Fluorination of Naturally Occurring N6-Benzyladenosine Remarkably Increased Its Antiviral Activity and Selectivity

 , ,
, ,
Abstract
:1. Introduction
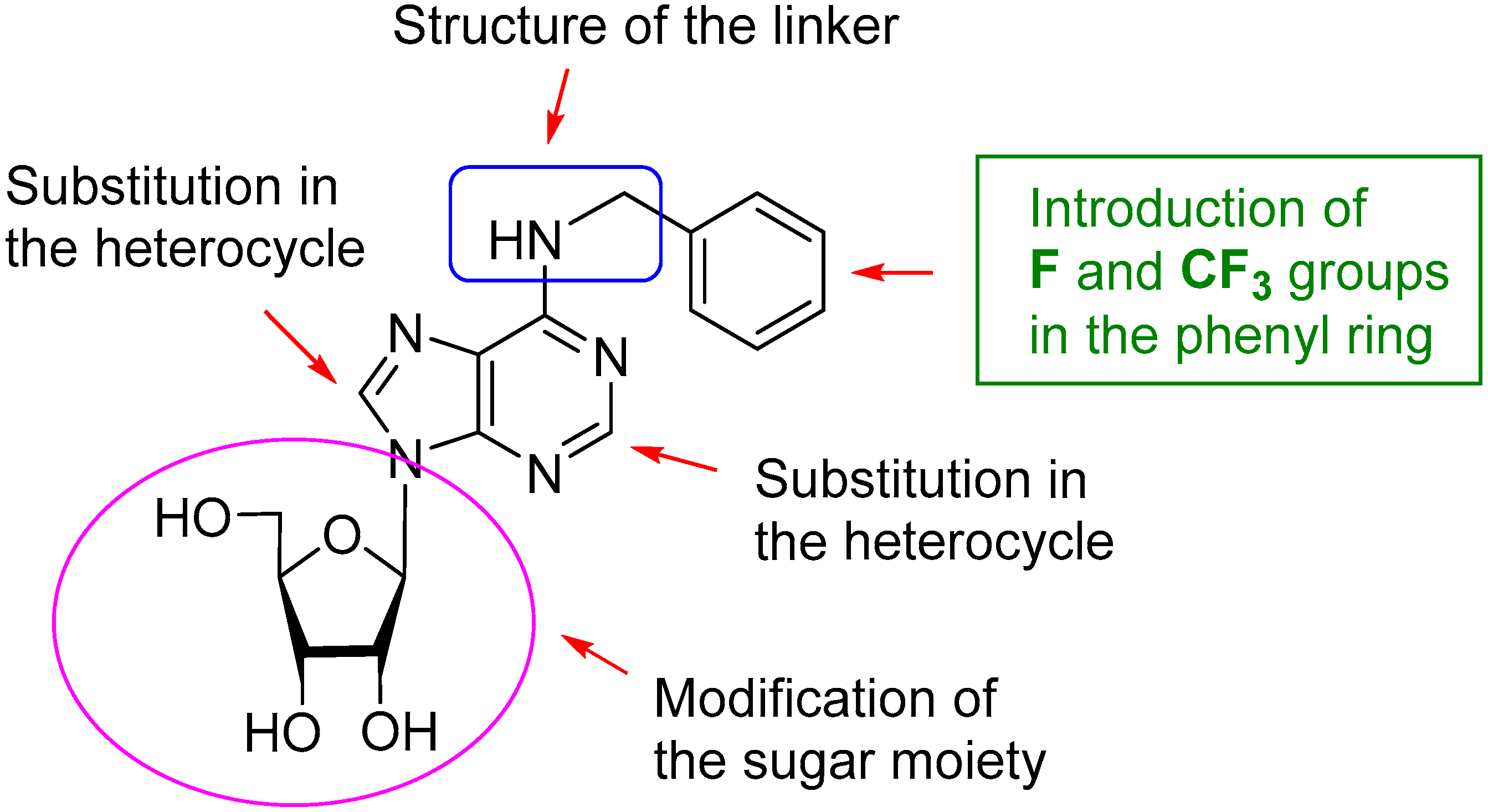
2. Results and Discussion
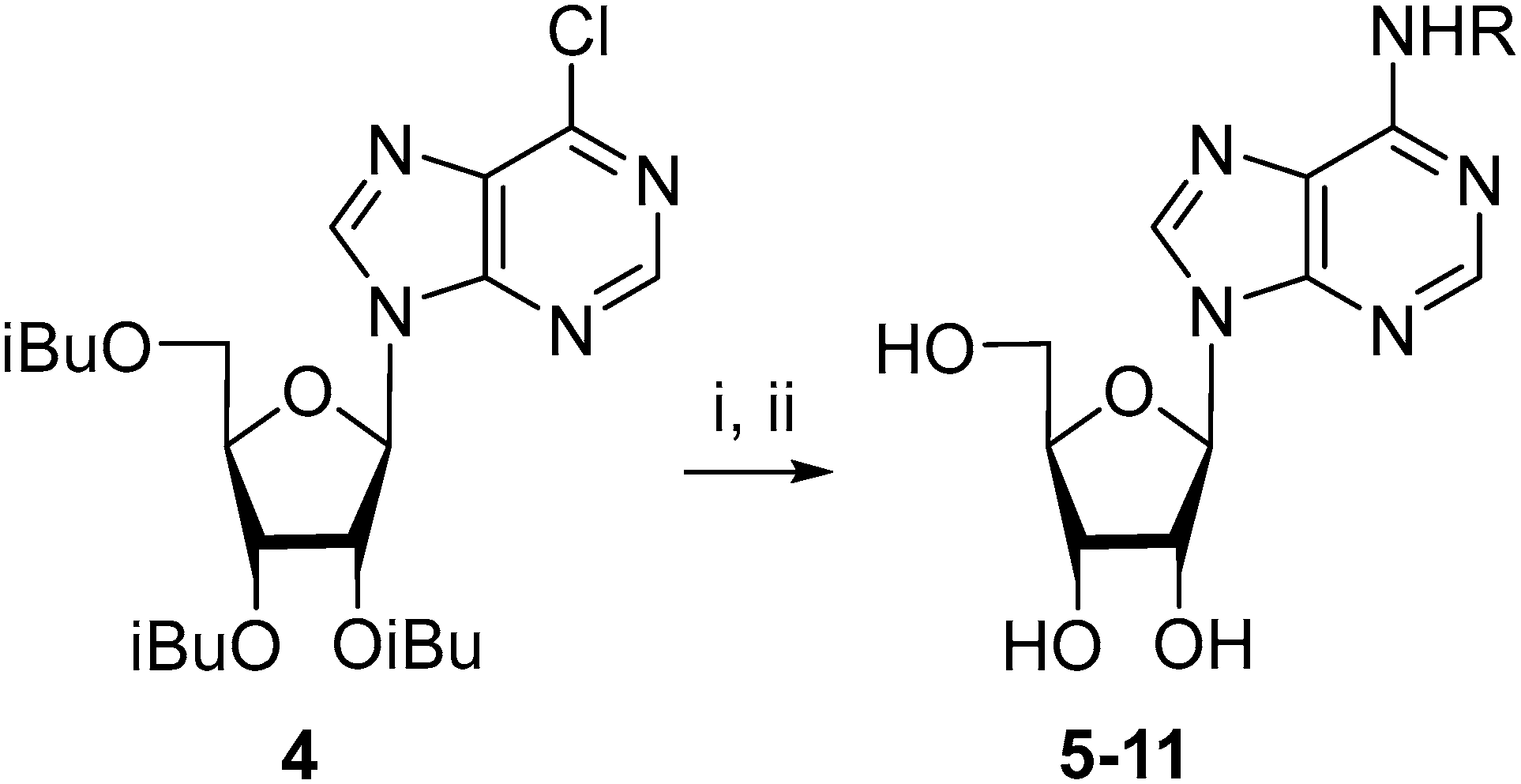
2.1. Chemistry
2.2. Biological Activity on EV71 and Structure-Activity Relationship (SAR)
3. Materials and Methods
3.1. General
3.2. 9-(2,3,5-Tri-O-isobutyroyl-β-d-ribofuranosyl)-6-chloropurine (4)
3.3. N6-(2-Fluorobenzyl)adenosine (5)
3.4. N6-(3-Fluorobenzyl)adenosine (6)
3.5. N6-(4-Fluorobenzyl)adenosine (7)
3.6. N6-(2,6-Difluorobenzyl)adenosine (8)
3.7. N6-(2-Trifluoromethylbenzyl)adenosine (9)
3.8. N6-(3-Trifluoromethylbenzyl)adenosine (10)
3.9. N6-(4-Trifluoromethylbenzyl)adenosine (11)
3.10. Antiviral Assay Against EV71 in RD Cells
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Butler, M.S.; Robertson, A.A.; Cooper, M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014, 31, 1612–1661. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.E. Design and synthesis of analogues of natural products. Org. Biomol. Chem. 2015, 13, 5302–5343. [Google Scholar] [CrossRef] [PubMed]
- Drenichev, M.S.; Oslovsky, V.E.; Mikhailov, S.N. Cytokinin nucleosides-natural compounds with a unique spectrum of biological activity. Curr. Top. Med. Chem. 2016, 16, 2562–2576. [Google Scholar] [CrossRef]
- Mikhailov, S.N.; Scotti, L.; Singla, R.K.; Scotti, M.T. Perspectives in Medicinal Chemistry. Curr. Top. Med. Chem. 2016, 16, 2725–2726. [Google Scholar] [CrossRef]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Rozenski, J.; Crain, P.F.; McCloskey, J.A. The RNA Modification Database: 1999 update. Nucleic Acids Res. 1999, 27, 196–197. [Google Scholar] [CrossRef]
- Agris, P.; Crain, P.F.; Rozenski, J.; Fabris, D.; Vendeix, F.A.P. The RNA Modification Database. The RNA Institute. Available online: http://mods.rna.albany.edu (accessed on 23 June 2017).
