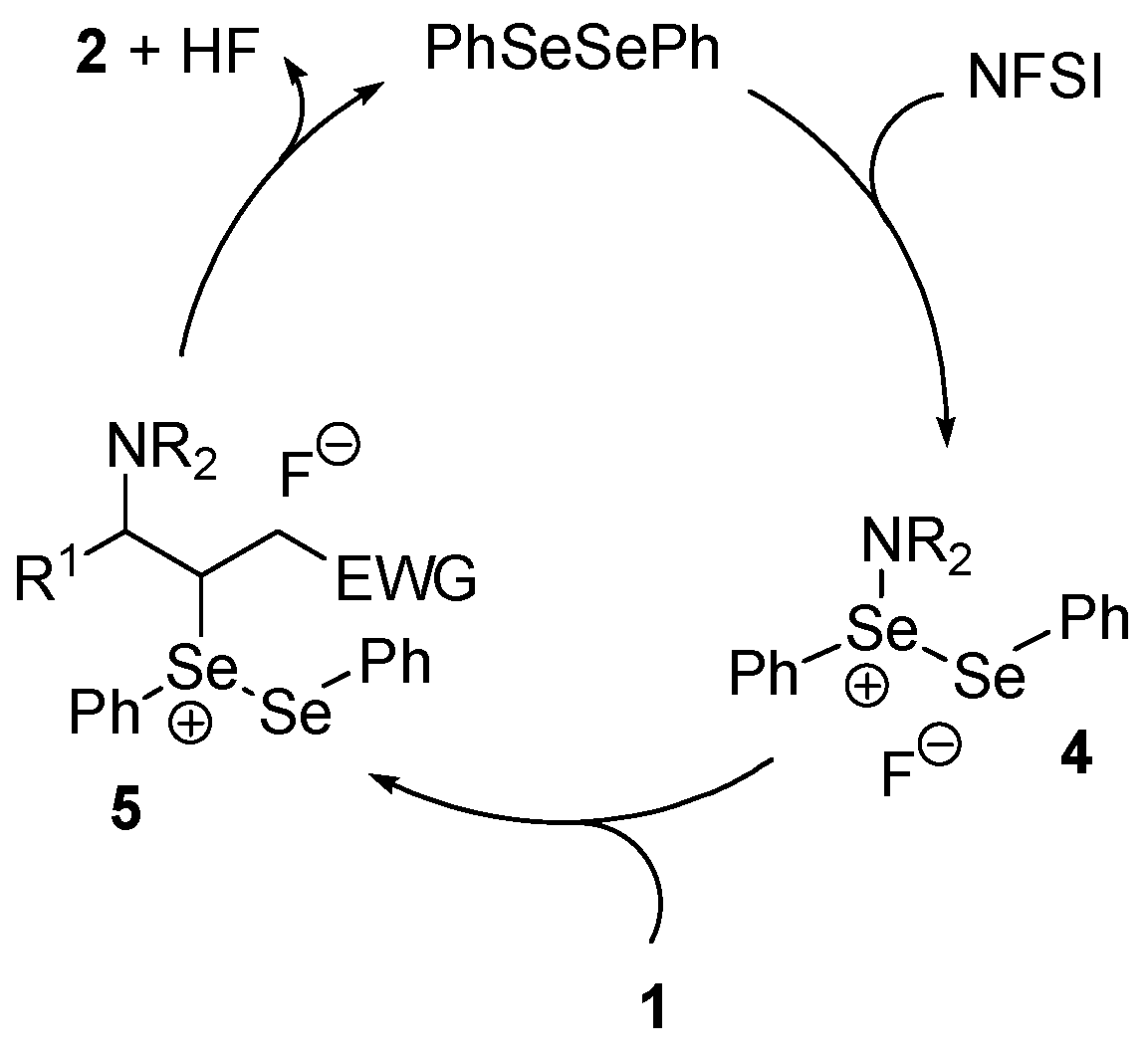
2.1. Allylic or Vinylic Imidation of Alkenes
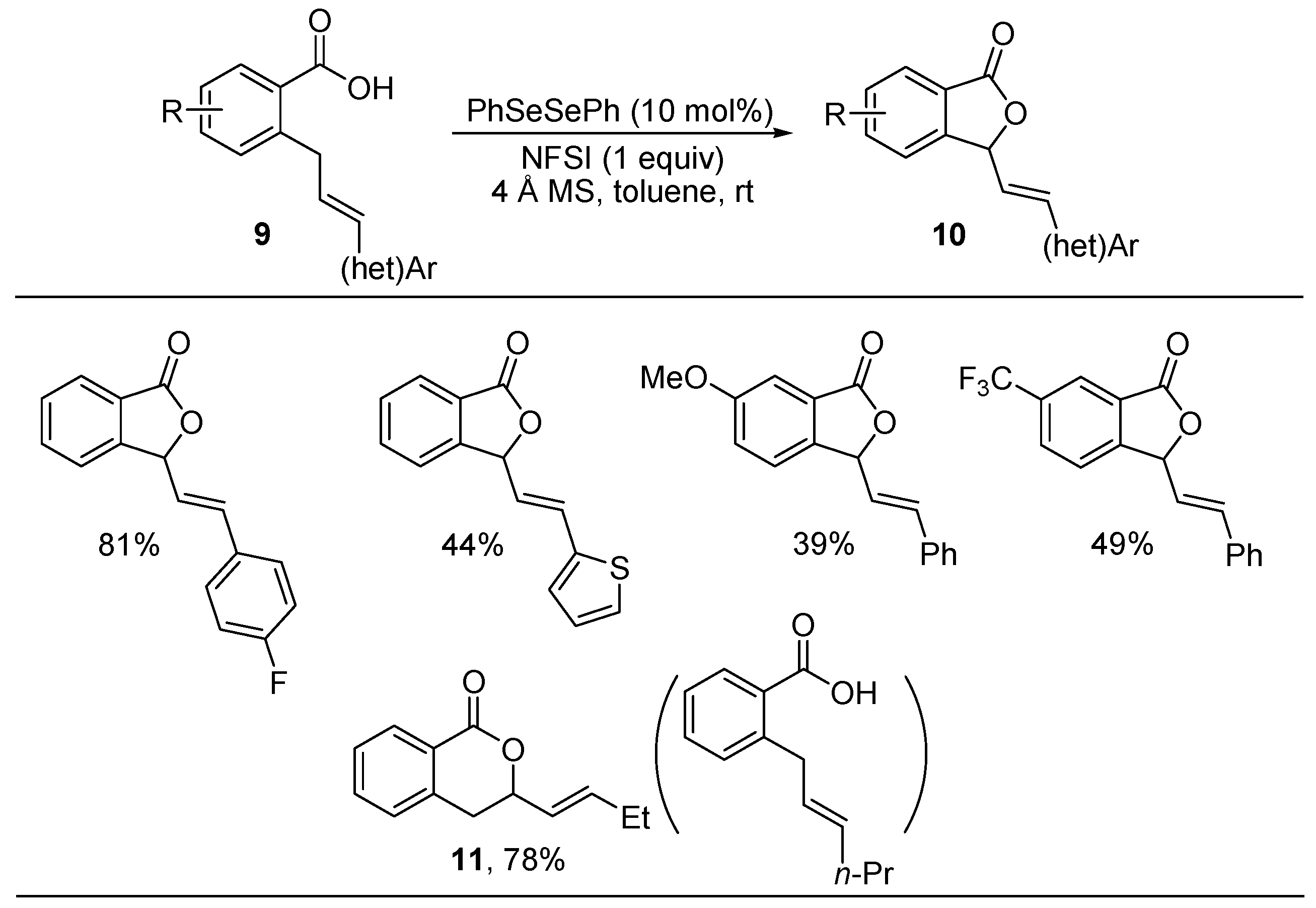
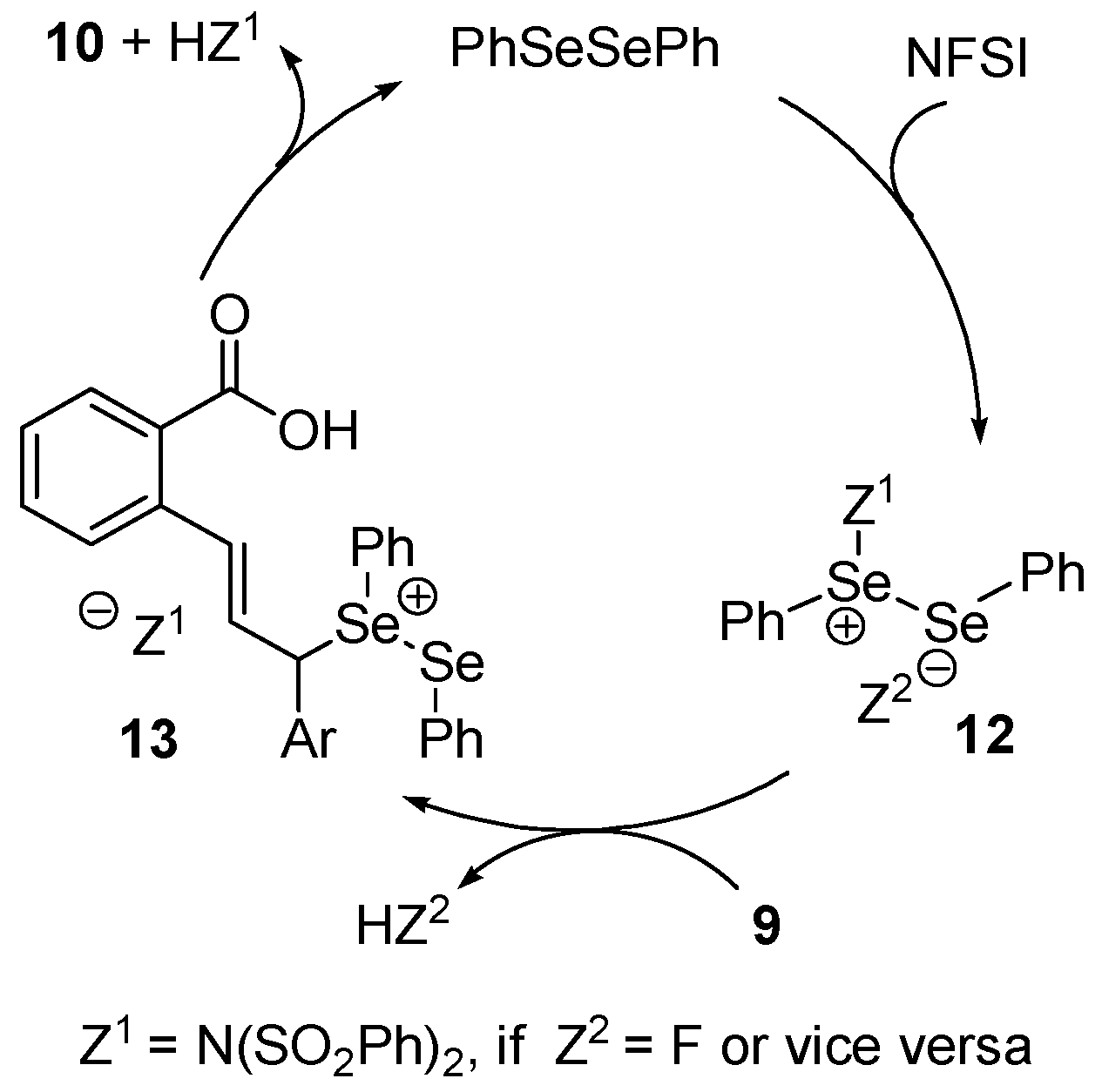
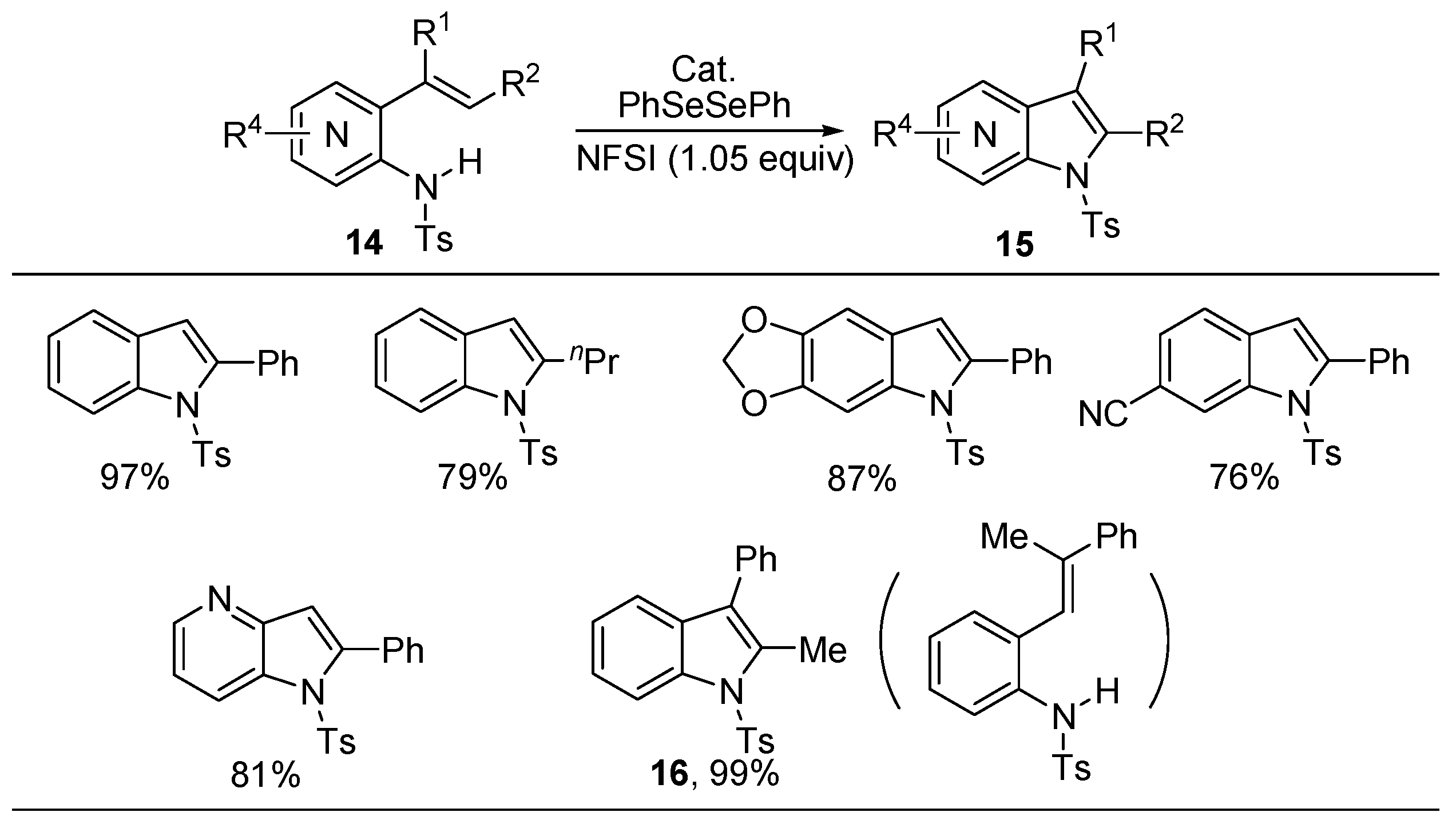
2.2. Synthesis of Isobenzofuranones and Indole Derivatives via Intramolecular C-H Functionalization
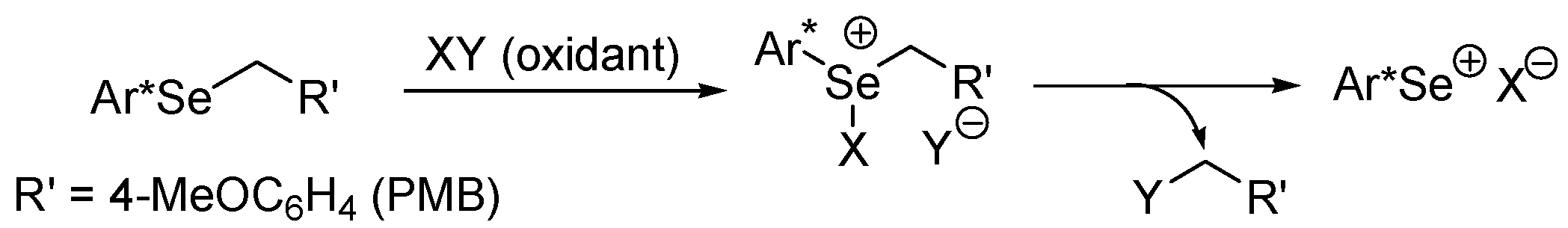
3. ESC with Fluoropyridinium Salts as the Oxidants
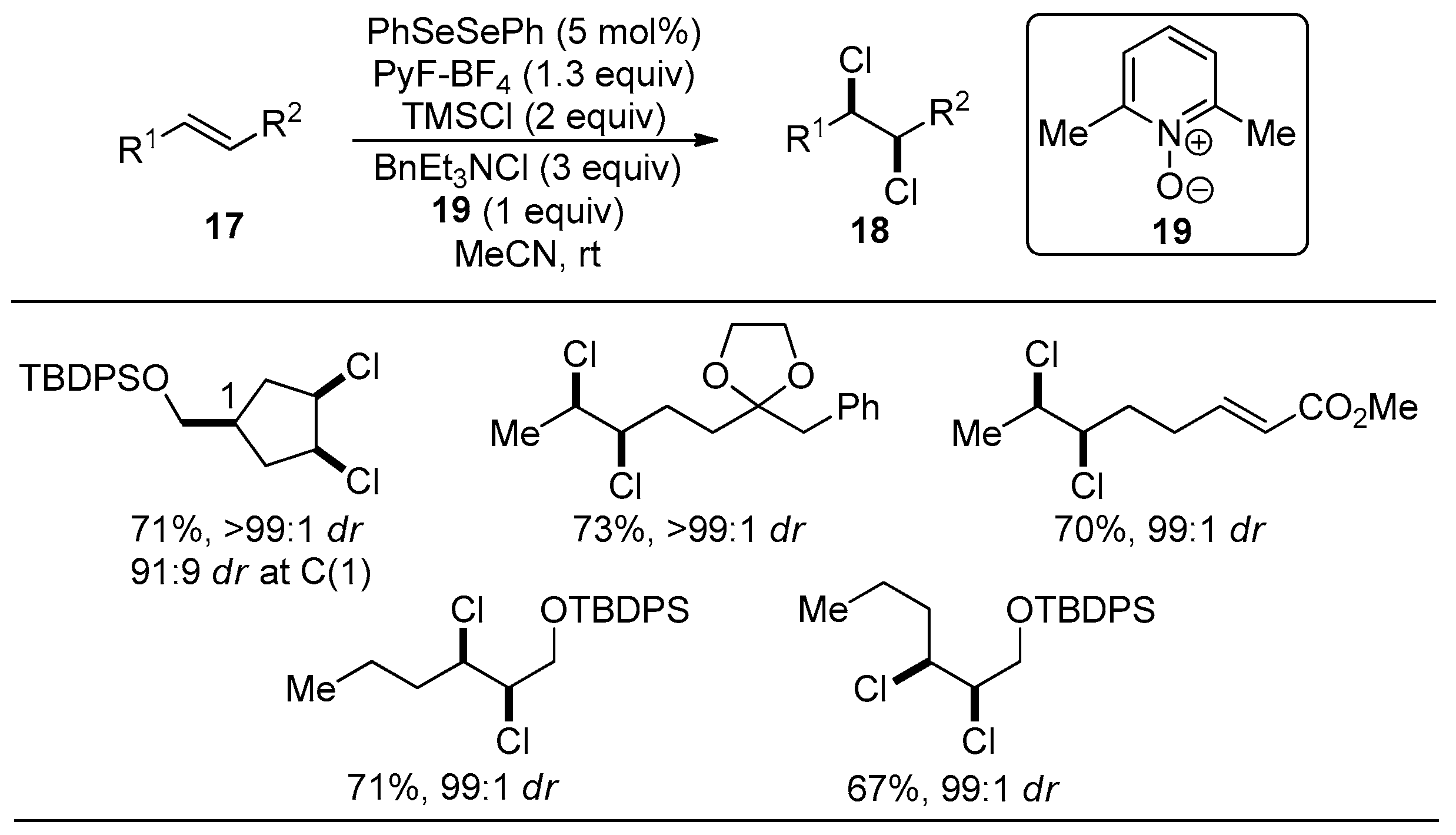
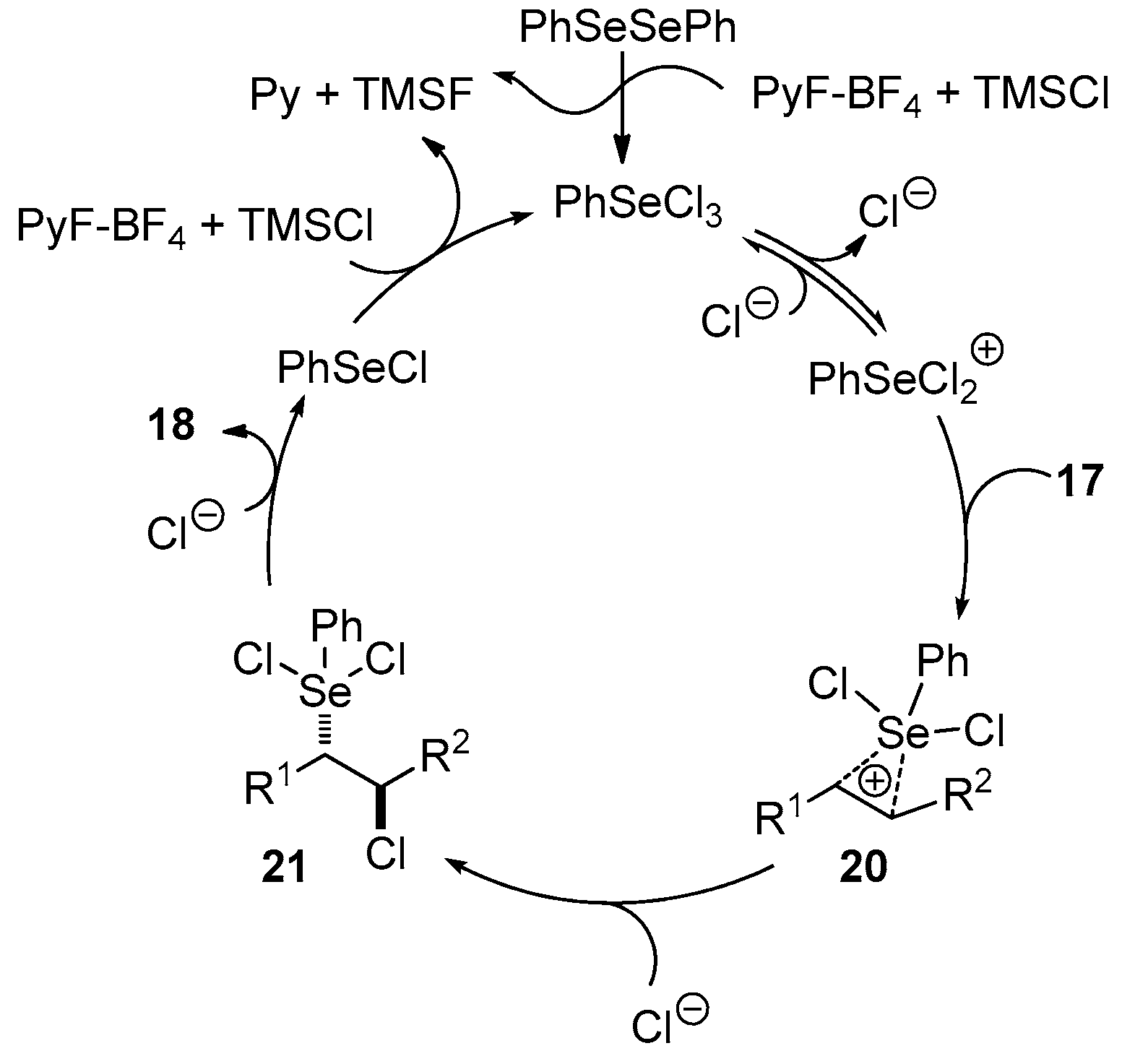
3.1. Stereospecific Syn-Dichlorination of Alkenes
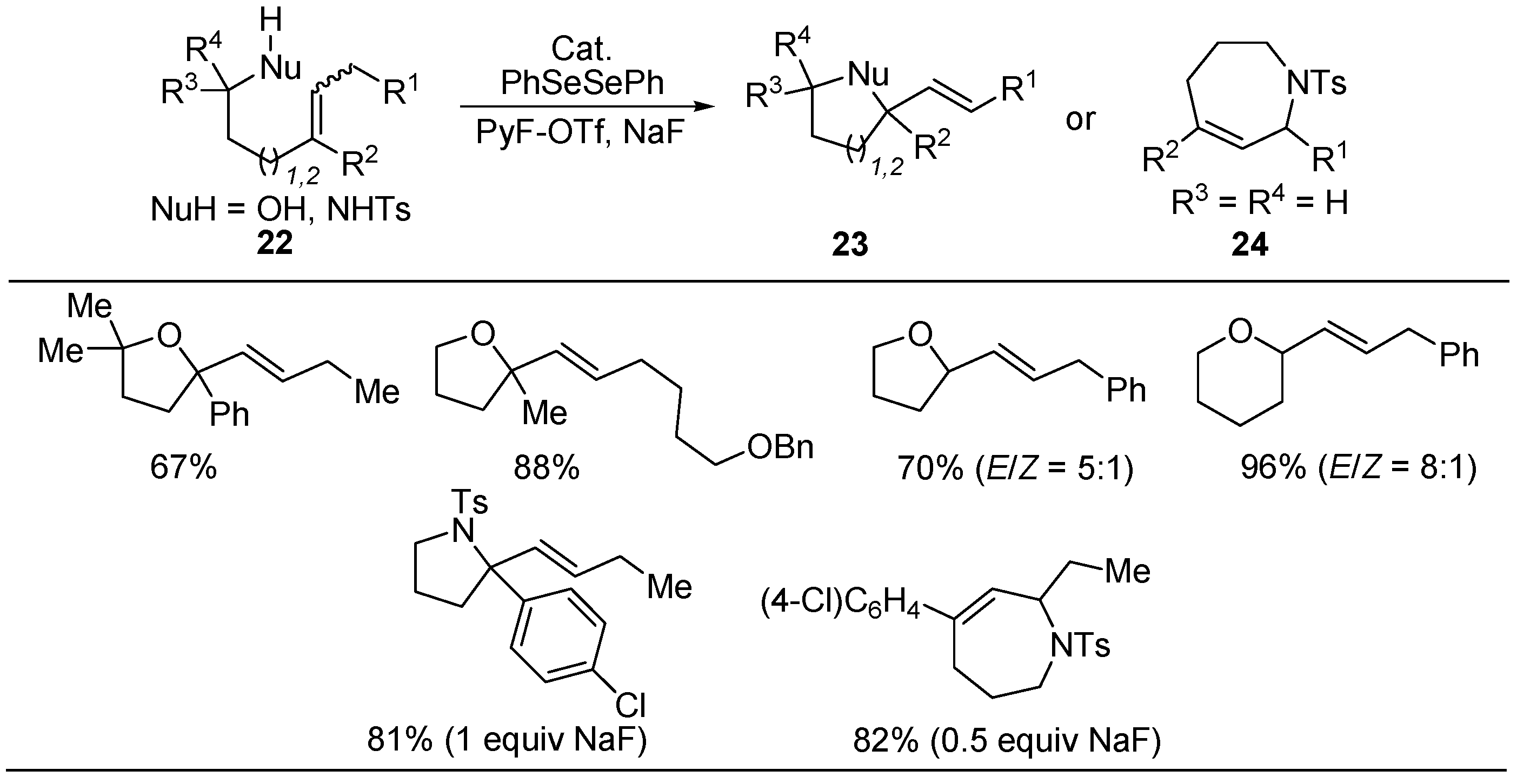
3.2. Synthesis of Oxygen- and Nitrogen-Containing Heterocycles via Oxidative Cyclization
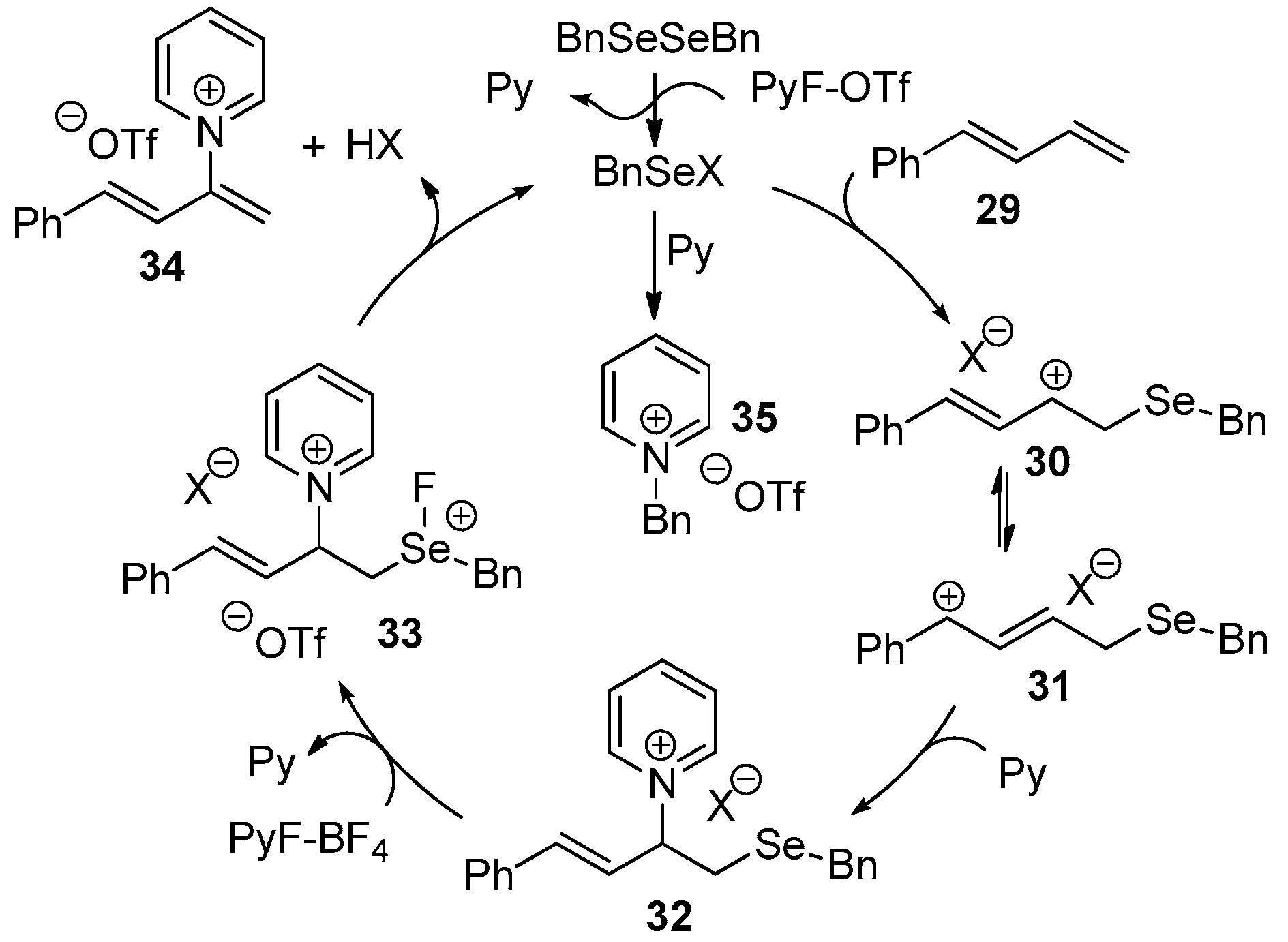
3.3. Regioselective Pyridination of 1,3-Dienes
4. Asymmetric Conversion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Patai, S.; Rappoport, Z. The Chemistry of Organic Selenium and Tellurium Compounds; John Wiley and Sons: Chichester, UK, 1986; pp. 275–337. [Google Scholar]
- Back, T.G. Organoselenium Chemistry—A Practical Approach; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Wirth, T. Organoselenium Chemistry: Synthesis and Reactions; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Clive, D.L.J. Modern organoselenium chemistry. Tetrahedron 1978, 34, 1049–1132. [Google Scholar] [CrossRef]
- Reich, H.J. Functional group manipulation using organoselenium reagents. Acc. Chem. Res. 1979, 12, 22–30. [Google Scholar] [CrossRef]
- Wirth, T. Chiral selenium compounds in organic synthesis. Tetrahedron 1999, 55, 1–28. [Google Scholar] [CrossRef]
- Wirth, T. Organoselenium chemistry in stereoselective reactions. Angew. Chem. Int. Ed. 2000, 39, 3740–3749. [Google Scholar] [CrossRef]
- Freudendahl, D.M.; Shahzad, S.A.; Wirth, T. Recent advances in organoselenium chemistry. Eur. J. Org. Chem. 2009, 1649–1664. [Google Scholar] [CrossRef]
- Santoro, S.; Azeredo, J.B.; Nascimento, V.; Sancineto, L.; Braga, A.L.; Santi, C. The green side of the moon: Ecofriendly aspects of organoselenium chemistry. RSC Adv. 2014, 4, 31521–31535. [Google Scholar] [CrossRef]
- Hori, T.; Sharpless, K.B. Selenium-catalyzed nonradical chlorination of olefins with N-chlorosuccinimide. J. Org. Chem. 1979, 44, 4204–4208. [Google Scholar] [CrossRef]
- Hori, T.; Sharpless, K.B. Conversion of allylic phenylselenides to the rearranged allylic chlorides by N-chlorosuccinimide. Mechanism of selenium-catalyzed allylic chlorination of β-pinene. J. Org. Chem. 1979, 44, 4208–4210. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Tingoli, M.; Bartoli, D. Selenium-catalyzed conversion of methyl ketones into α-keto acetals. J. Org. Chem. 1990, 55, 4523–4528. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Tingoli, M.; Chianelli, D.; Bartoli, D. Selenium-mediated conversion of alkynes into α-dicarbonyl compounds. J. Org. Chem. 1991, 56, 4529–4534. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Tingoli, M.; Bartoli, D.; Marini, F. Selenium-promoted conversion of β-diketones and β-keto esters into α,α-dimethoxy β-diketones and α,α-dimethoxy β-keto esters. J. Org. Chem. 1991, 56, 5207–5210. [Google Scholar] [CrossRef]
- lwaoka, M.; Tomoda, S. Catalytic conversion of alkenes into allylic ethers and esters using diselenides having internal tertiary amines. J. Chem. Soc. Chem. Commun. 1992, 1165–1167. [Google Scholar]
- Tiecco, M.; Testaferri, L.; Tingoli, M.; Bagnoli, L.; Santi, C. Catalytic conversion of β,γ-unsaturated esters, amides and nitriles into γ-alkoxy or γ-hydroxy α,β-unsaturated derivatives induced by persulfate anion oxidation of diphenyl diselenide. J. Chem. Soc. Chem. Commun. 1993, 637–639. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Tingoli, M.; Marini, F. Selenium promoted conversion of α-substituted β,γ-unsaturated ketones into 2,3,5-trisubstituted furans. Synlett 1994, 373–374. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferr, L.; Santi, C. Catalytic oxyselenenylation–deselenenylation reactions of alkenes-stereoselective one-pot conversion of 3-alkenols into 2,5-dihydrofurans. Eur. J. Org. Chem. 1999, 797–803. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Temperini, A.; Marini, F.; Bagnoli, L.; Santi, C. Selenium promoted stereospecific one-pot conversion of cinnamyl derivatives into oxazoleses. A simple synthetic route to racemic taxol side chain. Synth. Commun. 1999, 29, 1773–1778. [Google Scholar] [CrossRef]
- Ten Brink, G.-J.; Vis, J.-M.; Arends, I.W.C.E.; Sheldon, R.A. Selenium-catalyzed oxidations with aqueous hydrogen peroxide. 2. Baeyer-Villiger reactions in homogeneous solution. J. Org. Chem. 2001, 66, 2429–2433. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, H.; Usami, Y.; Arimoto, M. Synthesis of novel organoselenium as catalyst for Baeyer—Villiger oxidation with 30% H2O2. Tetrahedron Lett. 2005, 46, 8665–8668. [Google Scholar] [CrossRef]
- Santoro, S.; Santi, C.; Sabatini, M.; Testaferri, L.; Tiecco, M. Eco-friendly olefin dihydroxylation catalyzed by diphenyl diselenide. Adv. Synth. Catal. 2008, 350, 2881–2884. [Google Scholar] [CrossRef]
- Santi, C.; Lorenzo, R.D.; Tidei, C.; Bagnoli, L.; Wirth, T. Stereoselective selenium catalyzed dihydroxylation and hydroxymethoxylation of alkenes. Tetrahedron 2012, 68, 10530–10535. [Google Scholar] [CrossRef]
