α-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration


Abstract
:1. Introduction
2. Brain PUFAs: Structure, Metabolism and Function
2.1. Lipid Composition and Metabolism in the Brain
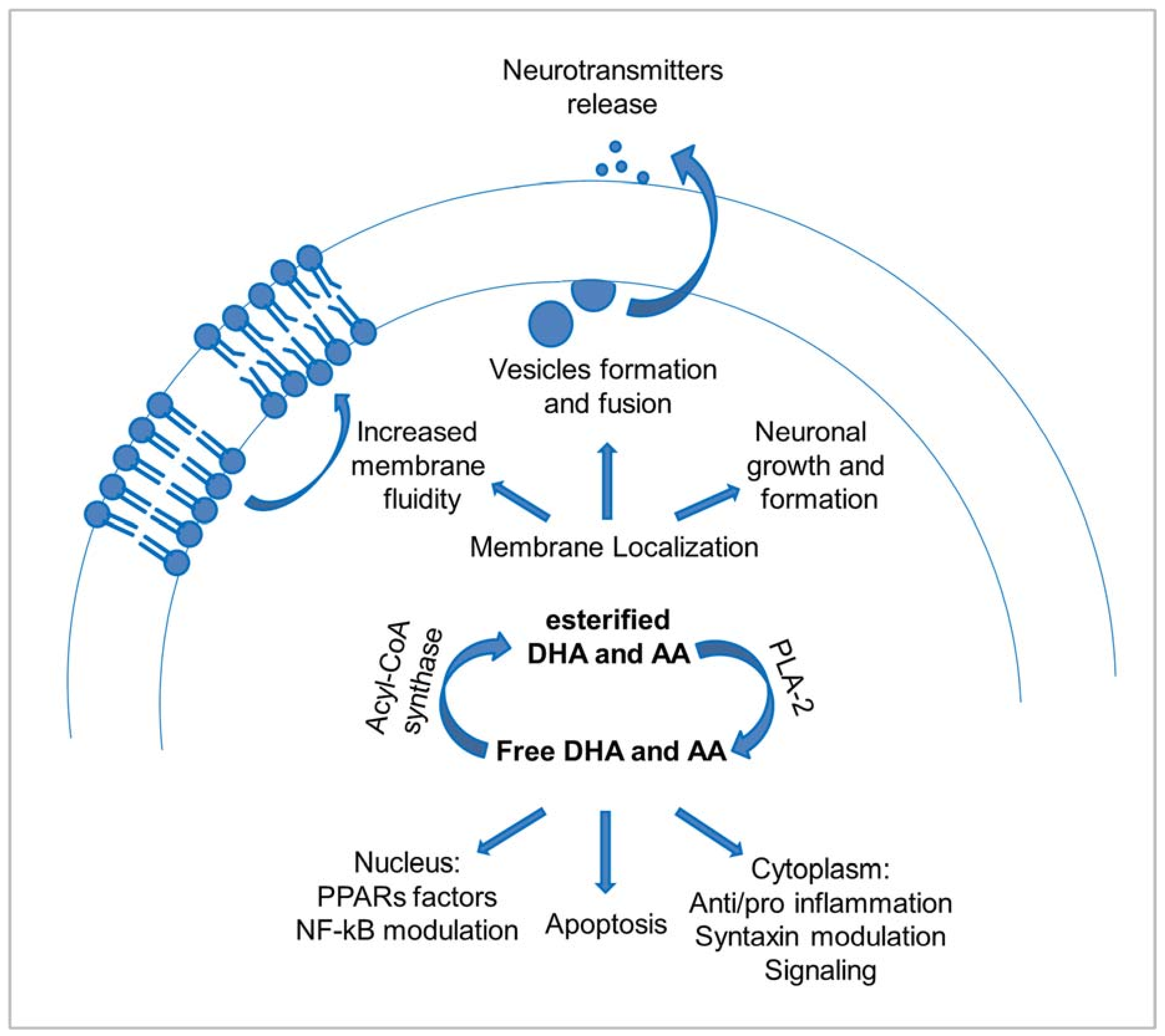
2.2. The Functions of PUFAs in the Brain
2.3. PUFAs in Aging and Disease
3. α-Synuclein: Structure and Physiological Role
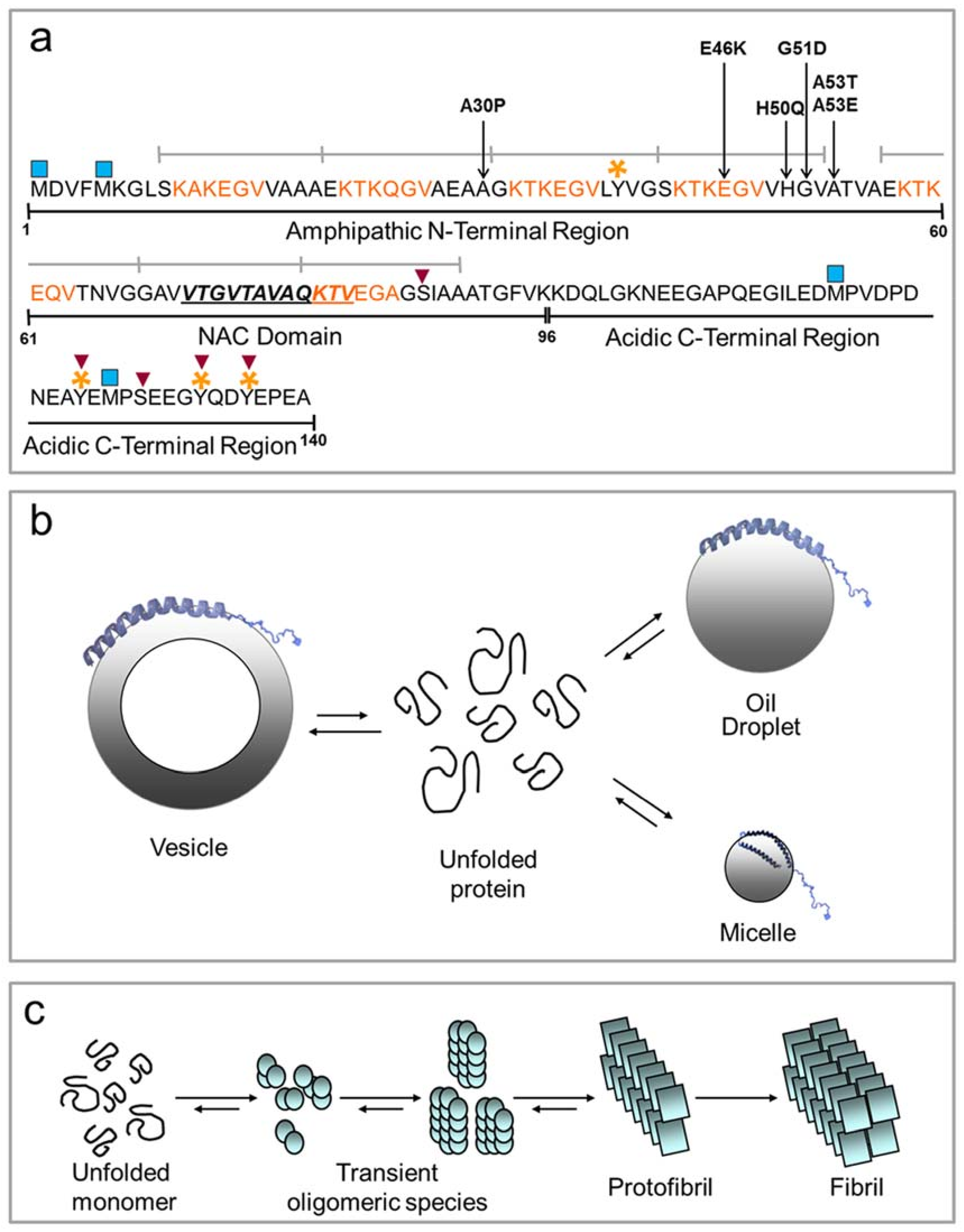
3.1. The Structure and Function of α-Synuclein
3.2. The Physiological and Pathological Folding of α-Synuclein
4. Molecular Interaction between α-Synuclein and PUFAs
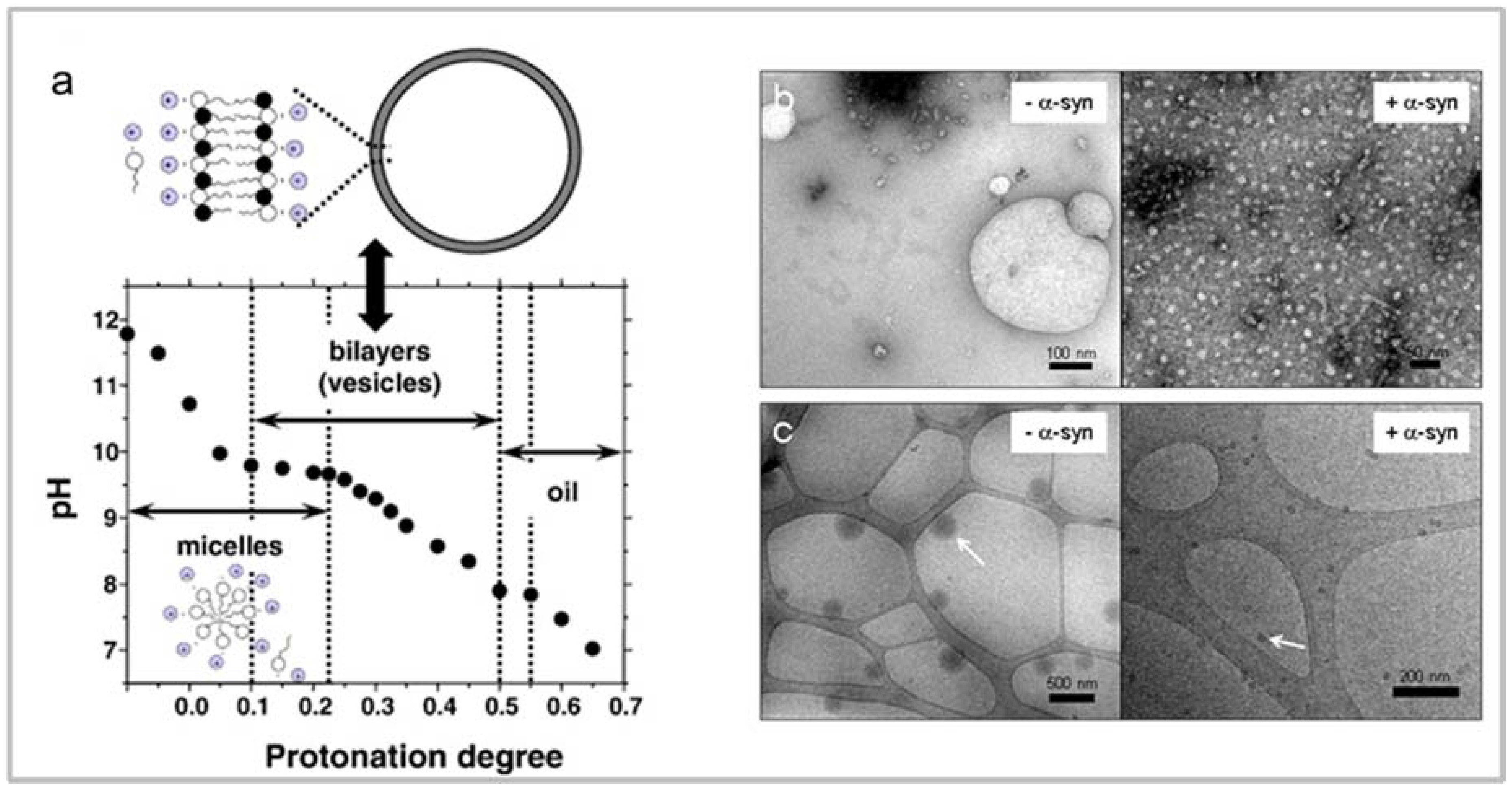
4.1. Fatty Acids Induce Changes in α-Synuclein Secondary Structure
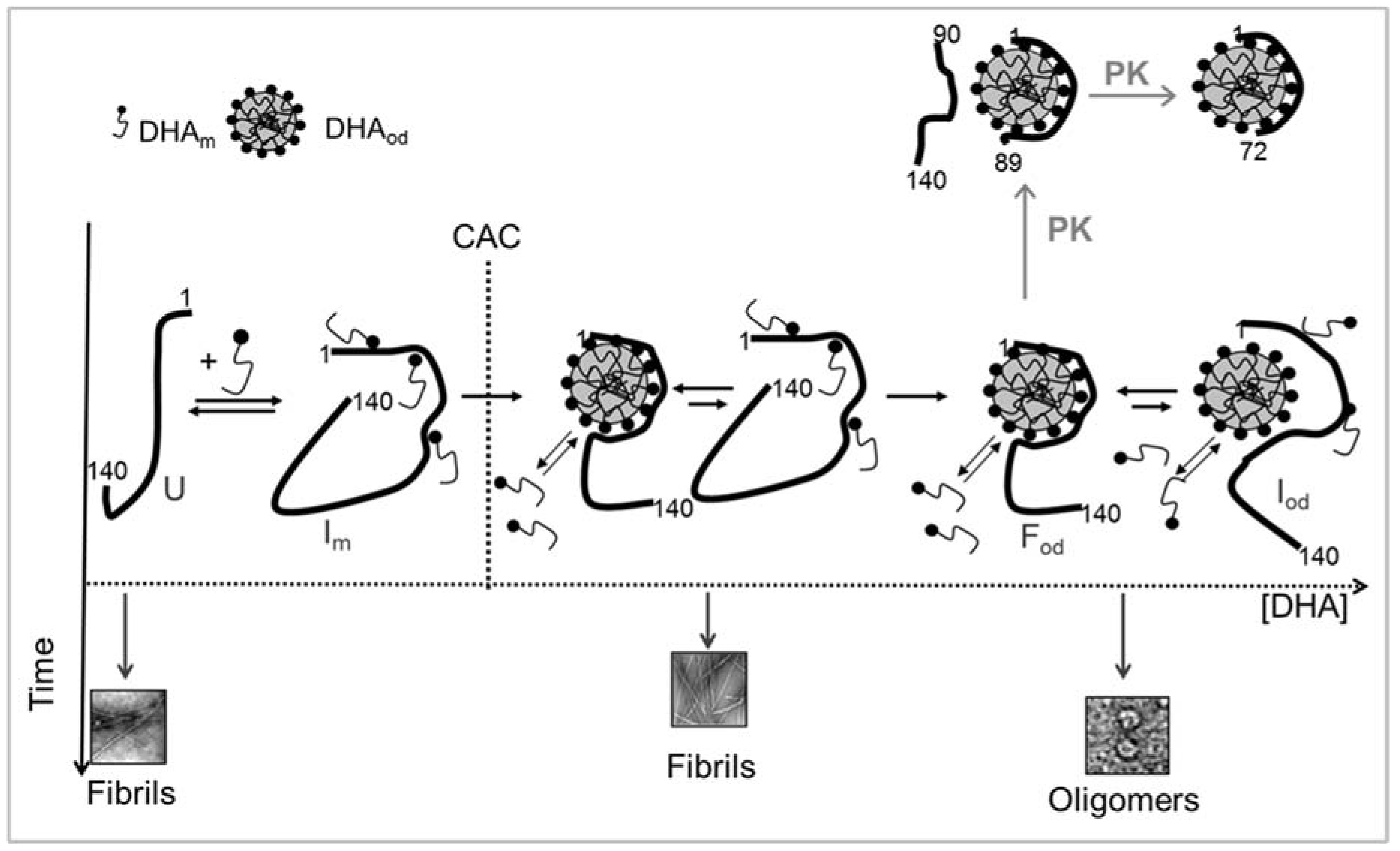
4.2. Fatty Acid-Induced α-Synuclein Oligomerization
5. Implication on Neurodegeneration
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giasson, B.I.; Jakes, R.; Goedert, M.; Duda, J.E.; Leight, S.; Trojanowski, J.Q.; Lee, V.M.Y. A panel of epitope-specific antibodies detects protein domains distributed throughout human α-synuclein in Lewy bodies of Parkinson's disease. J. Neurosci. Res. 2000, 59, 528–553. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, E.; Oliva, R.; Obach, V.; Martí, M.J.; Pastor, P.; Ballesta, F.; Tolosa, E. Identification of Spanish familial Parkinson's disease and screening for the Ala53Thr mutation of the α-synuclein gene in early onset patients. Neurosci. Lett. 1997, 235, 57–60. [Google Scholar] [CrossRef]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Mondal, M.; Mohite, G.M.; Singh, P.K.; Ranjan, P.; Anoop, A.; Ghosh, S.; Jha, N.N.; Kumar, A.; Maji, S.K. The Parkinson's disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry 2013, 52, 6925–6927. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A. Intracellular dynamics of synucleins: “Here, There and Everywhere”. Int. Rev. Cell Mol. Biol. 2015, 320, 103–169. [Google Scholar] [CrossRef] [PubMed]
- Ruipérez, V.; Darios, F.; Davletov, B. α-Synuclein, lipids and Parkinson’s disease. Prog. Lipid Res. 2010, 49, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson's disease: Dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-Joseph, I.; Mirick, G.E.; Serhan, C.N.; Selkoe, D.J. Altered fatty acid composition of dopaminergic neurons expressing α-synuclein and human brains with α-synucleinopathies. J. Biol. Chem. 2003, 278, 49874–49881. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C. The role of lipids interacting with α-synuclein in the pathogenesis of Parkinson’s disease. J. Parkinsons Dis. 2017, 7, 433–450. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, G.; Fecchio, C.; Sharon, R.; Schapira, A.H.V.; Proukakis, C.; Bellotti, V.; Polverino de Laureto, P. α-Synuclein structural features inhibit harmful polyunsaturated fatty acids oxidation, suggesting roles in neuroprotection. J. Biol. Chem. 2017, 292, 6927–6937. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Brunaldi, K. A model for fatty acid transport into the brain. J. Mol. Neurosci. 2007, 33, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The formation of highly soluble oligomers of α-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef]
