Characterization of Red Wine Proanthocyanidins Using a Putative Proanthocyanidin Database, Amide Hydrophilic Interaction Liquid Chromatography (HILIC), and Time-of-Flight Mass Spectrometry



Abstract
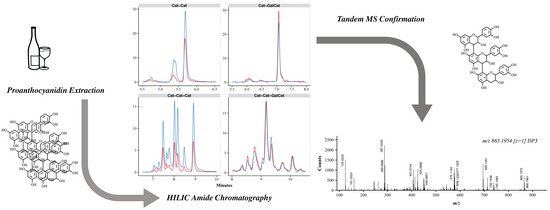
:
1. Introduction
2. Results and Discussion
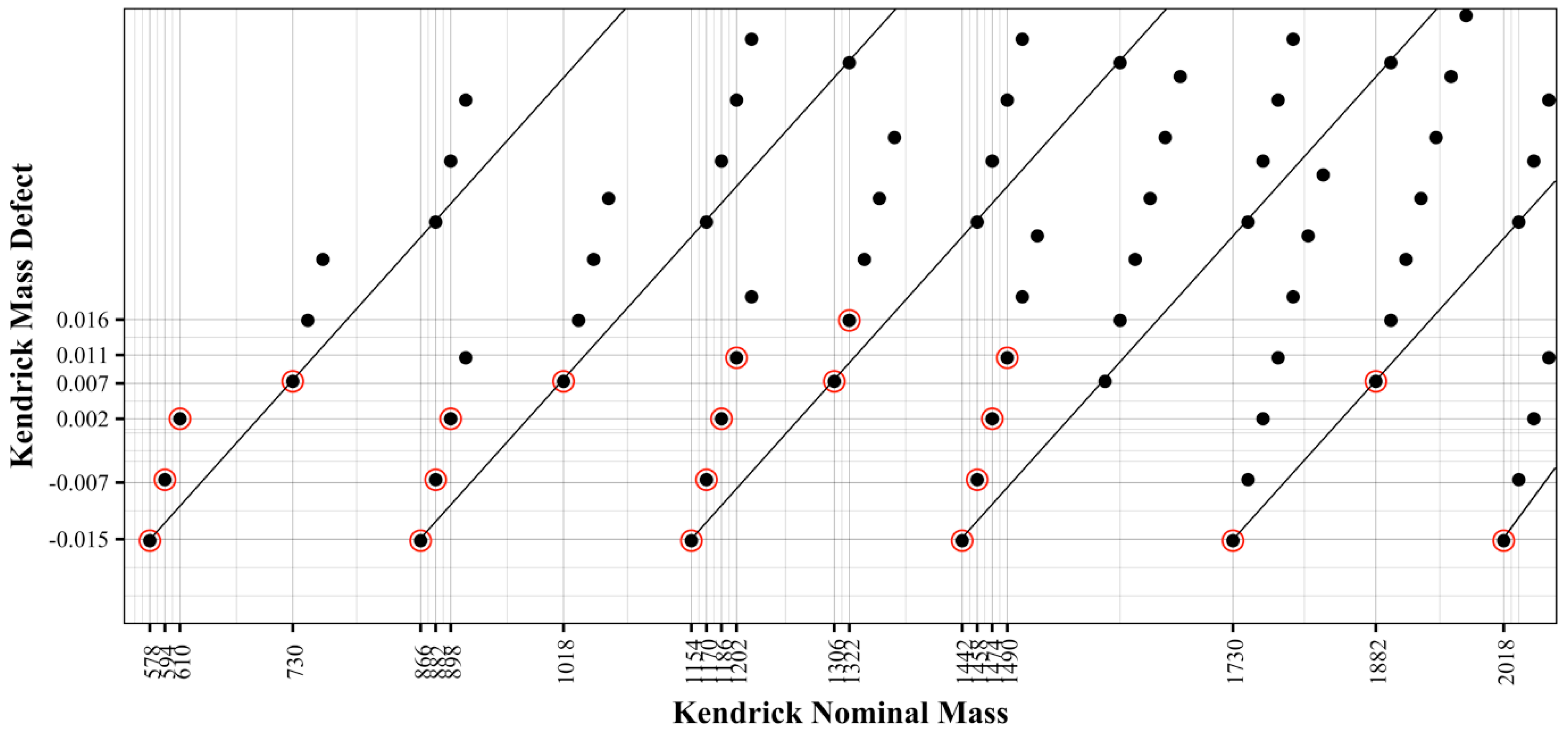
2.1. Initial Compound Identification: Agilent Profinder
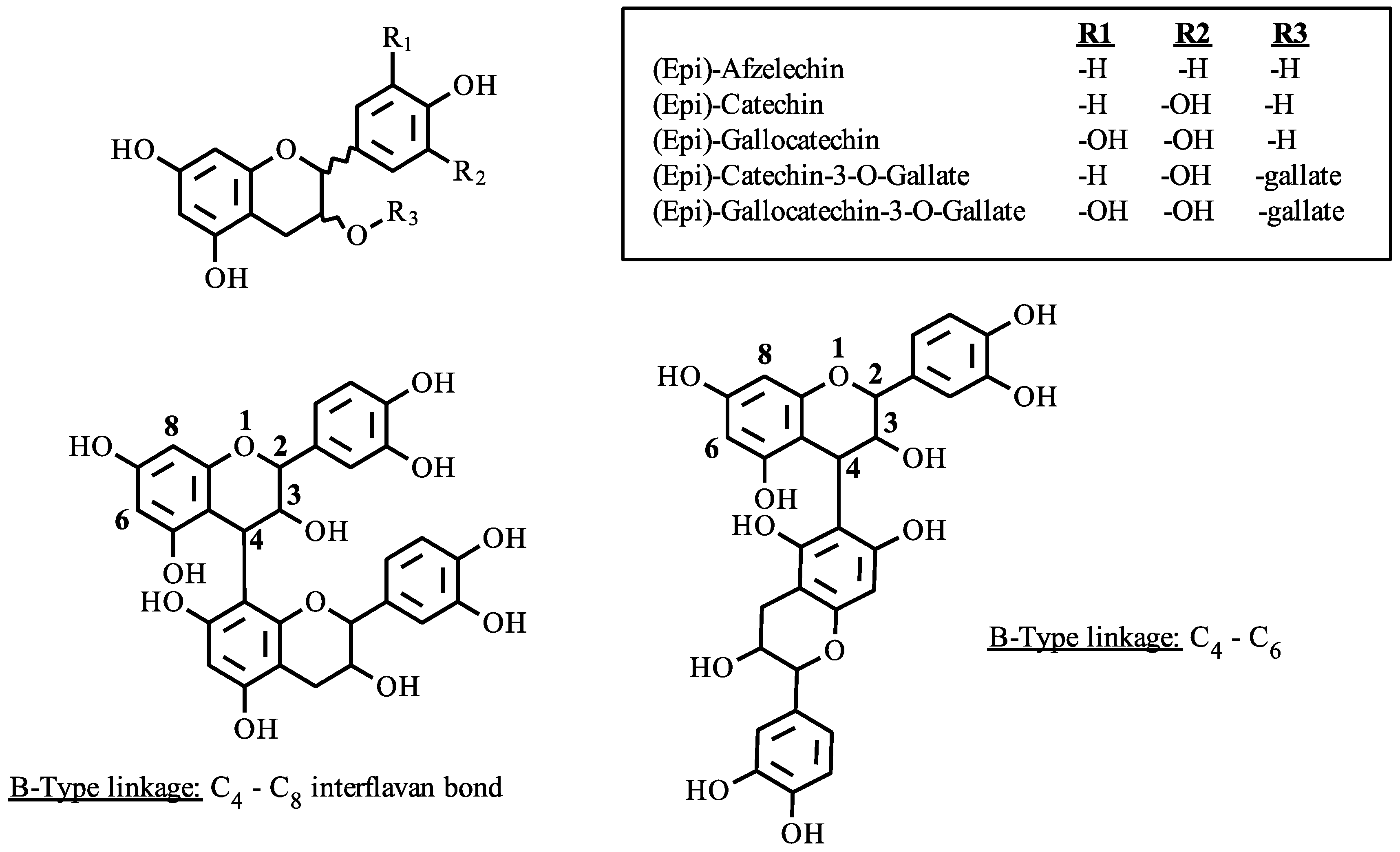
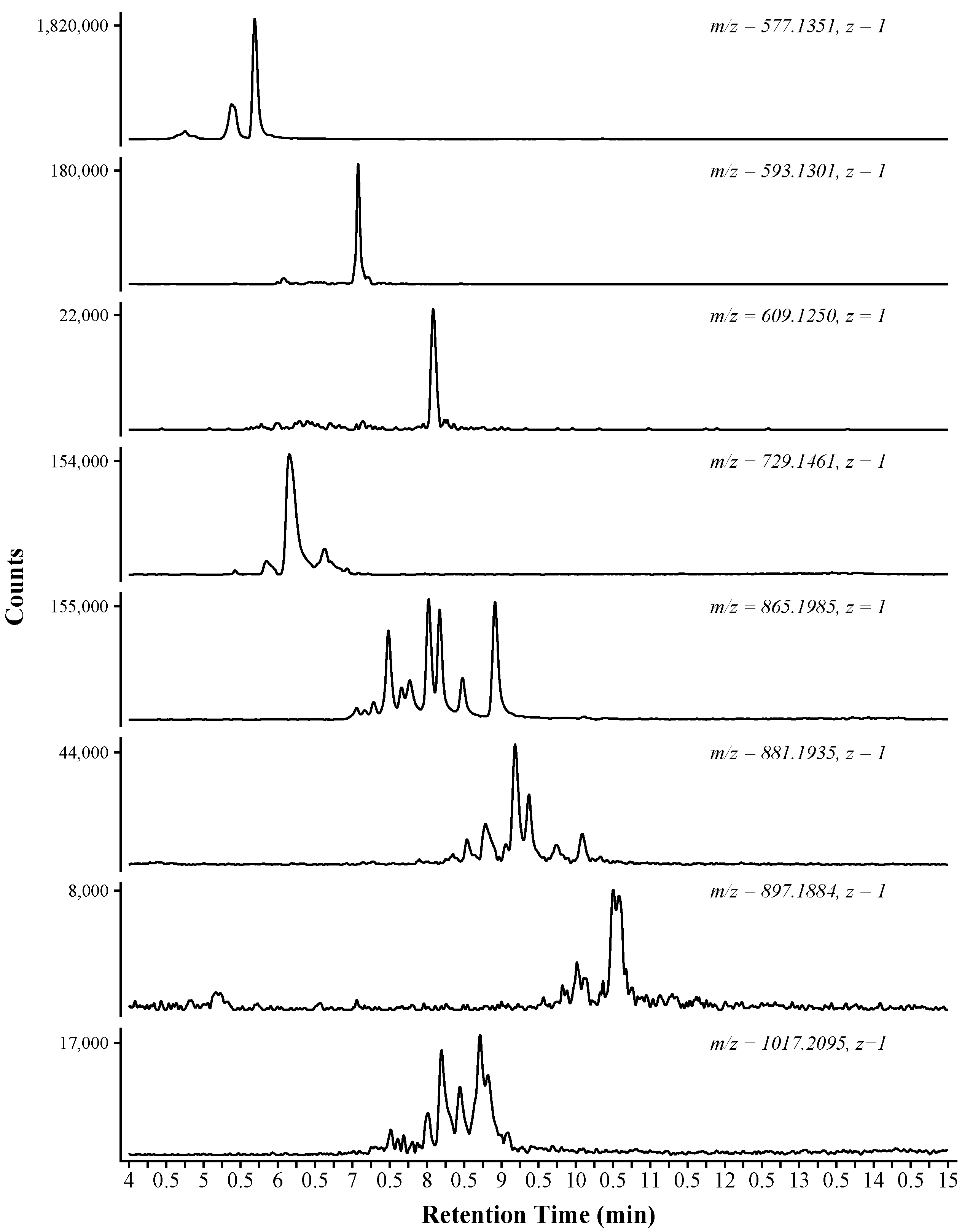
2.2. Chromatography
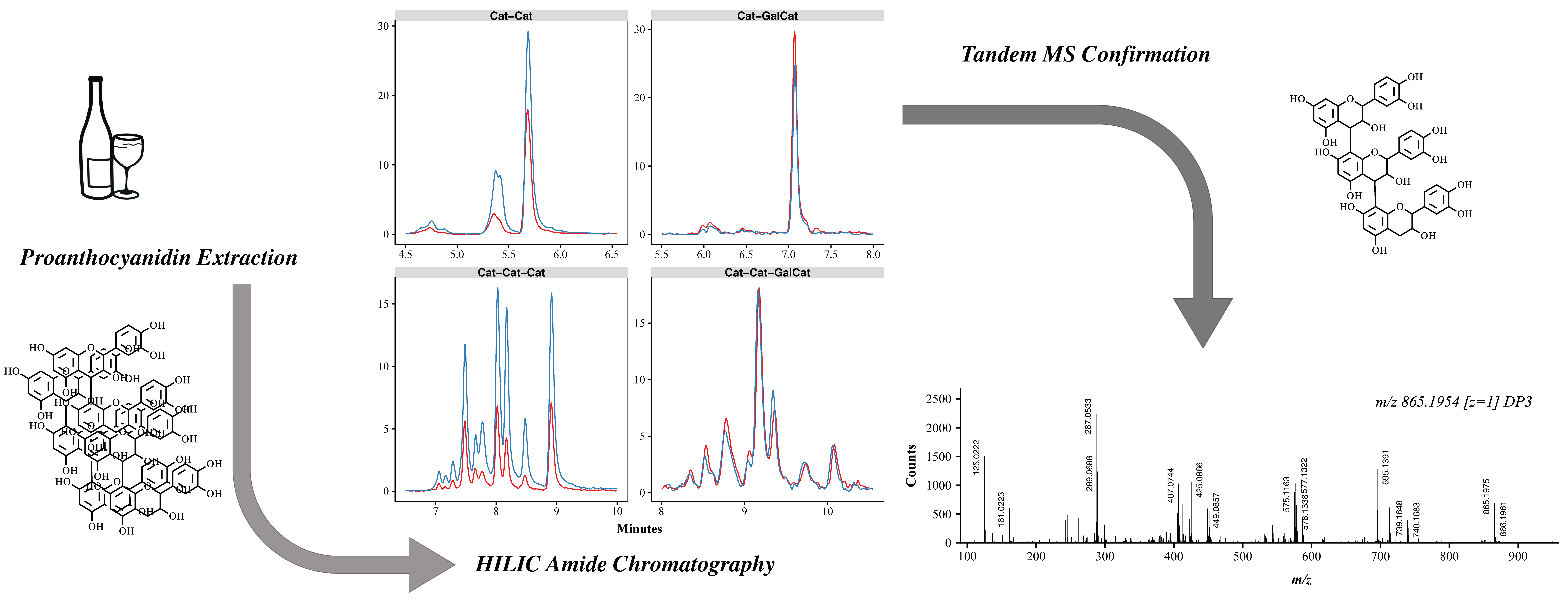
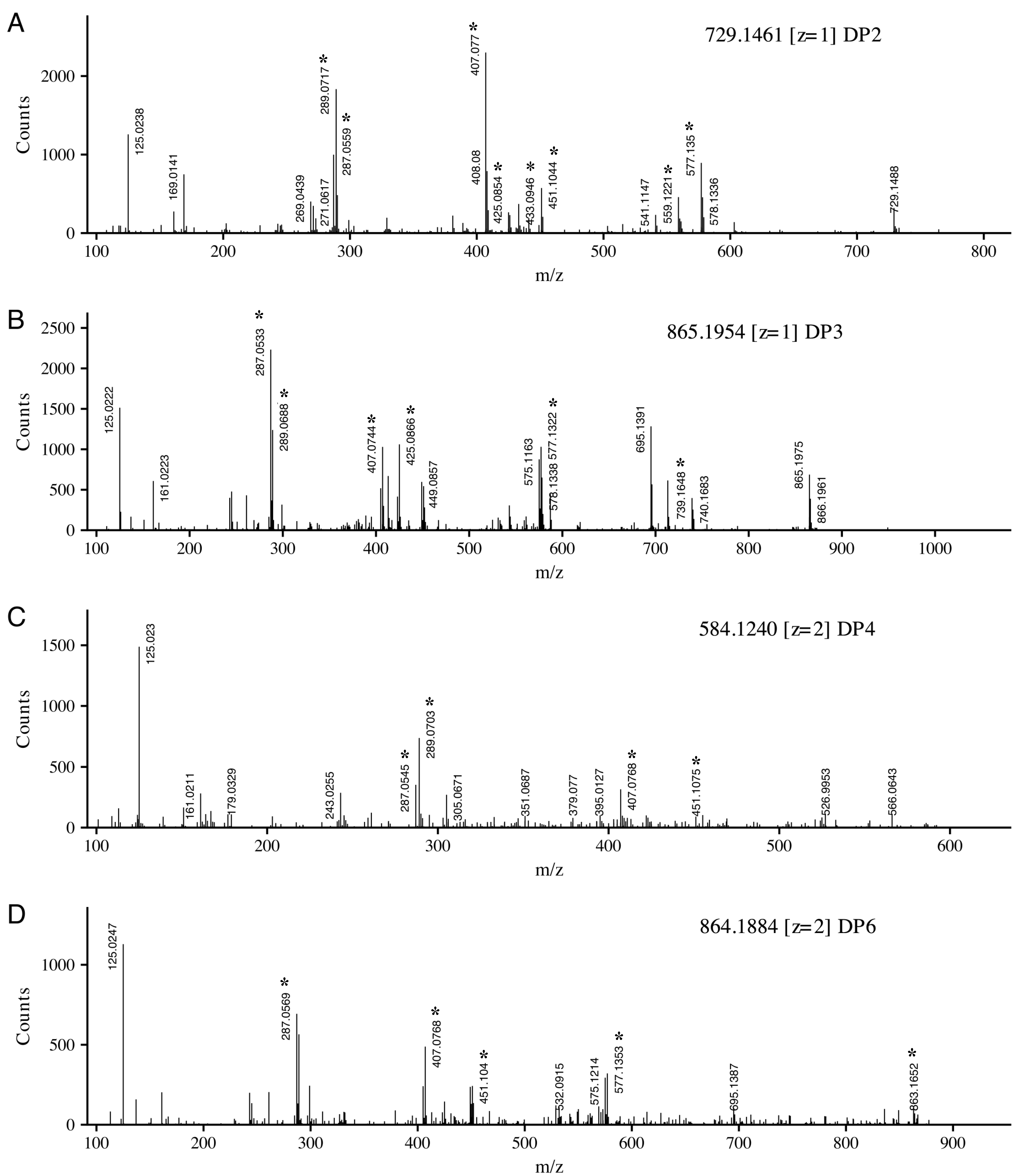
2.3. MS/MS Identification of Putative Compounds
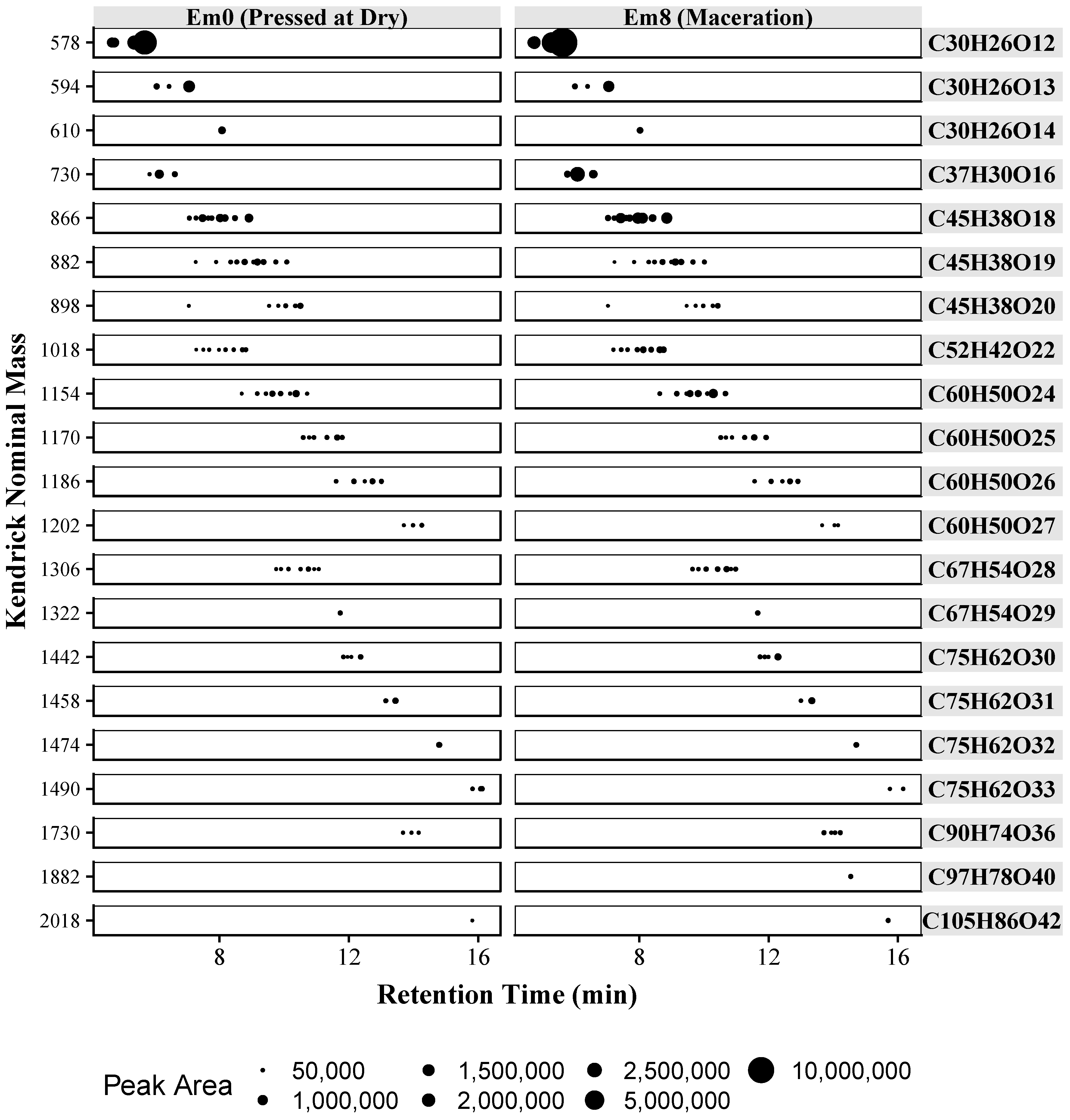
2.4. Analysis of Wine Samples
3. Materials and Methods
3.1. Chemical and Reagents
3.2. Wine Production
3.3. Sample Preparation
3.4. UHPLC-Q-TOF MS Analysis
3.5. Data Analysis and Workflow
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brillouet, J.-M.; Romieu, C.; Schoefs, B.; Solymosi, K.; Cheynier, V.; Fulcrand, H.; Verdeil, J.-L.; Conéjéro, G. The tannosome is an organelle forming condensed tannins in the chlorophyllous organs of Tracheophyta. Ann. Bot. 2013, 112, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Cala, O.; Fabre, S.; Fouquet, E.; Dufourc, E.J.; Pianet, I. NMR of human saliva protein/wine tannin complexes. Towards deciphering astringency with physico-chemical tools. C. R. Chim. 2010, 13, 449–452. [Google Scholar] [CrossRef]
- Harbertson, J.F.; Picciotto, E.A.; Adams, D.O. Measurement of polymeric pigments in grape berry extracts and wines using a protein precipitation assay combined with bisulfite bleaching. Am. J. Enol. Vitic. 2003, 54, 301–306. [Google Scholar]