- Yu, L.; Wu, Y.; Cao, H.; Zhang, X.; Shi, X.; Luan, J.; Chen, T.; Pan, Y.; Xu, Q. Facile synthesis of 2-methylenecyclobutanones via Ca(OH)2-catalyzed direct condensation of cyclobutanone with aldehydes and (PhSe)2-catalyzed Baeyer–Villiger oxidation to 4-methylenebutanolides. Green Chem. 2014, 16, 287–293. [Google Scholar] [CrossRef]
- Yu, L.; Li, H.; Zhang, X.; Ye, J.; Liu, J.; Xu, Q.; Lautens, M. Organoselenium-catalyzed mild dehydration of aldoximes: An unexpected practical method for organonitrile synthesis. Org. Lett. 2014, 16, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Browne, D.M.; Niyomura, O.; Wirth, T. Catalytic use of selenium electrophiles in cyclizations. Org. Lett. 2007, 9, 3169–3171. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, S.A.; Venin, C.; Wirth, T. Diselenide- and disulfide-mediated synthesis of isocoumarins. Eur. J. Org. Chem. 2010, 3465–3472. [Google Scholar] [CrossRef]
- Singh, F.V.; Wirth, T. Selenium-catalyzed regioselective cyclization of unsaturated carboxylic acids using hypervalent iodine oxidants. Org. Lett. 2011, 13, 6504–6507. [Google Scholar] [CrossRef] [PubMed]
- Tingoli, M.; Mazzella, M.; Panunzi, B.; Tuzi, A. Elemental iodine or diphenyl diselenide in the [bis(trifluoroacetoxy)-iodo] benzene-mediated conversion of alkynes into 1,2-diketones. Eur. J. Org. Chem. 2011, 2011, 399–404. [Google Scholar] [CrossRef]
- Torii, S.; Uneyama, K.; Ono, M.; Bannou, T. Generation and recycle use of selenenylating reagents in electrochemical oxyselenenylation-deselenenylation of olefins. J. Am. Chem. Soc. 1981, 103, 4606–4608. [Google Scholar] [CrossRef]
- Van der Toorn, J.C.; Kemperman, G.; Sheldon, R.A.; Arends, I.W.C.E. Electroreductive ring-opening of α,β-epoxy carbonyl compounds and their homologues through recyclable use of diphenyl diselenide or diphenyl ditelluride as a mediator. J. Org. Chem. 1990, 55, 1548–1553. [Google Scholar]
- Niyomura, O.; Cox, M.; Wirth, T. Electrochemical generation and catalytic use of selenium electrophiles. Synlett 2006, 251–254. [Google Scholar]
- Van der Toorn, J.C.; Kemperman, G.; Sheldon, R.A.; Arends, I.W.C.E. Diphenyldiselenide-catalyzed selective oxidation of activated alcohols with tert-butyl hydroperoxide: New mechanistic insights. J. Org. Chem. 2009, 74, 3085–3089. [Google Scholar] [CrossRef] [PubMed]
- Freudendahl, D.M.; Santoro, S.; Shahzad, S.A.; Santi, C.; Wirth, T. Green chemistry with selenium reagents: Development of efficient catalytic reactions. Angew. Chem. Int. Ed. 2009, 48, 8409–8411. [Google Scholar] [CrossRef] [PubMed]
- Santi, C.; Santoro, S.; Battistelli, B. Organoselenium compounds as catalysts in nature and laboratory. Curr. Org. Chem. 2010, 14, 2442–2462. [Google Scholar] [CrossRef]
- Breder, A.; Ortgies, S. Recent developments in sulfur- and selenium-catalyzed oxidative and isohypsic functionalization reactions of alkenes. Tetrahedron Lett. 2015, 56, 2843–2852. [Google Scholar] [CrossRef]
