
Synthesis and Excellent Duplex Stability of Oligonucleotides Containing 2′-Amino-LNA Functionalized with Galactose Units


Abstract
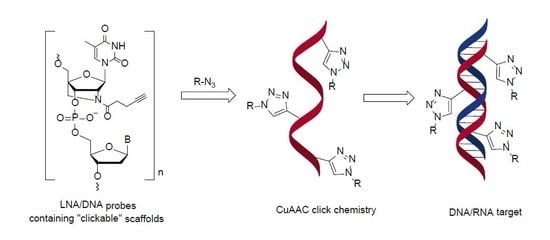
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Azido-Functionalized Galactose Derivatives
2.2. ON Synthesis
2.3. Post-Oligomerization CuAAC Click Chemistry
2.4. Thermal Denaturation Studies
2.4.1. Binding Affinity
2.4.2. Binding Specificity
3. Materials and Methods
3.1. General Information
3.2. Synthesis of 2-[2-(2-Tert-butyloxycarbonylamidoethoxy)ethoxy]ethoxy 2,3,4,6-tetra-O-acetyl-β-d-galactopyranoside (3)
3.3. Synthesis of 2-Azidoacetyl-N-{tris[3-(ethylcarboxylethoxy)methyl]methyl}-amine (5)
3.4. Synthesis of 2-Azidoacetyl-N-{tris[(2-carboxyethoxy)methyl]methyl}amine (6)
3.5. Synthesis of 2-Azidoacetyl-N-{tris[3-(pentafluorophenylcarboxylethoxy)methyl]methyl}amine (7)
3.6. Synthesis of Triantennary Azido 2,3,4,6-Tetra-O-Acetyl-β-d-Galactopyranoside (8)
3.7. Synthesis of Triantennary Azido 2-β-d-Galactopyranoside (9)
3.8. Synthesis and Purification of ON1–ON2
3.9. Thermal Denaturation Studies
3.10. Postsynthetic Click Procedure in Solution
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shabanpoor, F.; McClorey, G.; Saleh, A.F.; Jarver, P.; Wood, M.J.A.; Gait, M.J. Bi-specific splice-switching PMO oligonucleotides conjugated via a single peptide active in a mouse model of Duchenne muscular dystrophy. Nucleic Acids Res. 2015, 43, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Sessler, J.L.; Lawrence, C.M.; Jayawickramarajah, J. Molecular recognition via base-pairing. Chem. Soc. Rev. 2007, 36, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Boersma, A.J.; Megens, R.P.; Feringa, B.L.; Roelfes, G. DNA-based asymmetric catalysis. Chem. Soc. Rev. 2010, 39, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Keefe, A.D. Building oligonucleotide therapeutics using non-natural chemistries. Curr. Opin. Chem. Biol. 2006, 10, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P. An overview of sugar-modified oligonucleotides for antisense therapeutics. Chem. Biodivers. 2011, 8, 1616–1641. [Google Scholar] [CrossRef] [PubMed]
- Kool, E.T. Replacing the nucleobases in DNA with designer molecules. Acc. Chem. Res. 2002, 35, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.N.; Kool, E.T. Fluorescent DNA base replacements: Reporters and sensors for biological systems. Org. Biomol. Chem. 2006, 4, 4265–4274. [Google Scholar] [CrossRef] [PubMed]
- Sipa, K.; Sochacka, E.; Kazmierczak-Baranska, J.; Maszewska, M.; Janicka, M.; Nowak, G.; Nawrot, B. Effect of base modifications on structure, thermodynamic stability, and gene silencing activity of short interfering RNA. RNA 2007, 13, 1301–1316. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.H.; Lim, S.; Wong, W.S. Antisense oligonucleotides: From design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Micklefield, J. Backbone modification of nucleic acids: Synthesis, structure and therapeutic applications. Curr. Med. Chem. 2001, 8, 1157–1179. [Google Scholar] [CrossRef] [PubMed]
- Meints, G.A.; Karlsson, T.; Drobny, G.P. Modeling furanose ring dynamics in DNA. J. Am. Chem. Soc. 2001, 123, 10030–100038. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, G.M.; Gait, M.J.; Loakes, D.; Williams, D.M. DNA and RNA structure. In Nucleic Acids in Chemistry and Biology, 3rd ed.; RCS Publishing: Cambridge, UK, 2006; pp. 13–75. [Google Scholar]