- Ernst, D.; Scháfer, W.; Oesterhelt, D. Isolation and identification of a new, naturally occurring cytokinin (6-benzylaminopurineriboside) from an anise cell culture (Pimpinella anisum L.). Planta 1983, 159, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Horgan, R.; Hewett, E.W.; Horgan, J.M.; Purse, J.; Wareing, P.F. A new cytokinin from Populus x robusta. Phytochemistry 1975, 14, 1005–1008. [Google Scholar] [CrossRef]
- Ge, L.; Yong, J.W.H.; Goh, N.K.; Chia, L.S.; Tan, S.N.; Ong, E.S. Identification of kinetin and kinetin riboside in coconut (Cocos nucifera L.) water using a combined approach of liquid chromatography-tandem mass spectrometry, high performance liquid chromatography and gel electrophoresis. J. Chromatogr. B 2005, 829, 26–34. [Google Scholar]
- Young, D.C.; Layre, E.; Pan, S.-J.; Tapley, A.; Adamson, J.; Seshadri, C.; Wu, Z.; Buter, J.; Minnaard, A.J.; Coscolla, M.; et al. In vivo biosynthesis of terpene nucleosides provides unique chemical markers of Mycobacterium tuberculosis infection. Chem. Biol. 2015, 22, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Wakita, T.; Shimizu, H. Characterization of pharmacologically active compounds that inhibit poliovirus and enterovirus 71 infectivity. J. Gen. Virol. 2008, 89, 2518–2530. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.Y.; Lin, T.Y.; Hsu, K.H.; Huang, Y.C.; Lin, K.L.; Hsueh, C.; Shih, S.R.; Ning, H.C.; Hwang, M.S.; Wang, H.S.; et al. Clinical features and risk factors of pulmonary oedema after enterovirus-71-related hand, foot, and mouth disease. Lancet 1999, 354, 1682–1686. [Google Scholar] [CrossRef]
- Wong, K.T.; Munisamy, B.; Ong, K.C.; Kojima, H.; Noriyo, N.; Chua, K.B.; Ong, B.B.; Nagashima, K. The distribution of inflammation and virus in human enterovirus 71 encephalomyelitis suggests possible viral spread by neural pathways. J. Neuropathol. Exp. Neurol. 2008, 67, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- Xing, W.; Liao, Q.; Viboud, C.; Zhang, J.; Sun, J.; Wu, J.T.; Chang, Z.; Liu, F.; Fang, V.J.; Zheng, Y.; et al. Hand, foot, and mouth disease in China, 2008-12: An epidemiological study. Lancet Infect. Dis. 2014, 14, 308–318. [Google Scholar] [CrossRef]