- Strekowski, L.; Kiselyov, A.S. N-Fluoropyridinium salts. Adv. Heterocycl. Chem. 1995, 62, 1–17. [Google Scholar]
- Lal, G.S.; Pez, G.P.; Syvret, R.G. Electrophilic NF fluorinating agents. Chem. Rev. 1996, 96, 1737–1755. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.D.; Kotoris, C.C.; Hum, G. Recent advances in electrophilic fluorination. Tetrahedron 1999, 55, 12431–12477. [Google Scholar] [CrossRef]
- Kiselyov, A.S. Chemistry of N-fluoropyridinium salts. Chem. Soc. Rev. 2005, 34, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Baudoux, J.; Cahard, D. Electrophilic Fluorination with N-F Reagents. In Organic Reactions; John Wiley and Sons: Hoboken, NJ, USA, 2007; Volume 69, pp. 347–672. [Google Scholar]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed. 2013, 52, 2–53. [Google Scholar] [CrossRef] [PubMed]
- Trenner, J.; Depken, C.; Weber, T.; Breder, A. Direct oxidative allylic and vinylic amination of alkenes through selenium catalysis. Angew. Chem. Int. Ed. 2013, 52, 8952–8957. [Google Scholar] [CrossRef] [PubMed]
- Mellegaard, S.R.; Tunge, J.A. Selenium-catalyzed halolactonization: Nucleophilic activation of electro- philic halogenating reagents. J. Org. Chem. 2004, 69, 8979–8981. [Google Scholar] [CrossRef] [PubMed]
- Carrera, I.; Brovetto, M.C.; Seoane, G.A. Selenium-catalyzed iodohydrin formation from alkenes. Tetrahedron Lett. 2006, 47, 7849–7852. [Google Scholar] [CrossRef]
- Bloom, S.; Knippel, J.L.; Holl, M.G.; Barber, R.; Lectka, T. A cooperative allylic fluorination: Combination of nucleophilic and electrophilic fluorine sources. Tetrahedron Lett. 2014, 55, 4576–4580. [Google Scholar] [CrossRef]
- Zheng, G.; Zhao, J.; Li, Z.; Zhang, Q.; Sun, J.; Sun, H.; Zhang, Q. Highly regio- and stereoselective intermolecular seleno- and thioamination of alkynes. Chem. Eur. J. 2016, 22, 3513–3518. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wei, J.; Liao, L.; Huang, H.; Zhao, X. Organoselenium-catalyzed, hydroxy-controlled regio- and stereoselective amination of terminal alkenes: Efficient synthesis of 3-amino allylic alcohols. Org. Lett. 2015, 17, 1834–1837. [Google Scholar] [CrossRef] [PubMed]
- Engman, L. Acetoxyselenylation of olefins for the preparation of vinylic and allylic acetates. J. Org. Chem. 1989, 54, 884–890. [Google Scholar] [CrossRef]
- Liotta, D.; Zima, G. Synthetic applications of phenylselenenyl chloride additions. A simple 1,3-enone transposition sequence. J. Org. Chem. 1980, 45, 2551–2553. [Google Scholar] [CrossRef]