- Plashkevych, O.; Chatterjee, S.; Honcharenko, D.; Pathmasiri, W.; Chattopadhyaya, J. Chemical and structural implications of 1′,2′-versus 2′,4′-conformational constraints in the sugar moiety of modified thymine nucleosides. J. Org. Chem. 2007, 72, 4716–4726. [Google Scholar] [CrossRef] [PubMed]
- Meldgaard, M.; Wengel, J. Bicyclic nucleosides and conformational restriction of oligonucleotides. J. Chem. Soc. Perkin Trans. 1 2000, 353, 3539–3554. [Google Scholar] [CrossRef]
- Leumann, C.J. DNA analogues: From supramolecular principles to biological properties. Bioorg. Med. Chem. 2002, 10, 841–854. [Google Scholar] [CrossRef]
- Enderlin, G.; Nielsen, P. Synthesis of 6′-branched locked nucleic acid by a radical TEMPO-scavanged stereoselective mercury cyclization. J. Org. Chem. 2008, 73, 6891–6894. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Koshkin, A.A.; Wengel, J.; Nielsen, P. LNA (locked nucleic acids): Synthesis and high-affinity nucleic acid recognition. Chem. Commun. 1998, 455–456. [Google Scholar] [CrossRef]
- Obika, S.; Nanbu, D.; Hari, Y.; Andoh, J.; Morio, K.; Doi, T.; Imanishi, T. Stability and structural features of the duplex containing nucleoside analogues with a fixed N-type conformation, 2′-O,4′-C-methyleneribonucleotieds. Tetrahedron Lett. 1998, 39, 5401–5404. [Google Scholar] [CrossRef]
- Wengel, J. Synthesis of 3′-C- and 4′-C-branched oligodeoxynucleotides and the development of locked nucleic acid (LNA). Acc. Chem. Res. 1999, 32, 301–310. [Google Scholar] [CrossRef]
- Bell, N.M.; Micklefield, J. Chemical modification of oligonucleotides for therapeutic, bioanalytical and other applications. ChemBioChem 2009, 10, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakatani, M.; Narukawa, K.; Obika, S. Antisense drug discovery and development. Future Med. Chem. 2011, 3, 339–365. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.R.; Brown, T. Formation of stable DNA triplexes. Biochem. Soc. Trans. 2011, 39, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Deleavey, G.F.; Damha, M.J. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 2012, 19, 937–954. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.E.; Singh, S.K.; Wengel, J.; Jacobsen, J.P. Solution structure of an LNA hybridized to DNA: NMR study of the d(CT(L)GCT(L)T(L)CT(L)GC):d(GCAGAAGCAG) duplex containing four locked nucleotides. Bioconjugate Chem. 2000, 11, 228–238. [Google Scholar] [CrossRef]
- Jespen, J.S.; Wengel, J. LNA-antisense rivals siRNA for gene silencing. Curr. Opin. Drug Discov. Dev. 2004, 7, 188–194. [Google Scholar]
- Frieden, M.; Ørum, H. The application of locked nucleic acids in the treatment of cancer. IDrugs 2006, 9, 706–711. [Google Scholar] [PubMed]
- Grunweller, A.; Hartmann, R.K. Locked nucleic acid oligonucleotides: The next generation of antisense agents? Biodrugs 2007, 21, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Silahtaroglu, A.N.; Lindow, M.; Fimen, J.; Kauppinen, S. The utility of LNA in microRNA-based cancer diagnostics and therapeutics. Semin. Cancer Biol. 2008, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Kumar, R.; Wengel, J. Synthesis of 2′-amino-LNA: A novel conformationally restricted high-affinity oligonucleotide analogue with a handle. J. Org. Chem. 1998, 63, 10035–10039. [Google Scholar] [CrossRef]