- Brenna, J.T.; Diau, G.Y. The influence of dietary docosahexaenoic acid and arachidonic acid on central nervous system polyunsaturated fatty acid composition. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.T.; Green, J.T.; Orr, S.K.; Bazinet, R.P. Regulation of brain polyunsaturated fatty acid uptake and turnover. Prostaglandins Leukot. Essent. Fat. Acids 2008, 79, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Crawford, M.A.; Hassam, A.G.; Williams, G. Essential fatty acids and fetal brain growth. Lancet 1976, 1, 452–453. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Allsop, D. α-Synuclein interacts with lipoproteins in plasma. J. Mol. Neurosci. 2017, 63, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A. Fatty acid activation. Prog. Lipid Res. 1997, 36, 55–83. [Google Scholar] [CrossRef]
- Darios, F.; Connell, E.; Davletov, B. Phospholipases and fatty acid signalling in exocytosis. J. Physiol. 2007, 585, 699–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef] [PubMed]
- Green, J.T.; Orr, S.K.; Bazinet, R.P. The emerging role of group VI calcium-independent phospholipase A2 in releasing docosahexaenoic acid from brain phospholipids. J. Lipid Res. 2008, 49, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamaraa, R.K.; Carlson, S.E. Role of omega-3 fatty acids in brain development and function: Potential implications for the pathogenesis and prevention of psychopathology. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 329–349. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, G.; Ecker, J. The opposing effects of n-3 and n-6 fatty acids. Prog. Lipid Res. 2008, 47, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.T.; Brenna, J.T. Dietary docosahexaenoic acid but not arachidonic acid influences central nervous system fatty acid status in baboon neonates. Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Tyburczy, C.; Kothapalli, K.S.D.; Park, W.J.; Blank, B.S.; Bradford, K.L.; Zimmer, J.P.; Butt, C.M.; Salem, N.; Brenna, J.T. Heart arachidonic acid is uniquely sensitive to dietary arachidonic acid and docosahexaenoic acid content in domestic piglets. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connell, E.; Darios, F.; Broersen, K.; Gatsby, N.; Peak-Chew, S.Y.; Rickmanw, C.; Davletov, B. Mechanism of arachidonic acid action on syntaxin–Munc1. EMBO Rep. 2007, 8, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G.; Tu, B.; Rodriguez de Turco, E.B. What synaptic lipid signaling tells us about seizure-induced damage and epileptogenesis. Prog. Brain Res. 2002, 135, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Rickman, C.; Davletov, B. Arachidonic acid allows SNARE complex formation in the presence of Munc18. Chem. Biol. 2005, 12, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Davletov, B. Omega-3 and omega-6 fatty acids stimulate cell membrane expansion by acting on syntaxin 3. Nature 2006, 440, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsinde, J.; Perez, R.; Balboa, M.A. Calcium-independent phospholipase A2 and apoptosis. Biochim. Biophys. Acta 2006, 1761, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Monjazeb, A.M.; Clay, C.E.; High, K.P.; Chilton, F.H. Antineoplastic properties of arachidonic acid and its metabolites. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Herbert, S.P.; Walker, J.H. Group VIA calcium-independent phospholipase A2 mediates endothelial cell S phase progression. J. Biol. Chem. 2006, 281, 35709–35716. [Google Scholar] [CrossRef] [PubMed]
- Bagga, D.; Wang, L.; Farias-Eisner, R.; Glaspy, T.A.; Reddy, S.T. Differential effects of prostaglandin derived from omega-6 and omega-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc. Natl. Acad. Sci. USA 2003, 4, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Stone, N.J. Anti-atherosclerotic and antithrombotic effects of omega-3 fatty acids. Am. J. Cardiol. 2006, 98, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, F. Arachidonic acid metabolism in brain physiology and pathology: Lessons from genetically altered mouse models. J. Neurochem. 2007, 102, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Orr, S.K.; Palumbo, S.; Bosetti, F.; Mount, H.T.; Kang, J.X.; Greenwood, C.E.; Ma, D.W.; Serhan, C.N.; Bazinet, R.P. Unesterified docosahexaenoic acid is protective in neuroinflammation. J. Neurochem. 2013, 127, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-κB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Jump, D.B.; Clarke, S.D. Regulation of gene expression by dietary fat. Annu. Rev. Nutr. 1999, 19, 63–90. [Google Scholar] [CrossRef] [PubMed]
- Dawson, G. Measuring brain lipids. Biochim. Biophys. Acta 2015, 1851, 1026–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svennerholm, L. Distribution and fatty acid composition of phosphoglycerides in normal human brain. J. Lipid Res. 1968, 9, 570–579. [Google Scholar] [PubMed]
- Anderton, B.H. Changes in the ageing brain in health and disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997, 352, 1781–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dexter, D.; Carter, C.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Lipid peroxidation as cause of nigral cell death in Parkinson’s disease. Lancet 1986, 2, 639–640. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Crawford, M.A. Energetic and nutritional constraints on infant brain development: Implications for brain expansion during human evolution. J. Hum. Evol. 2014, 77, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Ames, A. CNS energy metabolism as related to function. Brain Res. Rev. 2000, 34, 42–68. [Google Scholar] [CrossRef]