- Liotta, D.; Zima, G.; Saindane, M. Origins of regio- and stereoselectivity in additions of phenylselenenyl chloride to allylic alcohols and the applicability of these additions to a simple 1,3-enone transposition sequence. J. Org. Chem. 1982, 47, 1258–1267. [Google Scholar] [CrossRef]
- Cooper, M.A.; Ward, A.D. Hydroxyselenation of allylic alcohols. Tetrahedron Lett. 1995, 36, 2327–2730. [Google Scholar] [CrossRef]
- Cooper, M.A.; Ward, A.D. Formation of dihydroxyselenides from allylic alcohols and their conversion to β-hydroxy epoxides via substitution of a phenylselenonyl group. Tetrahedron 2004, 60, 7963–7972. [Google Scholar] [CrossRef]
- Duhamel, L.; Chauvin, J.; Messier, A. β-Lithioenamines by metalation and reduction of β-(phenylthio)enamines. J. Chem. Res. (S) 1982, 48–49. [Google Scholar]
- Kashima, C.; Hibi, S.; Shimizu, M.; Tajima, T.; Omote, Y. The regioselective preparation of 1-alken-3-ones by the reaction of 3-(1-imidazolyl)-2-alken-1-ones with organometallic compounds. Heterocycles 1986, 24, 429–436. [Google Scholar] [CrossRef]
- Kraetzschmar, F.; Kassel, M.; Delony, D.; Breder, A. Selenium-catalyzed C(sp3)-H acyloxylation: Application in the expedient synthesis of isobenzofuranones. Chem. Eur. J. 2015, 21, 7030–7035. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.-Q.; Liu, K.; Zhang, C. Effective oxidation of benzylic and alkane C-H bonds catalyzed by sodium o-iodobenzenesulfonate with oxone as a terminal oxidant under phase-transfer conditions. Org. Biomol. Chem. 2011, 9, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Liang, Y.; Hong, X.; Sun, H.; Sun, W.-Y.; Houk, K.N.; Shi, Z. Iodoarene-catalyzed stereospecific intramolecular sp3 C-H amination: Reaction development and mechanistic insights. J. Am. Chem. Soc. 2015, 137, 7564–7567. [Google Scholar] [CrossRef] [PubMed]
- Ortgies, S.; Breder, A. Selenium-catalyzed oxidative C(sp2)-H amination of alkenes exemplified in the expedient synthesis of (aza-)indoles. Org. Lett. 2015, 17, 2748–2751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, R.; Zhao, X. Organoselenium-catalyzed synthesis of indoles through intramolecular C-H amination. Org. Chem. Front. 2015, 2, 9562–9566. [Google Scholar] [CrossRef]
- Maity, S.; Zheng, N. A visible-light-mediated oxidative C-N bond formation/aromatization cascade: Photocatalytic preparation of N-arylindoles. Angew. Chem. Int. Ed. 2012, 51, 8952–8957. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.H.; Youn, S.W. Metal-free C-H amination for indole synthesis. Org. Lett. 2014, 16, 3720–3723. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Li, J.; Ma, A.-L.; Huang, Y.-N.; Deng, J. Metal-free synthesis of indole via NIS-mediated cascade C-N bond formation/aromatization. J. Org. Chem. 2015, 80, 3841–3851. [Google Scholar] [CrossRef] [PubMed]