- Madsen, A.S.; Jørgensen, A.S.; Jensen, T.B.; Wengel, J. Large scale synthesis of 2′-amino-LNA thymine and 5-methylcytosine nucleosides. J. Org. Chem. 2012, 77, 10718–10728. [Google Scholar] [CrossRef] [PubMed]
- Astakhova, I.K.; Wengel, J. Scaffolding along nucleic acid duplexes using 2′-amino-locked nucleic acids. Acc. Chem. Res. 2014, 47, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, X.-L.; Li, X.R. Research progress on siRNA delivery with nonviral carriers. Int. J. Nanomed. 2011, 6, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.J.; Kalra, N.; Wengel, J.; Vester, B. Aptamers as a model for functional evaluation of LNA and 2′-amino LNA. Bioorg. Med. Chem. Lett. 2009, 19, 6585–6587. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, K.K.; Wengel, J. Locked nucleic acid and aptamers. Nucleic Acid Ther. 2012, 22, 366–370. [Google Scholar] [PubMed]
- Weisbrod, S.H.; Marx, A. Novel strategies for the site-specific covalent labelling of nucleic acids. Chem. Commun. 2008, 5675–5685. [Google Scholar] [CrossRef] [PubMed]
- Lönnberg, H. Solid-phase synthesis of oligonucleotide conjugates useful for delivery and targeting of potential nucleic acid therapeutics. Bioconjugate Chem. 2009, 20, 1065–1094. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, G.; Harford, J. Carbohydrate-specific receptors of the liver. Annu. Rev. Biochem. 1982, 51, 531–554. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, N.; Defrancq, E.; Morvan, F. Glycoclusters on oligonucleotide and PNA scaffolds: Synthesis and applications. Chem. Soc. Rev. 2013, 42, 4557–4573. [Google Scholar] [CrossRef] [PubMed]
- Sliedregt, L.A.J.M.; Rensen, P.C.N.; Rump, E.T.; van Santbrink, P.J.; Bijsterbosch, M.K.; Valentijn, A.R.P.M.; van der Marel, G.A.; van Boom, J.H.; van Berkel, T.J.C.; Biessen, E.A.L. Design and synthesis of novel amphiphilic dendritic galactosides for selective targeting of liposomes to the hepatic asialoglycoprotein receptor. J. Med. Chem. 1999, 42, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Westerlind, U.; Westman, J.; Törnquist, E.; Smith, C.I.E.; Oscarson, S.; Lahmann, M.; Norberg, T. Ligands of the asialoglycoprotein receptor for targeted gene delivery, part 1: Synthesis of and binding studies with biotinylated cluster glycosides containing N-acetylgalactosamine. Glycoconj. J. 2004, 21, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Khorev, O.; Stokmaier, D.; Schwardt, O.; Cutting, B.; Ernst, B. Trivalent, Gal/GalNAc-containing ligands designed for the asialoglycoprotein receptor. Bioorg. Med. Chem. 2008, 16, 5216–5231. [Google Scholar] [CrossRef] [PubMed]
- Mäkilä, J.; Jadhav, S.; Kiviniemi, A.; Käkelä, M.; Liljenbäck, H.; Poijärvi-Virta, P.; Laitala-Leinonen, T.; Lönnberg, H.; Roivainen, A.; Virta, P. Synthesis of multi-galactose-conjugated 2′-O-methyl oligoribonucleotides and their in vivo imaging with positron emission tomography. Bioorg. Med. Chem. 2014, 22, 6806–6813. [Google Scholar] [CrossRef] [PubMed]
- Positive Initial Phase 2 Data with Revusiran (ALN-TTRsc). Available online: http://www.alnylam.com/capella/presentations/positive-initial-revusiran-phase-2-data/ (accessed on 25 April 2017).