- Kennedy, J.A.; Taylor, A.W. Analysis of proanthocyanidins by high-performance gel permeation chromatography. J. Chromatogr. A. 2003, 995, 99–107. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Jones, G.P. Analysis of proanthocyanidin cleavage products following acid-catalysis in the presence of excess phloroglucinol. J. Agric. Food Chem. 2001, 49, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Iland, P.G.; Oberholster, A.; Sefton, M.A.; Waters, E.J. Analysis of pigmented polymers in red wine by reverse phase HPLC. Aust. J. Grape Wine Res. 2002, 8, 70–75. [Google Scholar] [CrossRef]
- Gu, L.; Kelm, M.A.; Hammerstone, J.F.; Zhang, Z.; Beecher, G.; Holden, J.; Haytowitz, D.; Prior, R.L. Liquid chromatographic/electrospray ionization mass spectrometric studies of proanthocyanidins in foods. J. Mass Spectrom. 2003, 38, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Kallio, H.; Yang, W.; Liu, P.; Yang, B. Proanthocyanidins in wild sea buckthorn (Hippophaë rhamnoides) berries analyzed by reversed-phase, normal-phase, and hydrophilic interaction liquid chromatography with UV and MS detection. J. Agric. Food Chem. 2014, 62, 7721–7729. [Google Scholar] [CrossRef] [PubMed]
- Kalili, K.M.; Vestner, J.; Stander, M.A.; de Villiers, A. Toward unraveling grape tannin composition: Application of online hydrophilic interaction chromatography × reversed-phase liquid chromatography-time-of-flight mass spectrometry for grape seed analysis. Anal. Chem. 2013, 85, 9107–9115. [Google Scholar] [CrossRef] [PubMed]
