
Development of Optimized Inhibitor RNAs Allowing Multisite-Targeting of the HCV Genome




Abstract
:1. Introduction
2. Results
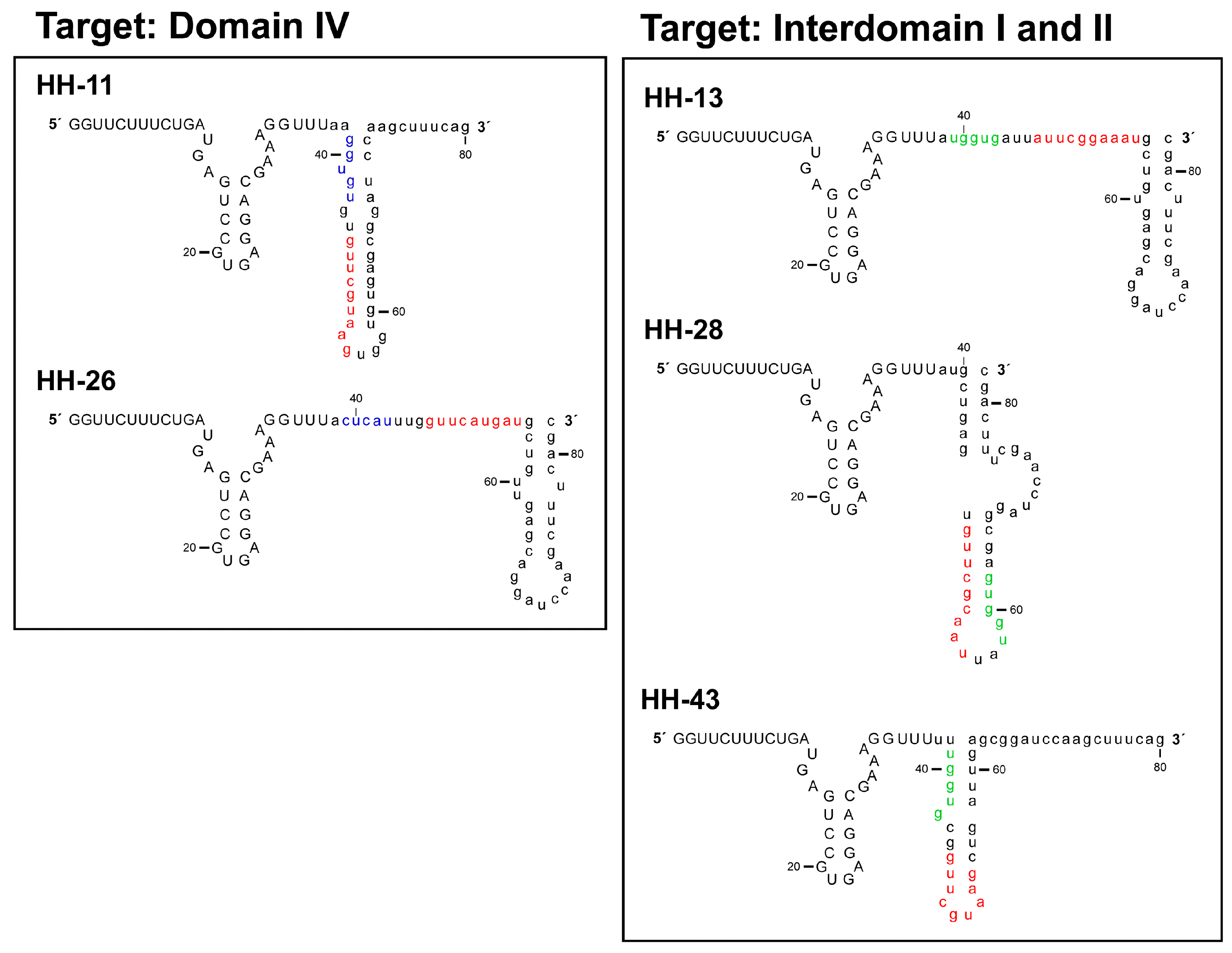
2.1. Isolation of Improved RNA Molecules that Interfere with HCV IRES Function In Vitro
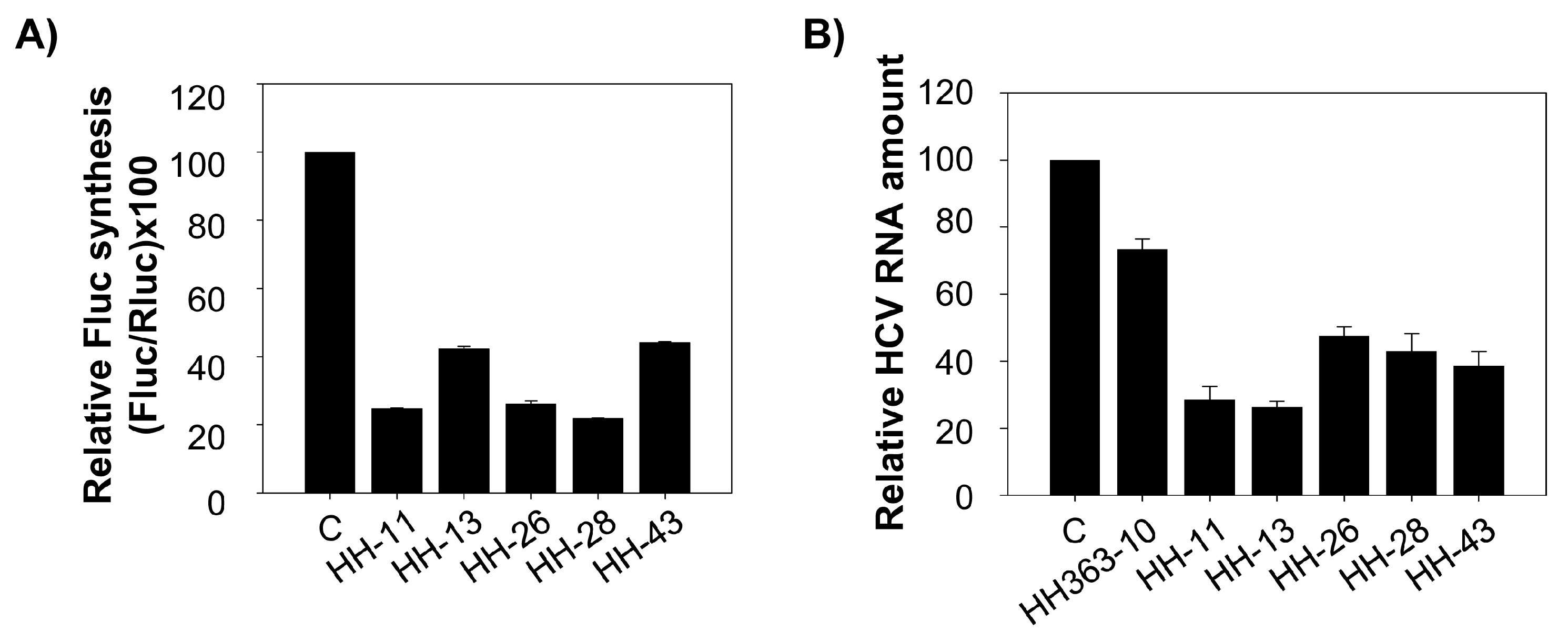
2.2. The Chimeric Inhibitor RNAs Inhibit HCV IRES-Dependent Translation in Cell Culture
2.3. Interference with HCV Replication by the Chimeric Inhibitors
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. DNA Templates and RNA Synthesis
4.3. In Vitro Translation Assays
4.4. RNA Transfection
4.5. Relative Quantification of HCV Subgenomic RNA
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Diafa, S.; Hollenstein, M. Generation of aptamers with an expanded chemical repertoire. Molecules 2015, 20, 16643–16671. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In Vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Nimjee, S.M.; Keys, J.R.; Pitoc, G.A.; Quick, G.; Rusconi, C.P.; Sullenger, B.A. A novel antidote-controlled anticoagulant reduces thrombin generation and inflammation and improves cardiac function in cardiopulmonary bypass surgery. Mol. Ther. 2006, 14, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Oney, S.; Lam, R.T.; Bompiani, K.M.; Blake, C.M.; Quick, G.; Heidel, J.D.; Liu, J.Y.; Mack, B.C.; Davis, M.E.; Leong, K.W.; et al. Development of universal antidotes to control aptamer activity. Nat. Med. 2009, 15, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Bompiani, K.M.; Woodruff, R.S.; Becker, R.C.; Nimjee, S.M.; Sullenger, B.A. Antidote control of aptamer therapeutics: The road to a safer class of drug agents. Curr. Pharm. Biotechnol. 2012, 13, 1924–1934. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Savory, N.; Abe, K.; Ikebukuro, K. Methods for improving aptamer binding affinity. Molecules 2016, 21, 421. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Huang, R.; Deng, Y.; He, N. Progress in selection and biomedical applications of aptamers. J. Biomed. Nanotechnol. 2014, 10, 3043–3062. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zheng, X.; Jiao, B.; Wang, L. Post-SELEX optimization of aptamers. Anal. Bioanal. Chem. 2016, 408, 4567–4573. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, V.M.; Martin, M.E.; Fernandez, G.; García-Sacristán, A. Use of aptamers as diagnostics tools and antiviral agents for human viruses. Pharmaceuticals (Basel) 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Jijakli, K.; Khraiwesh, B.; Fu, W.; Luo, L.; Alzahmi, A.; Koussa, J.; Chaiboonchoe, A.; Kirmizialtin, S.; Yen, L.; Salehi-Ashtiani, K. The In Vitro selection world. Methods 2016, 106, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Gawande, B.N.; Rohloff, J.C.; Carter, J.D.; von Carlowitz, I.; Zhang, C.; Schneider, D.J.; Janjic, N. Selection of DNA aptamers with two modified bases. Proc. Natl. Acad. Sci. USA 2017, 114, 2898–2903. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Barroso-delJesus, A.; Puerta-Fernández, E.; Berzal-Herranz, A. Interfering with hepatitis C virus IRES activity using RNA molecules identified by a novel In Vitro selection method. Biol. Chem. 2005, 386, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Díaz-González, R.; Berzal-Herranz, A. Inhibition of hepatitis C virus internal ribosome entry site-mediated translation by an RNA targeting the conserved IIIf domain. Cell Mol. Life Sci. 2007, 64, 2994–3006. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Díaz-González, R.; Barroso-delJesus, A.; Berzal-Herranz, A. Inhibition of HCV replication and IRES-dependent translation by an RNA molecule. J. Gen. Virol. 