- Østergaard, M.E.; Yu, J.; Kinberger, G.A.; Wan, W.B.; Migawa, M.T.; Vasquez, G.; Schmidt, K.; Gaus, H.J.; Murray, H.M.; Low, A.; et al. Efficient synthesis and biological evaluation of 5′-GalNAc conjugated antisense oligonucleotides. Bioconjugate Chem. 2015, 26, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Wan, W.B.; Low, A.; Yu, J.; Chappell, A.E.; Gaus, H.; Kinberger, G.A.; Østergaard, M.E.; Migawa, M.T.; Swayze, E.E.; et al. Solid-phase synthesis of 5′-triantennary N-acetylgalactosamine conjugated antisense oligonucleotides using phosphoramidite chemistry. Bioorg. Med. Chem. Lett. 2015, 25, 4127–4130. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Keiser, K.; Nair, J.K.; Charisse, K.; Manoharan, R.M.; Kretschmer, P.; Peng, C.G.; Kel’in, A.V.; Kandasamy, P.; Willoughby, J.L.S.; et al. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem. Biol. 2015, 10, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Rajeev, K.G.; Nair, J.K.; Jayaraman, M.; Charisse, K.; Taneja, N.; O′Shea, J.; Willoughby, J.L.; Yucius, K.; Nguyen, T.; Shulga-Morskaya, S.; et al. Hepatocyte-specific delivery of siRNAs conjugated to novel non-nucleosidic trivalent N-acetylgalactosamine elicits robust gene silencing in vivo. ChemBioChem 2015, 16, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Hong, V.; Presolski, S.I.; Ma, C.; Finn, M.G. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed. 2009, 48, 9879–9883. [Google Scholar] [CrossRef] [PubMed]
- Ustinov, A.V.; Stepanova, I.A.; Dubnyakova, V.V.; Zatsepin, T.S.; Nozhevnikova, E.V.; Korshun, V.A. Modification of nucleic acids using [3 + 2]-dipolar cycloaddition of azides and alkynes. Rus. J. Bioorg. Chem. 2010, 36, 401–445. [Google Scholar] [CrossRef]
- El-Sagheer, A.H.; Brown, T. Click chemistry with DNA. Chem. Soc. Rev. 2010, 39, 1388–1405. [Google Scholar] [CrossRef] [PubMed]
- Kiviniemi, A.; Virta, P.; Lönnberg, H. Utilization of intrachain 4′-C-azidomethylthymidine for preparation of oligodeoxyribonucleotide conjugates by click chemistry in solution and on a solid support. Bioconjugate Chem. 2008, 19, 1726–1734. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, P.M.E.; Warncke, S.; Gierlich, J.; Carell, T. Click-click-click: Single to triple modification of DNA. Angew. Chem. 2008, 47, 349–3493. [Google Scholar] [CrossRef] [PubMed]