- Brenna, J.T.; Carlson, S.E. Docosahexaenoic acid and human brain development: Evidence that a dietary supply is needed for optimal development. J. Hum. Evol. 2014, 77, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Purdon, A.D.; Rosenberger, T.A.; Shetty, H.U.; Rapoport, S.I. Energy consumption by phospholipid metabolism in mammalian brain. Neurochem Res. 2002, 27, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Yoshimoto, M.; Masliah, E.; Saitoh, T. Non-A. beta. Component of Alzheimer's disease amyloid (NAC) is amyloidogenic. Biochemistry 1995, 34, 10139–10145. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; Engel, W.K.; Alvarez, R.B.; McFerrin, J.; Broccolini, A. Novel immunolocalization of α-synuclein in human muscle of inclusion-body myositis, regenerating and necrotic muscle fibers, and at neuromuscular junctions. J. Neuropathol. Exp. Neurol. 2000, 59, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.C.; Cho, S.E.; Lee, D.K.; Hur, M.W.; Paik, S.R.; Park, J.H.; Kim, J. Expression patterns of α-synuclein in human hematopoietic cells and in Drosophila at different developmental stages. Mol. Cells 2000, 10, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jeon, B.S.; Heo, C.; Im, P.S.; Ahn, T.B.; Seo, J.H.; Kim, H.S.; Park, C.H.; Choi, S.H.; Cho, S.H.; et al. α-Synuclein induces apoptosis by altered expression in human peripheral lymphocyte in Parkinson's disease. FASEB J. 2004, 18, 1615–1617. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Fink, A.L. Amino acid determinants of α-synuclein aggregation: Putting together pieces of the puzzle. FEBS Lett. 2002, 522, 9–13. [Google Scholar] [CrossRef]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Polverino de Laureto, P.; Tosatto, L.; Frare, E.; Marin, O.; Uversky, V.N.; Fontana, A. Conformational properties of the SDS-bound state of α-synuclein probed by limited proteolysis: Unexpected rigidity of the acidic C-terminal tail. Biochemistry 2006, 45, 11523–11531. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular link between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.B.; Ait-Bouziad, N.; Dikiy, I.; Mbefo, M.K.; Jovičić, A.; Kiely, A.; Holton, J.L.; Lee, S.J.; Gitler, A.D.; Eliezer, D.; Lashuel, H.A. The novel Parkinson's disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 2014, 23, 4491–4509. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Sahay, S.; Ranjan, P.; Salot, S.; Mohite, G.M.; Singh, P.K.; Dwivedi, S.; Carvalho, E.; Banerjee, R.; Kumar, A.; et al. The newly discovered Parkinson's disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry 2014, 53, 6419–6421. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective membrane interactions of familial Parkinson's disease mutant A30P α-synuclein. J. Mol. Biol. 2002, 315, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feany, M.B. A-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Snead, D.; Eliezer, D. α-Synuclein function and dysfunction on cellular membranes. Exp. Neurobiol. 2014, 23, 292–313. [Google Scholar] [CrossRef] [PubMed]