- Cresswell, A.J.; Eey, S.T.-C.; Denmark, S.E. Catalytic, stereospecific syn-dichlorination of alkenes. Nat. Chem. 2015, 7, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Garratt, D.G.; Schmid, G.H. The addition of arylselenium trichlorides vs. areneselenenyl chlorides to cis- and trans-1-phenylpropene. Can. J. Chem. 1974, 52, 3599–3606. [Google Scholar] [CrossRef]
- Morella, A.M.; Ward, D.A. The cis chlorination of alkenes using selenium reagents. Tetrahedron Lett. 1984, 25, 1197–1200. [Google Scholar] [CrossRef]
- Morella, A.M.; Ward, D.A. Cis 1,2-functionalization of cyclohexane using selenium intermediates. Tetrahedron Lett. 1985, 26, 2899–2900. [Google Scholar] [CrossRef]
- Engman, L. Phenylselenium trichloride in organic synthesis. Reaction with unsaturated compounds. Preparation of vinylic chlorides via selenoxide elimination. J. Org. Chem. 1987, 52, 4086–4094. [Google Scholar] [CrossRef]
- Paulmier, C. Inter and intramolecular nucleophilic substitution of activated phenylselanyl groups. Phosphorus Sulfur Silicon Relat. Elem. 2001, 172, 25–54. [Google Scholar] [CrossRef]
- Guo, R.; Huang, J.; Huang, H.; Zhao, X. Organoselenium-catalyzed synthesis of oxygen- and nitrogen-containing heterocycles. Org. Lett. 2016, 18, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, T. Palladium-catalysed arylation of 1,3-dienes: A highly chemo, regio and stereoselective synthesis of (E,E) conjugated dienic aromatics. Tetrahedron Lett. 1992, 33, 1989–1992. [Google Scholar] [CrossRef]
- Kirai, N.; Iguchi, S.; Ito, T.; Takaya, J.; Iwasawa, N. PSiP-pincer type palladium-catalyzed dehydro-genative borylation of alkenes and 1,3-dienes. Bull. Chem. Soc. Jpn. 2013, 86, 784–799. [Google Scholar] [CrossRef]
- Kohn, B.L.; Rovis, T. Cobaltate anion couples terminal dienes with trifluoroacetic anhydride: A direct fluoroacylation of 1,3-dienes. Chem. Sci. 2014, 5, 2889–2892. [Google Scholar] [CrossRef]
- Szudkowska-Frątczak, J.; Marciniec, B.; Hreczycho, G.; Kubicki, M.; Pawluć, P. Ruthenium-catalyzed silylation of 1,3-butadienes with vinylsilanes. Org. Lett. 2015, 17, 2366–2369. [Google Scholar] [CrossRef] [PubMed]
- Tanaskov, M.M. Reaction of benzenesulfonic acid haloanhydrides with 1-phenyl-1,3-butadiene. Zh. Org. Khim. 1981, 17, 1115. [Google Scholar]
- Besset, T.; Kuhl, N.; Patureau, F.W.; Glorius, F. RhIII-catalyzed oxidative olefination of vinylic C-H bonds: Efficient and selective access to di-unsaturated α-amino acid derivatives and other linear 1,3-butadienes. Chem. Eur. J. 2011, 17, 7167–7171. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Shimizu, R.; Usui, Y.; Sodeoka, M. Oxy-trifluoromethylation of alkenes and its application to the synthesis of β-trifluoromethylstyrene derivatives. J. Fluorine Chem. 2014, 167, 172–178. [Google Scholar] [CrossRef]