2009, 90, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Berzal-Herranz, B.; Gomez, J.; Berzal-Herranz, A. An engineered inhibitor RNA that efficiently interferes with hepatitis C virus translation and replication. Antivir. Res. 2012, 94, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Vorobyeva, M.; Vorobjev, P.; Venyaminova, A. Multivalent aptamers: Versatile tools for diagnostic and therapeutic applications. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Berry, K.E.; Waghray, S.; Doudna, J.A. The HCV IRES pseudoknot positions the initiation codon on the 40S ribosomal subunit. RNA 2010, 16, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Berry, K.E.; Waghray, S.; Mortimer, S.A.; Bai, Y.; Doudna, J.A. Crystal structure of the HCV IRES central domain reveals strategy for start-codon positioning. Structure 2011, 19, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Mueckstein, U.; Tafer, H.; Hackermueller, J.; Bernhart, S.H.; Stadler, P.F.; Hofacker, I.L. Thermodynamics of RNA-RNA binding. Bioinformatics. 2006, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Grace, K.; Gartland, M.; Karayiannis, P.; McGarvey, M.J.; Clarke, B. The 5′ untranslated region of GB virus B shows functional similarity to the internal ribosome entry site of hepatitis C virus. J. Gen. Virol. 1999, 80, 2337–2341. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Korner, F.; Dobierzewska, A.; Bartenschlager, R. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J. Virol. 2001, 75, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Larrea, E.; Aldabe, R.; Molano, E.; Fernandez-Rodriguez, C.M.; Ametzazurra, A.; Civeira, M.P.; Prieto, J. Altered expression and activation of signal transducers and activators of transcription (STATs) in hepatitis C virus infection: In Vivo and In Vitro studies. Gut 2006, 55, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Lapa, S.A.; Chudinov, A.V.; Timofeev, E.N. The toolbox for modified aptamers. Mol. Biotechnol. 