- Willemse, C.M.; Stander, M.A.; Vestner, J.; Tredoux, A.G.J.; de Villiers, A. Comprehensive two-dimensional hydrophilic interaction chromatography (HILIC) × reversed-phase liquid chromatography coupled to high-resolution mass spectrometry (RP-LC-UV-MS) analysis of anthocyanins and derived pigments in red wine. Anal. Chem. 2015, 87, 12006–12015. [Google Scholar] [CrossRef] [PubMed]
- Xian, F.; Hendrickson, C.L.; Marshall, A.G. High resolution mass spectrometry. Anal. Chem. 2012, 84, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Junot, C.; Fenaille, F.; Colsch, B.; Bécher, F. High resolution mass spectrometry based techniques at the crossroads of metabolic pathways. Mass Spectrom. Rev. 2014, 33, 471–500. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, R.D.; Lucio, M.; Frommberger, M.; Peyron, D.; Chassagne, D.; Alexandre, H.; Feuillat, F.; Voilley, A.; Cayot, P.; Gebefügi, I.; et al. The chemodiversity of wines can reveal a metabologeography expression of cooperage oak wood. Proc. Natl. Acad. Sci. USA 2009, 106, 9174–9179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendrick, E. A mass scale based on CH2=14.0000 for high resolution mass spectrometry of organic compounds. Anal. Chem. 1963, 35, 2146–2154. [Google Scholar] [CrossRef]
- Lerno, L.A.; German, J.B.; Lebrilla, C.B. Method for the identification of lipid classes based on referenced Kendrick mass analysis. Anal. Chem. 2010, 82, 4236–4245. [Google Scholar] [CrossRef] [PubMed]
- Dier, T.K.F.; Egele, K.; Fossog, V.; Hempelmann, R.; Volmer, D.A. Enhanced mass defect filtering to simplify and classify complex mixtures of lignin degradation products. Anal. Chem. 2016, 88, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Vallverdú-Queralt, A.; Meudec, E.; Eder, M.; Lamuela-Raventos, R.M.; Sommerer, N.; Cheynier, V. Targeted