- Yip, C.C.Y.; Lau, S.K.P.; Woo, P.C.Y.; Yuen, K.-Y. Human enterovirus 71 epidemics: What’s next? Emerg. Health Threats J. 2013, 6, e19780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunaseelan, S.; Chu, J.J.H. Identifying novel antiviral targets against enterovirus 71: Where are we? Future Virol. 2017, 12, 171–191. [Google Scholar] [CrossRef]
- Bauer, L.; Lyoo, H.; van der Schaar, H.M.; Strating, J.R.; van Kuppeveld, F.J. Direct-acting antivirals and host-targeting strategies to combat enterovirus infections. Curr. Opin. Virol. 2017, 24, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Tararov, V.I.; Tijsma, A.; Kolyachkina, S.V.; Oslovsky, V.E.; Neyts, J.; Drenichev, M.S.; Leyssen, P.; Mikhailov, S.N. Chemical modification of the plant isoprenoid cytokinin N6-isopentenyladenosine yields a selective inhibitor of human enterovirus 71 replication. Eur. J. Med. Chem. 2015, 90, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Drenichev, M.S.; Oslovsky, V.E.; Sun, L.; Tijsma, A.; Kurochkin, N.N.; Tararov, V.I.; Chizhov, A.O.; Neyts, J.; Pannecouque, C.; Leyssen, P.; et al. The length and structure of the linker in N6-benzyladenosine derivatives affects the antiviral potency against enterovirus 71. Eur. J. Med. Chem. 2016, 111, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Tararov, V.I.; Kolyachkina, S.V.; Alexeev, C.S.; Mikhailov, S.N. N6-acetyl-2′,3′,5′-tri-O-acetyladenosine, a convenient, missed out substrate for regioselective N6-alkylations. Synthesis 2011, 2483–2489. [Google Scholar] [CrossRef]
- Kolyachkina, S.V.; Tararov, V.I.; Alexeev, C.S.; Krivosheev, D.M.; Romanov, G.A.; Stepanova, E.V.; Solomko, E.S.; Inshakov, A.N.; Mikhailov, S.N. N6-Substituted adenosines. Cytokinin and antitumor activities. Collect. Czech. Chem. Commun. 2011, 76, 1361–1378. [Google Scholar] [CrossRef]