- Berndl, S.; Herzig, N.; Kele, P.; Lachmann, D.; Li, X.; Wolfbeis, O.S.; Wagenknecht, H.-A. Comparison of a nucleosidic vs. non-nucleosidic postsynthetic “click” modification of DNA with base-labile fluorescent probes. Bioconjugate Chem. 2009, 20, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, A.S.; Gupta, P.; Wengel, J.; Astakhova, I.K. “Clickable” LNA/DNA probes for fluorescence sensing of nucleic acids and autoimmune antibodies. Chem. Commun. 2013, 49, 10751–10753. [Google Scholar] [CrossRef] [PubMed]
- Astakhova, K.; Wengel, J. Interfacing click chemistry with automated oligonucleotide synthesis for the preparation of fluorescent DNA probes containing internal xanthene and cyanine dyes. Chem. Eur. J. 2013, 19, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Astakhova, K.; Kumar, T.S.; Campbell, M.A.; Ustinov, A.V.; Korshun, V.A.; Wengel, J. Branched DNA nanostructures efficiently stabilised and monitored by novel pyrene-perylene 2′-α-L-amino-LNA FRET pairs. Chem. Commun. 2013, 49, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Astakhova, K.; Hansen, L.H.; Vester, B.; Wengel, J. Peptide-LNA oligonucleotide conjugates. Org. Biomol. Chem. 2013, 11, 4240–4249. [Google Scholar] [CrossRef] [PubMed]
- Aertner, L.M.; Merkel, L.; Bohlke, N.; Braun, F.B.; Weise, C.; Dernedde, J.; Budisa, N.; Hackenberger, C.P.R. Site-selective modification of proteins for the synthesis of structurally defined multivalent scaffolds. Chem. Commun. 2012, 48, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Lai, C.; Chien, C.; Liang, C.; Adak, A.K.; Chuang, Y.; Chen, Y.; Lin, C. Synthesis and evaluation of a photoactive probe with a multivalent carbohydrate for capturing carbohydrate-lectin interactions. Bioconjugate Chem. 2013, 24, 1895–1906. [Google Scholar] [CrossRef] [PubMed]
- Kikkeri, R.; Liu, X.; Adibekian, A.; Tsaia, Yu-H.; Seeberger, P.H. Facile synthesis of size dependent Ru(II)-carbohydrate dendrimers via click chemistry. Chem. Commun. 2010, 46, 2197–2199. [Google Scholar] [CrossRef] [PubMed]