filtering reduces the complexity of UHPLC-Orbitrap-HRMS data to decipher polyphenol polymerization. Food Chem. 2017, 227, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.C.; Blackman, J.W.; Hjelmeland, A.K.; Ebeler, S.E.; Heymann, H. Extended maceration and cap management impacts on the phenolic, volatile, and sensory profile of Merlot wine. Am. J. Enol. Vitic. 2018, in press. [Google Scholar] [CrossRef]
- Awad, H.; Khamis, M.M.; El-Aneed, A. Mass spectrometry, review of the basics: Ionization. Appl. Spectrosc. Rev. 2015, 50, 158–175. [Google Scholar] [CrossRef]
- Montero, L.; Herrero, M.; Prodanov, M.; Ibáñez, E.; Cifuentes, A. Characterization of grape seed procyanidins by comprehensive two-dimensional hydrophilic interaction × reversed phase liquid chromatography coupled to diode array detection and tandem mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 4627–4638. [Google Scholar] [CrossRef] [PubMed]
- Tarascou, I.; Barathieu, K.; Simon, C.; Ducasse, M.-A.; André, Y.; Fouquet, E.; Dufourc, E.J.; de Freitas, V.; Laguerre, M.; Pianet, I. A 3D structural and conformational study of procyanidin dimers in water and hydro-alcoholic media as viewed by NMR and molecular modeling. Magn. Reson. Chem. 2006, 44, 868–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikberg, E.; Sparrman, T.; Viklund, C.; Jonsson, T.; Irgum, K. A 2H nuclear magnetic resonance study of the state of water in neat silica and zwitterionic stationary phases and its influence on the chromatographic retention characteristics in hydrophilic interaction high-performance liquid chromatography. J. Chromatogr. A 2011, 1218, 6630–6638. [Google Scholar] [CrossRef] [PubMed]