- Doležal, K.; Popa, I.; Hauserová, E.; Sṕıchal, L.; Chakrabarty, K.; Novák, O.; Kryštof, V.; Voller, J.; Holub, J.; Strnad, M. Preparation, biological activity and endogenous occurrence of N6-benzyladenosines. Bioorg. Med. Chem. 2007, 15, 3737–3747. [Google Scholar] [CrossRef] [PubMed]
- Fleysher, M.H.; Bloch, A.; Hakala, M.T.; Nichol, C.A. Synthesis and biological activity of some new N6-substituted purine nucleosides. J. Med. Chem. 1969, 12, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Žemlička, J.; Owens, J. 6-Chloro-9-β-d-ribofuranosylpurine. A versatile intermediate in the synthesis of purine ribonucleosides. In Nucleic Acid Chemistry, Part 2; Townsend, L.B., Tipson, R.S., Eds.; Wiley: New York, NY, USA, 1978; pp. 611–614. [Google Scholar]
- Marek, R.; Sklenàř, V. NMR studies of purines. Annu. Rep. NMR Spectrosc. 2004, 54, 201–242. [Google Scholar]
- Dolezel, P.; Koudelkova, P.; Mlejnek, P. Halogenation of N6-benzyladenosine decreases its cytotoxicity in human leukemia cells. Toxicol. In Vitro 2010, 24, 2079–2083. [Google Scholar] [CrossRef] [PubMed]
- Tijsma, A.; Franco, D.; Tucker, S.; Hilgenfeld, R.; Froeyen, M.; Leyssen, P.; Neyts, J. The capsid binder vapendavir and the novel protease inhibitor SG85 inhibit enterovirus 71 replication. Antimicrob. Agents Chemother. 2014, 58, 6990–6992. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
| Substrate | O-Deacylation Conditions, 20 °C a | t½. h | Complete O-Deacylation, h |
|---|---|---|---|
 | CH3NH2/C2H5OH (4M) | 0.25 | 2 |
| NH3/MeOH (4M) | 1 | 5 | |
 | CH3NH2/C2H5OH (4M) | 3 | 15 |
| NH3/MeOH (4M) | 15 | 75 | |
 | CH3NH2/C2H5OH (4M) | 6 | 26 |
| NH3/MeOH (4M) | 19 | 96 |

| No. | Compound Name | Substituent (R) | CC50 ± SD a,b | EC50 ± SD a,b | SI с |
|---|---|---|---|---|---|
| 1 | N6-benzyladenosine (BAPR) |  | 4.3 ± 1.6 | 0.28 ± 0.05 | 15 |
| 2 | N6-isopenthenyladenosine (iPR) |  | 6.0 ± 0.6 | 1.0 ± 0.2 | 6.0 |