2016, 58, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Cao, Z.; Tan, W. Molecular assembly for high-performance bivalent nucleic acid inhibitor. Proc. Natl. Acad. Sci. USA 2008, 105, 5664–5669. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Brown, E.A.; Lemon, S.M. Stability of a stem-loop involving the initiator AUG controls the efficiency of internal initiation of translation on hepatitis C virus RNA. RNA 1996, 2, 955–968. [Google Scholar] [PubMed]
- Beguiristain, N.; Robertson, H.D.; Gomez, J. RNase III cleavage demonstrates a long range RNA: RNA duplex element flanking the hepatitis C virus internal ribosome entry site. Nucleic Acids Res. 2005, 33, 5250–5261. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schuttler, C.G.; Fehr, C.; Junemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.P.; Lewis, A.P.; Jopling, C.L. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res. 2011, 39, 7716–7729. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sanles, A.; Berzal-Herranz, B.; González-Matamala, R.; Ríos-Marco, P.; Romero-López, C.; Berzal-Herranz, A. RNA aptamers as molecular tools to study the functionality of the hepatitis C virus CRE region. Molecules 2015, 20, 16030–16047. [Google Scholar] [CrossRef] [PubMed]
- Martell, M.; Gomez, J.; Esteban, J.I.; Sauleda, S.; Quer, J.; Cabot, B.; Esteban, R.; Guardia, J. High-throughput real-time reverse transcription-PCR quantitation of hepatitis C virus RNA. J. Clin. Microbiol. 1999, 37, 327–332. [Google Scholar] [PubMed]