- Okochi, M.; Walter, J.; Koyama, A.; Nakajo, S.; Baba, M.; Iwatsubo, T.; Meijer, L.; Kahle, P.J.; Haass, C. Constitutive phosphorylation of the Parkinson's disease associated α-synuclein. J. Biol. Chem. 2000, 275, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef] [PubMed]
- Fauvet, B.; Fares, M.B.; Samuel, F.; Dikiy, I.; Tandon, A.; Eliezer, D.; Lashuel, H.A. Characterization of semisynthetic and naturally Nα-acetylated α-synuclein in vitro and in intact cells: Implications for aggregation and cellular properties of α-synuclein. J. Biol. Chem. 2012, 287, 28243–28262. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamashita, H.; Nakamura, T.; Nagano, Y.; Nakamura, S. Tyrosine 125 of α-synuclein plays a critical role for dimerization following nitrative stress. Brain Res. 2002, 938, 73–80. [Google Scholar] [CrossRef]
- Danielson, S.R.; Andersen, J.K. Oxidative and nitrative protein modifications in Parkinson’s disease. Free Radic. Biol. Med. 2008, 44, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Dorval, V.; Fraser, P.E. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and α-synuclein. J. Biol. Chem. 2006, 281, 9919–9924. [Google Scholar] [CrossRef] [PubMed]
- Münch, G.; Lüth, H.J.; Wong, A.; Arendt, T.; Hirsch, E.; Ravid, R.; Riederer, P. Crosslinking of α-synuclein by advanced glycation endproducts–An early pathophysiological step in Lewy body formation? J. Chem. Neuroanat. 2000, 20, 253–257. [Google Scholar] [CrossRef]
- Liu, C.W.; Giasson, B.I.; Lewis, K.A.; Lee, V.M.; Demartino, G.N.; Thomas, P.J. A precipitating role for truncated α-synuclein and the proteasome in α-synuclein aggregation: Implications for pathogenesis of Parkinson disease. J. Biol. Chem. 2005, 280, 22670–22678. [Google Scholar] [CrossRef] [PubMed]
- Muntané, G.; Ferrer, I.; Martinez-Vicente, M. α-synuclein phosphorylation and truncation are normal events in the adult human brain. Neuroscience 2012, 200, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, A.S.; Chen, J.; Levine, R.L.; Bax, A. Site-specific interaction between α-synuclein and membranes probed by NMR-observed methionine oxidation rates. J. Am. Chem. Soc. 2013, 135, 2943–2946. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J. Neurochem. 2007, 103, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Qin, Z.J.; Hu, D.; Munishkina, L.A.; Fink, A.L. A-synuclein can function as an antioxidant preventing oxidation of unsaturated lipid in vesicles. Biochemistry 2006, 45, 8135–8142. [Google Scholar] [CrossRef] [PubMed]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ninan, I.; Antonova, I.; Battaglia, F.; Trinchese, F.; Narasanna, A.; Kolodilov, N.; Dauer, W.; Hawkins, R.D.; Arancio, O. A-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004, 23, 4506–4516. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Caraveo, G.; Lindquist, S. α-Synuclein: Membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 2010, 26, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A role for α-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Ouberai, M.M.; Wang, J.; Swann, M.J.; Galvagnion, C.; Guilliams, T.; Dobson, C.M.; Welland, M.E. α-Synuclein senses lipid packing defects and induces lateral expansion of lipids leading to membrane remodeling. J. Biol. Chem. 2013, 288, 20883–20895. [Google Scholar] [CrossRef] [PubMed]