- Lou, C.; Vester, B.; Wengel, J. Oligonucleotides containing a piperazino-modified 2′-amino-LNA monomer exhibit very high duplex stability and remarkable nuclease resistance. Chem. Commun. 2015, 51, 4024–4027. [Google Scholar] [CrossRef] [PubMed]
- Nabo, L.J.; Madsen, C.S.; Jensen, K.J.; Kongsted, J.; Astakhova, K. Ultramild protein-mediated click chemistry creates efficient oligonucleotide probes for targeting and detecting nucleic acids. ChemBioChem 2015, 16, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Ries, A.; Kumar, R.; Lou, C.; Kosbar, T.; Vengut-Climent, E.; Jørgensen, P.T.; Morales, J.C.; Wengel, J. Synthesis and biophysical investigations of oligonucleotides containing galactose-modified DNA, LNA, and 2′-amino-LNA monomers. J. Org. Chem. 2016, 81, 10845–10856. [Google Scholar] [CrossRef] [PubMed]
- Lou, C.; Samuelsen, S.V.; Christensen, N.J.; Vester, B.; Wengel, J. Oligonucleotides containing aminated 2′-amino-LNA nucleotides: Synthesis and strong binding to complementary DNA and RNA. Bioconjugate Chem. 2017, 28, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lindhorst, T.K. A modular approach for the synthesis of oligosaccharide mimetics. J. Org. Chem. 2001, 66, 2674–2680. [Google Scholar] [CrossRef] [PubMed]
- Kværnø, L.; Kumar, R.; Dahl, B.M.; Olsen, C.E.; Wengel, J. Synthesis of abasic locked nucleic acid and two seco-LNA derivatives and evaluation of their hybridization properties compared with their more flexible DNA counterparts. J. Org. Chem. 2000, 65, 5167–5176. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
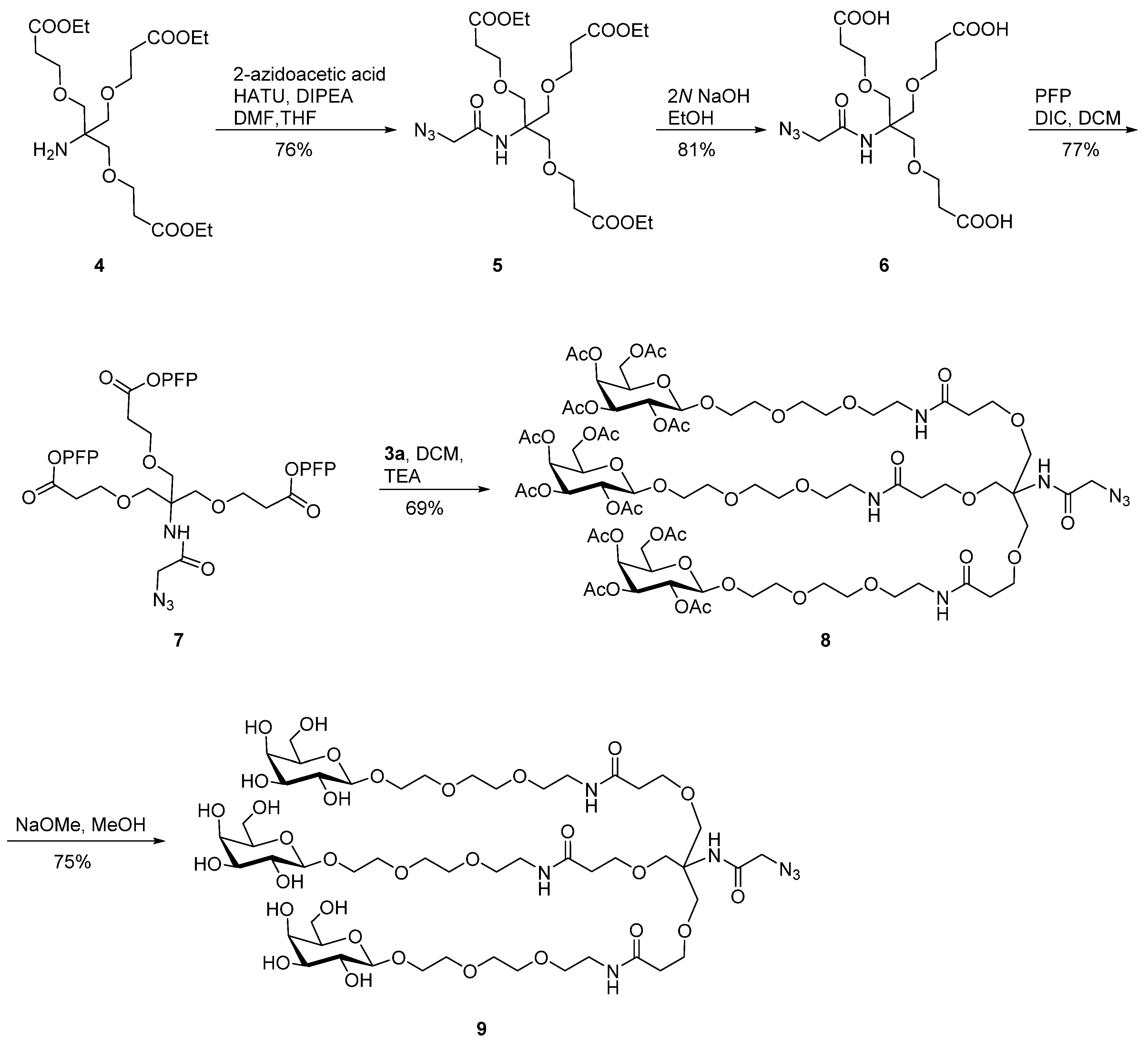{kind=link}
{kind=link}
| ON | Sequence | DNA: 3′-CACTATACG Tm/ΔTm (°C) | RNA: 3′-CACUAUACG Tm/ΔTm (°C) |
|---|---|---|---|
| ONref | 5′-GTGATATGC | 29.0 | 27.0 |
| ON3 | 5′-GTGAM2ATGC | 33.0/+4.0 | 35.5/+8.5 |
| ON4 | 5′-GM2GAM2AM2GC | 40.0/+11.0 | 50.5/+23.5 |
| ON5 | 5′-GTGAM3ATGC | 31.0/+2.0 | 34.5/+7.5 |
| ON6 | 5′-GM3GAM3AM3GC | 37.5/+8.5 | 48.5/+21.5 |
| ON7 | 5′-GTGAM4ATGC | 33.0/+4.0 | 35.0/+8.0 |
| ON8 | 5′-GM4GAM4AM4GC | 38.5/+9.5 | 47.0/+20.0 |