- Greco, G.; Letzel, T. Main interactions and influences of the chromatographic parameters in HILIC separations. J. Chromatogr. Sci. 2013, 51, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Jandera, P. Stationary and mobile phases in hydrophilic interaction chromatography: A review. Anal. Chim. Acta 2011, 692, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Kelm, M.A.; Hammerstone, J.F.; Beecher, G.; Holden, J.; Haytowitz, D.; Gebhardt, S.; Prior, R.L. Concentrations of proanthocyanidins in common foods and estimations of normal consumption. J. Nutr. 2004, 134, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Tarascou, I.; Mazauric, J.-P.; Meudec, E.; Souquet, J.-M.; Cunningham, D.; Nojeim, S.; Cheynier, V.; Fulcrand, H. Characterisation of genuine and derived cranberry proanthocyanidins by LC-ESI-MS. Food Chem. 2011, 128, 802–810. [Google Scholar] [CrossRef]
- Bindon, K.A.; Smith, P.A.; Kennedy, J.A. Interaction between grape-derived proanthocyanidins and cell wall material. 1. Effect on proanthocyanidin composition and molecular mass. J. Agric. Food Chem. 2010, 58, 2520–2528. [Google Scholar] [CrossRef] [PubMed]
- Bindon, K.A.; Smith, P.A.; Holt, H.; Kennedy, J.A. Interaction between grape-derived proanthocyanidins and cell wall material. 2. Implications for vinification. J. Agric. Food Chem. 2010, 58, 10736–10746. [Google Scholar] [CrossRef] [PubMed]
- Lerno, L.; Reichwage, M.; Panprivech, S.; Ponangi, R.; Hearne, L.; Oberholster, A.; Block, D.E. Chemical gradients in pilot-scale Cabernet Sauvignon fermentations and their effect on phenolic extraction. Am. J. Enol. Vitic. 2017, 68, 401–411. [Google Scholar] [CrossRef]