- Ben Gedalya, T.; Loeb, V.; Israeli, E.; Altschuler, Y.; Selkoe, D.J.; Sharon, R. α-Synuclein and polyunsaturated fatty acids promote clathrin-mediated endocytosis and synaptic vesicle recycling. Traffic 2009, 10, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Golovko, M.Y.; Faergeman, N.J.; Cole, N.B.; Castagnet, P.I.; Nussbaum, R.L.; Murphy, E.J. α-Synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of α-synuclein palmitate binding. Biochemistry 2005, 44, 8251–8259. [Google Scholar] [CrossRef] [PubMed]
- Golovko, M.Y.; Rosenberger, T.A.; Faergeman, N.J.; Feddersen, S.; Cole, N.B.; Pribill, I.; Berger, J.; Nussbaum, R.L.; Murphy, E.J. Acyl-CoA synthetase activity links wild-type but not mutant α-synuclein to brain arachidonate metabolism. Biochemistry 2006, 45, 6956–6966. [Google Scholar] [CrossRef] [PubMed]
- Golovko, M.Y.; Rosenberger, T.A.; Feddersen, S.; Faergeman, N.J.; Murphy, E.J. α-Synuclein gene ablation increases docosahexaenoic acid incorporation and turnover in brain phospholipids. J. Neurochem. 2007, 101, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T.Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T.Jr. Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000, 39, 2552–2563. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; McLaurin, J.; Yip, C.M.; St George-Hyslop, P.; Fraser, P.E. α-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000, 275, 34328–34334. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.R.; Rhoades, E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 2010, 99, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J. Biol. Chem. 2001, 276, 41958–41962. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed]
- Jao, C.C.; Hegde, B.G.; Chen, J.; Haworth, I.S.; Langen, R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, E.R.; Ramlall, T.F.; Borbat, P.P.; Freed, J.H.; Eliezer, D. Membrane-bound α-synuclein forms an extended helix: Long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Am. Chem. Soc. 2008, 130, 12856–12857. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.; Gambin, Y.; Lemke, E.A.; Deniz, A.A. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 2009, 106, 5645–5650. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of native brain α-synuclein. Nature 2013, 498, E4. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 30, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef] [PubMed]