| 3 | N6-furfuryladenosine (KINR) |  | 7.8 ± 3.4 | 1.4 ± 0.3 | 5.6 |
| 5 | N6-(2-fluorobenzyl) adenosine |  | 13.3 ± 3.7 | 0.30 ± 0.05 | 44 |
| 6 | N6-(3-fluorobenzyl) adenosine |  | 6.2 ± 1.8 | 0.24 ± 0.09 | 26 |
| 7 | N6-(4-fluorobenzyl) adenosine |  | 2.7 ± 0.9 | 0.14 ± 0.05 | 19 |
| 8 | N6-(2,6-difluorobenzyl) adenosine |  | >254 | 0.21 ± 0.01 | >1210 |
| 9 | N6-(2-trifluoromethylbenzyl) adenosine |  | >235 | 1.0 ± 0.1 | >235 |
| 10 | N6-(3-trifluoromethylbenzyl) adenosine |  | >235 | 0.068 ± 0.001 | >3456 |
| 11 | N6-(4-trifluoromethylbenzyl) adenosine |  | >235 | 1.0 ± 0.1 | >235 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oslovsky, V.E.; Drenichev, M.S.; Sun, L.; Kurochkin, N.N.; Kunetsky, V.E.; Mirabelli, C.; Neyts, J.; Leyssen, P.; Mikhailov, S.N. Fluorination of Naturally Occurring N6-Benzyladenosine Remarkably Increased Its Antiviral Activity and Selectivity. Molecules 2017, 22, 1219. https://doi.org/10.3390/molecules22071219
Oslovsky VE, Drenichev MS, Sun L, Kurochkin NN, Kunetsky VE, Mirabelli C, Neyts J, Leyssen P, Mikhailov SN. Fluorination of Naturally Occurring N6-Benzyladenosine Remarkably Increased Its Antiviral Activity and Selectivity. Molecules. 2017; 22(7):1219. https://doi.org/10.3390/molecules22071219
Chicago/Turabian StyleOslovsky, Vladimir E., Mikhail S. Drenichev, Liang Sun, Nikolay N. Kurochkin, Vladislav E. Kunetsky, Carmen Mirabelli, Johan Neyts, Pieter Leyssen, and Sergey N. Mikhailov. 2017. "Fluorination of Naturally Occurring N6-Benzyladenosine Remarkably Increased Its Antiviral Activity and Selectivity" Molecules 22, no. 7: 1219. https://doi.org/10.3390/molecules22071219
APA StyleOslovsky, V. E., Drenichev, M. S., Sun, L., Kurochkin, N. N., Kunetsky, V. E., Mirabelli, C., Neyts, J., Leyssen, P., & Mikhailov, S. N. (2017). Fluorination of Naturally Occurring N6-Benzyladenosine Remarkably Increased Its Antiviral Activity and Selectivity. Molecules, 22(7), 1219. https://doi.org/10.3390/molecules22071219