- Li, H.-J.; Deinzer, M.L. Tandem mass spectrometry for sequencing proanthocyanidins. Anal. Chem. 2007, 79, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-J.; Deinzer, M.L. The mass spectral analysis of isolated hops A-type proanthocyanidins by electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 2008, 43, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.C.; Blackman, J.W.; Ebeler, S.E.; Heymann, H. Analysis of temporal dominance of sensation data using correspondence analysis on Merlot wine with differing maceration and cap management regimes. Food Qual. Prefer. 2018, 64, 245–252. [Google Scholar] [CrossRef]
- Cerpa-Calderón, F.K.; Kennedy, J.A. Berry integrity and extraction of skin and seed proanthocyanidins during red wine fermentation. J. Agric. Food Chem. 2008, 56, 9006–9014. [Google Scholar] [CrossRef] [PubMed]
- Casassa, L.F.; Larsen, R.C.; Beaver, C.W.; Mireles, M.S.; Keller, M.; Riley, W.R.; Harbertson, J.F. Impact of extended maceration and regulated deficit irrigation (RDI) in Cabernet Sauvignon wines: Characterization of proanthocyanidin distribution, anthocyanin extraction, and chromatic properties. J. Agric. Food Chem. 2013, 61, 6446–6457. [Google Scholar] [CrossRef] [PubMed]
- Lerno, L.; Reichwage, M.; Ponangi, R.; Hearne, L.; Block, D.E.; Oberholster, A. Effects of cap and overall fermentation temperature on phenolic extraction in Cabernet Sauvignon fermentations. Am. J. Enol. Vitic. 2015, 66, 444–453. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Retention Time (min) | Mean Retention Time (min) | Formula | Polymer | Mean Experimental Mass 1 | Mean Mass Error (ppm) |