- Gould, N.; Mor, D.E.; Lightfoot, R.; Malkus, K.; Giasson, B.; Ischiropoulos, H. Evidence of native α-synuclein conformers in the human brain. J. Biol. Chem. 2014, 289, 7929–7934. [Google Scholar] [CrossRef] [PubMed]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T.Jr. α-Synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Lansbury, P.T., Jr. Is there a cause-and-effect relationship between α-synuclein fibrillization and Parkinson's disease? Nat. Cell Biol 2000, 2, E115. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, G.; Frare, E.; Bubacco, L.; Mammi, S.; Fontana, A.; Polverino de Laureto, P. Molecular insights into the interaction between α-synuclein and docosahexaenoic acid. J. Mol. Biol. 2009, 394, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. α-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115. [Google Scholar] [CrossRef] [PubMed]
- Lücke, C.; Gantz, D.L.; Klimtchuk, E.; Hamilton, J.A. Interactions between fatty acids and α-synuclein. J. Lipid Res. 2006, 47, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Broersen, K.; van den Brink, D.; Fraser, G.; Goedert, M.; Davletov, B. α-Synuclein adopts an α-helical conformation in the presence of polyunsaturated fatty acids to hinder micelle formation. Biochemistry 2006, 45, 15610–15616. [Google Scholar] [CrossRef] [PubMed]
- Namani, T.; Walde, P. From decanoate micelles to decanoic acid/dodecylbenzenesulfonate vesicles. Langmuir 2005, 21, 6210–6219. [Google Scholar] [CrossRef] [PubMed]
- Namani, T.; Ishikawa, T.; Morigaki, K.; Walde, P. Vesicles from docosahexaenoic acid. Colloids Surf. B Biointerfaces 2007, 54, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Walde, P. Surfactant assemblies and their various possible roles for the origin(s) of life. Orig. Life Evol. Biosph. 2006, 36, 109–150. [Google Scholar] [CrossRef] [PubMed]
- Morigaki, K.; Walde, P. Fatty Acid Vesicles. Curr. Opin. Colloid Interface Sci. 2007, 12, 75–80. [Google Scholar] [CrossRef]
- De Franceschi, G.; Frare, E.; Pivato, M.; Relini, A.; Penco, A.; Greggio, E.; Bubacco, L.; Fontana, A.; Polverino de Laureto, P. Structural and morphological characterization of aggregated species of α-synuclein induced by docosahexaenoic acid. J. Biol. Chem. 2011, 286, 22262–22274. [Google Scholar] [CrossRef] [PubMed]
- Necula, M.; Chirita, C.N.; Kuret, J. Rapid anionic micelle-mediated α-synuclein fibrillization in vitro. J. Biol. Chem. 2003, 278, 46674–46680. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Buell, A.K.; Meisl, G.; Michaels, T.C.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücke, C.; Gutiérrez-González, L.H.; Hamilton, J.A. Intracellular lipid binding. In Cellular Proteins and Their Fatty Acids in Health and Disease; Duttaroy, A.K., Spener, F., Eds.; Wiley-VCH: Weinheim, Germany, 2003; pp. 95–118. ISBN 9783527601547. [Google Scholar]
- De Franceschi, G.; Polverino de Laureto, P. Role of different regions of α-synuclein in the interaction with the brain fatty acid DHA. J. Chromatograph. Separat. Techniq. 2014, 5, 219–226. [Google Scholar] [CrossRef]