- Liao, L.; Guo, R.; Zhao, X. Organoselenium-catalyzed regioselective C-H pyridination of 1,3-dienes and alkenes. Angew. Chem. Int. Ed. 2017, 56, 3201–3205. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. Power and structure-variable fluorinating agents. The N-fluoropyridinium salt system. J. Am. Chem. Soc. 1990, 112, 8563–8575. [Google Scholar] [CrossRef]
- Borisov, A.V.; Matsulevich, Z.V.; Tyurin, Y.M. Pathways of reactions of 3,4-dihydropyran with hetarene-selanyl chlorides. Chem. Heterocycl. Compd. 2011, 47, 649–650. [Google Scholar] [CrossRef]
- Borisov, A.V.; Matsulevich, Z.V.; Osmanov, V.K.; Borisova, G.N. Synthesis of 2,3-dihydroselenazolo [3,2-α]pyridinium salts based on reactions of pyeidine-2-selanyl chloride with alkenes and dienes. Chem. Heterocycl. Compd. 2012, 48, 492–496. [Google Scholar] [CrossRef]
- Borisov, A.V.; Matsulevich, Z.V.; Osmanov, V.K.; Borisova, G.N.; Mammadova, G.Z.; Maharramov, A.M.; Khrustalev, V.N. Heterocyclization of cycloalkenes with di(2-pyridyl) diselenide in the presence of antimony pentachloride. Chem. Heterocycl. Compd. 2012, 48, 963–968. [Google Scholar] [CrossRef]
- Denmark, S.E.; Kornfilt, D.J.P.; Vogler, T. Catalytic asymmetric thiofunctionalization of unactivated alkenes. J. Am. Chem. Soc. 2011, 133, 15308–15311. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Jaunet, A. Catalytic, enantioselective, intramolecular carbosulfenylation of olefins. J. Am. Chem. Soc. 2013, 135, 6419–6422. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tan, C.K.; Yeung, Y.Y. C2-Symmetric cyclic selenium catalyzed enantioselective bromoamino-cyclization. J. Am. Chem. Soc. 2013, 135, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Chi, H.M. Catalytic, enantioselective, intramolecular carbosulfenylation of olefins. Mechanistic Aspects: A remarkable case of negative catalysis. J. Am. Chem. Soc. 2014, 136, 3655–3663. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Chi, H.M. Catalytic, enantioselective sulfenylation of ketone-derived enoxysilanes. J. Am. Chem. Soc. 2014, 136, 13016–13028. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Chi, H.M. Lewis base catalyzed, enantioselective, intramolecular sulfenoamination of olefins. J. Am. Chem. Soc. 2014, 136, 8915–8918. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Hartmann, E.; Kornfilt, D.J.P.; Wang, H. Mechanistic, crystallographic, and computational studies on the catalytic, enantioselective sulfenofunctionalization of alkenes. Nat. Chem. 2014, 6, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Jaunet, A. Catalytic, enantioselective, intramolecular carbosulfenylation of olefins. Preparative and stereochemical aspects. J. Org. Chem. 2014, 79, 140–171. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Kornfilt, D.J.P. Catalytic, enantioselective, intramolecular sulfenofunctionalization of alkenes with phenols. J. Org. Chem. 2017, 82, 3192–3222. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Chi, H.M. Catalytic, enantioselective, intramolecular sulfenoamination of alkenes with anilines. J. Org. Chem. 2017, 82, 3826–3843. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Iwaoka, M.; Tomoda, S. Synthesis of diaryl diselenides having chiral pyrrolidine rings with C2 symmetry. Their application to the asymmetric methoxyselenenylation of trans-β-methylstyrenes. Chem. Lett. 1994, 923–926. [Google Scholar] [CrossRef]
- Fukuzawa, S.-I.; Takahashi, K.; Kato, H.; Yamazaki, H. Asymmetric methoxyselenenylation of alkenes with chiral ferrocenylselenium reagents. J. Org. Chem. 1997, 62, 7711–7716. [Google Scholar] [CrossRef]
- Wirth, T.; Häuptli, S.; Leuenberger, M. Catalytic asymmetric oxyselenenylation–elimination reactions using chiral selenium compounds. Tetrahedron Asymmetry 1998, 9, 547–550. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Santi, C.; Tomassini, C.; Marini, F.; Bagnoli, L.; Temperini, A. New nitrogen containing chiral diselenides: Synthesis and asymmetric addition reactions to olefins. Tetrahedron Asymmetry 2000, 11, 4645–4650. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Santi, C.; Tomassini, C.; Marini, F.; Bagnoli, L.; Temperini, A. Preparation of a new chiral non-racemic sulfur-containing diselenide and applications in asymmetric synthesis. Chem. Eur. J. 2002, 8, 1118–1124. [Google Scholar] [CrossRef]
- Kawamata, Y.; Hashimoto, T.; Maruoka, K. A chiral electrophilic selenium catalyst for highly enantioselective oxidative cyclization. J. Am. Chem. Soc. 2016, 138, 5205–5209. [Google Scholar] [CrossRef] [PubMed]














© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, R.; Liao, L.; Zhao, X. Electrophilic Selenium Catalysis with Electrophilic N-F Reagents as the Oxidants. Molecules 2017, 22, 835. https://doi.org/10.3390/molecules22050835
Guo R, Liao L, Zhao X. Electrophilic Selenium Catalysis with Electrophilic N-F Reagents as the Oxidants. Molecules. 2017; 22(5):835. https://doi.org/10.3390/molecules22050835
Chicago/Turabian StyleGuo, Ruizhi, Lihao Liao, and Xiaodan Zhao. 2017. "Electrophilic Selenium Catalysis with Electrophilic N-F Reagents as the Oxidants" Molecules 22, no. 5: 835. https://doi.org/10.3390/molecules22050835
APA StyleGuo, R., Liao, L., & Zhao, X. (2017). Electrophilic Selenium Catalysis with Electrophilic N-F Reagents as the Oxidants. Molecules, 22(5), 835. https://doi.org/10.3390/molecules22050835