|---|---|---|---|---|---|
| 4.68, 4.72, 5.31, 5.63 | 5.09 | C30H26O12 | Cat-Cat | 578.1401 | 4.0 |
| 6.01, 6.38, 7.05 | 6.48 | C30H26O13 | Cat-GalCat | 594.1308 | 11.0 |
| 8.03 | 8.03 | C30H26O14 | GalCat-GalCat | 610.1264 | 9.6 |
| 5.78, 6.09, 6.59 | 6.15 | C37H30O16 | Cat-Cat:Gal | 730.1480 | 7.4 |
| 7.04, 7.24, 7.44, 7.61, 7.72, 7.97, 8.12, 8.42, 8.86 | 7.82 | C45H38O18 | Cat-Cat-Cat | 866.2011 | 5.4 |
| 7.24, 7.85, 8.30, 8.49, 8.74, 9.01, 9.13, 9.32, 9.69, 10.04 | 8.78 | C45H38O19 | Cat-Cat-GalCat | 882.1940 | 7.6 |
| 7.03, 9.49, 9.77, 10.00, 10.30, 10.46 | 9.51 | C45H38O20 | Cat-GalCat-GalCat | 898.1860 | 10.7 |
| 7.22, 7.46, 7.65, 7.95, 8.15, 8.39, 8.66, 8.77 | 8.03 | C52H42O22 | Cat-Cat-Cat:Gal | 1018.2106 | 6.1 |
| 8.65, 9.13, 9.41, 9.59, 9.85, 10.14, 10.32, 10.69 | 9.72 | C60H50O24 | Cat-Cat-Cat-Cat | 1154.2613 | 6.8 |
| 10.54, 10.70, 10.89, 11.29, 11.58, 11.86 | 11.14 | C60H50O25 | Cat-Cat-Cat-GalCat 2 | 1170.2537 | 8.9 |
| 11.62, 12.10, 12.45, 12.69, 12.94 | 12.36 | C60H50O26 | Cat-Cat-GalCat-GalCat 2 | 1186.2434 | 13.2 |
| 13.69, 13.95, 14.20 | 13.95 | C60H50O27 | Cat-GalCat-GalCat-GalCat 2 | 1202.2393 | 12.2 |
| 9.70, 9.86, 10.09, 10.45, 10.72, 10.88, 11.02 | 10.31 | C67H54O28 | Cat-Cat-Cat-Cat:Gal 2 | 1306.2720 | 6.2 |
| 11.68 | 11.68 | C67H54O29 | Cat-Cat-Cat-GalCat:Gal 2 | 1322.2583 | 12.7 |
| 11.77, 11.91, 12.02, 12.31 | 12.00 | C75H62O30 | Cat-Cat-Cat-Cat-Cat 2 | 1442.3256 | 4.8 |
| 13.07, 13.38 | 13.23 | C75H62O31 | Cat-Cat-Cat-Cat-GalCat 2 | 1458.3216 | 4.0 |
| 14.75 | 14.75 | C75H62O32 | Cat-Cat-Cat-GalCat-GalCat 2 | 1474.3129 | 6.5 |
| 15.8, 16.06 | 15.93 | C75H62O33 | Cat-Cat-GalCat-GalCat-GalCat 2 | 1490.2938 | 15.8 |