| ON | Sequence X = | DNA: 3′-CACTXTACG | RNA: 3′-CACUXUACG | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Tm/°C | ΔTm/°C | Tm/°C | ΔTm/°C | ||||||
| A | T | G | C | A | U | G | C | ||
| ONref | 5′-GTGATATGC | 29.0 | −15.0 | −9.5 | −16.0 | 27.0 | −16.0 | −5.0 | −21.0 |
| ON3 | 5′-GTGAM2ATGC | 33.0 | −16.0 | −15.0 | −18.0 | 35.5 | −17.5 | −10.5 | −17.5 |
| ON4 | 5′-GM2GAM2AM2GC | 40.0 | −15.5 | −17.5 | −18.0 | 50.5 | −16.5 | −11.0 | −15.5 |
| ON5 | 5′-GTGAM3ATGC | 31.0 | −16.5 | −16.0 | −18.0 | 34.5 | −17.5 | −10.0 | −18.0 |
| ON6 | 5′-GM3GAM3AM3GC | 37.5 | −15.5 | −15.0 | −18.0 | 48.5 | −15.5 | −10.0 | −15.0 |
| ON7 | 5′-GTGAM4ATGC | 33.0 | −16.0 | −14.5 | −18.5 | 35.0 | −17.0 | −9.0 | −17.0 |
| ON8 | 5′-GM4GAM4AM4GC | 38.5 | −16.0 | −15.0 | −18.5 | 47.0 | −15.5 | −9.0 | −15.5 |
| ON | Sequence | Calculated | Found |
|---|---|---|---|
| ON1 | 5′-GTGAM1ATGC | 2861.18 | 2858.69 |
| ON2 | 5′GM1GAM1AM1GC | 3075.74 | 3075.78 |
| ON | Sequence | Calculated | Found |
|---|---|---|---|
| ON3 | 5′-GTGAM2ATGC | 3198.33 | 3198.25 |
| ON4 | 5′-GM2GAM2AM2GC | 4087.19 | 4087.93 |
| ON5 | 5′-GTGAM3ATGC | 4160.77 | 4160.66 |
| ON6 | 5′-GM3GAM3AM3GC | 6974.51 | 6974.76 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, R.; Ries, A.; Wengel, J. Synthesis and Excellent Duplex Stability of Oligonucleotides Containing 2′-Amino-LNA Functionalized with Galactose Units. Molecules 2017, 22, 852. https://doi.org/10.3390/molecules22050852
Kumar R, Ries A, Wengel J. Synthesis and Excellent Duplex Stability of Oligonucleotides Containing 2′-Amino-LNA Functionalized with Galactose Units. Molecules. 2017; 22(5):852. https://doi.org/10.3390/molecules22050852
Chicago/Turabian StyleKumar, Rajesh, Annika Ries, and Jesper Wengel. 2017. "Synthesis and Excellent Duplex Stability of Oligonucleotides Containing 2′-Amino-LNA Functionalized with Galactose Units" Molecules 22, no. 5: 852. https://doi.org/10.3390/molecules22050852
APA StyleKumar, R., Ries, A., & Wengel, J. (2017). Synthesis and Excellent Duplex Stability of Oligonucleotides Containing 2′-Amino-LNA Functionalized with Galactose Units. Molecules, 22(5), 852. https://doi.org/10.3390/molecules22050852