- Barroso-delJesus, A.; Romero-Lopez, C.; Lucena-Aguilar, G.; Melen, G.J.; Sanchez, L.; Ligero, G.; Berzal-Herranz, A.; Menendez, P. Embryonic stem cell-specific miR302–367 cluster: Human gene structure and functional characterization of its core promoter. Mol. Cell Biol. 2008, 28, 6609–6619. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the different chimeric inhibitory RNAs are available from the authors. |






{kind=link}
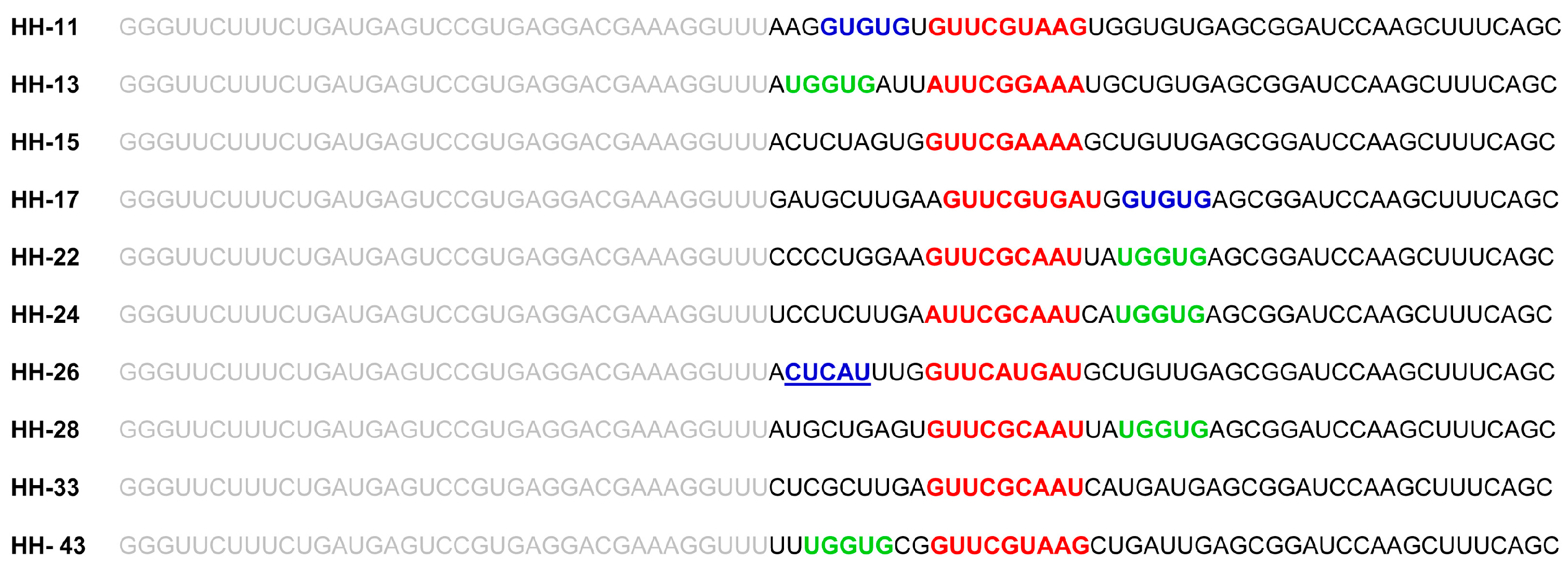{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 (µM) a | Relative Fluc Synthesis (%) b |
|---|---|---|
| HH363-10 [14] | 0.15 ± 0.04 | 11.22 ± 2.77 |
| HH-11 | 0.17 ± 0.02 | 0.01 ± 1.10 |
| HH-13 | 0.91 ± 0.31 | 8.78 ± 2.21 |
| HH-26 | 1.46 ± 0.24 | 9.25 ± 1.47 |
| HH-28 | 0.44 ± 0.06 | 2.56 ± 1.34 |
| HH-43 | 1.02 ± 0.15 | 1.00 ± 0.40 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-López, C.; Lahlali, T.; Berzal-Herranz, B.; Berzal-Herranz, A. Development of Optimized Inhibitor RNAs Allowing Multisite-Targeting of the HCV Genome. Molecules 2017, 22, 861. https://doi.org/10.3390/molecules22050861
Romero-López C, Lahlali T, Berzal-Herranz B, Berzal-Herranz A. Development of Optimized Inhibitor RNAs Allowing Multisite-Targeting of the HCV Genome. Molecules. 2017; 22(5):861. https://doi.org/10.3390/molecules22050861
Chicago/Turabian StyleRomero-López, Cristina, Thomas Lahlali, Beatriz Berzal-Herranz, and Alfredo Berzal-Herranz. 2017. "Development of Optimized Inhibitor RNAs Allowing Multisite-Targeting of the HCV Genome" Molecules 22, no. 5: 861. https://doi.org/10.3390/molecules22050861
APA StyleRomero-López, C., Lahlali, T., Berzal-Herranz, B., & Berzal-Herranz, A. (2017). Development of Optimized Inhibitor RNAs Allowing Multisite-Targeting of the HCV Genome. Molecules, 22(5), 861. https://doi.org/10.3390/molecules22050861