| 13.66, 13.88, 14.09, 14.27 | 13.98 | C90H74O36 | Cat-Cat-Cat-Cat-Cat-Cat 2 | 1730.3864 | 5.5 |
| 14.6 | 14.60 | C97H78O40 | Cat-Cat-Cat-Cat-Cat-Cat:Gal 2 | 1882.3969 | 5.3 |
| 15.77 | 15.77 | C105H86O42 | Cat-Cat-Cat-Cat-Cat-Cat-Cat 2 | 2018.4477 | 5.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frost, S.; Lerno, L.A.; Zweigenbaum, J.; Heymann, H.; Ebeler, S.E. Characterization of Red Wine Proanthocyanidins Using a Putative Proanthocyanidin Database, Amide Hydrophilic Interaction Liquid Chromatography (HILIC), and Time-of-Flight Mass Spectrometry. Molecules 2018, 23, 2687. https://doi.org/10.3390/molecules23102687
Frost S, Lerno LA, Zweigenbaum J, Heymann H, Ebeler SE. Characterization of Red Wine Proanthocyanidins Using a Putative Proanthocyanidin Database, Amide Hydrophilic Interaction Liquid Chromatography (HILIC), and Time-of-Flight Mass Spectrometry. Molecules. 2018; 23(10):2687. https://doi.org/10.3390/molecules23102687
Chicago/Turabian StyleFrost, Scott, Larry A. Lerno, Jerry Zweigenbaum, Hildegarde Heymann, and Susan E. Ebeler. 2018. "Characterization of Red Wine Proanthocyanidins Using a Putative Proanthocyanidin Database, Amide Hydrophilic Interaction Liquid Chromatography (HILIC), and Time-of-Flight Mass Spectrometry" Molecules 23, no. 10: 2687. https://doi.org/10.3390/molecules23102687
APA StyleFrost, S., Lerno, L. A., Zweigenbaum, J., Heymann, H., & Ebeler, S. E. (2018). Characterization of Red Wine Proanthocyanidins Using a Putative Proanthocyanidin Database, Amide Hydrophilic Interaction Liquid Chromatography (HILIC), and Time-of-Flight Mass Spectrometry. Molecules, 23(10), 2687. https://doi.org/10.3390/molecules23102687