- Karube, H.; Sakamoto, M.; Arawaka, S.; Hara, S.; Sato, H.; Ren, C.H.; Goto, S.; Koyama, S.; Wada, M.; Kawanami, T.; et al. N-terminal region of α-synuclein is essential for the fatty acid-induced oligomerization of the molecules. FEBS Lett. 2008, 582, 3693–3700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodner, C.R.; Maltsev, A.S.; Dobson, C.M.; Bax, A. Differential phospholipid binding of α-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry 2010, 49, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Iljina, M.; Tosatto, L.; Choi, M.L.; Sang, J.C.; Ye, Y.; Hughes, C.D.; Bryant, C.E.; Gandhi, S.; Klenerman, D. Arachidonic acid mediates the formation of abundant α-helical multimers of α-synuclein. Sci. Rep. 2016, 6, 33928. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, M. α-Synuclein oligomers-neurotoxic molecules in Parkinson's disease and other Lewy body disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- Fecchio, C.; De Franceschi, G.; Relini, A.; Greggio, E.; Dalla Serra, M.; Bubacco, L.; Polverino de Laureto, P. α-Synuclein oligomers induced by docosahexaenoic acid affect membrane integrity. PLoS ONE 2013, 8, e82732. [Google Scholar] [CrossRef] [PubMed]
- Assayag, K.; Yakunin, E.; Loeb, V.; Selkoe, D.J.; Sharon, R. Polyunsaturated fatty acids induce α-synuclein-related pathogenic changes in neuronal cells. Am. J. Pathol. 2007, 171, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Riedel, M.; Goldbaum, O.; Wille, M.; Richter-Landsberg, C. Membrane lipid modification by docosahexaenoic acid (DHA) promotes the formation of α-synuclein inclusion bodies immunopositive for SUMO-1 in oligodendroglial cells after oxidative stress. J. Mol. Neurosci. 2011, 43, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, M.; Gue, K.; Emond, V.; Julien, P.; Kang, J.X.; Cicchetti, F.; Calon, F. Transgenic conversion of omega-6 into omega-3 fatty acids in a mouse model of Parkinson’s disease. J. Lipid Res. 2011, 52, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Alcalay, R.N. Genetic forms of Parkinson’s disease. Semin. Neurol. 2017, 37, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Fecchio, C. α-Synuclein oligomers induced by docosahexaenoic acid: A study of activity and molecular characterization. PhD Thesis, Università degli Studi di Padova, Padova, Italy, 2014. [Google Scholar]
- Schildknecht, S.; Gerding, H.R.; Karreman, C.; Drescher, M.; Lashuel, H.A.; Outeiro, T.F.; Di Monte, D.A.; Leist, M. Oxidative and nitrative α-synuclein modifications and proteostatic stress: Implications for disease mechanisms and interventions in synucleinopathies. J. Neurochem. 2013, 125, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Bazan, N.G. Docosahexaenoic acid and the aging brain. J. Nutr. 2008, 138, 2510–2514. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, L.; Brambilla, P.; Mazzocchi, A.; Harsløf, L.B.; Ciappolino, V.; Agostoni, C. DHA effects in brain development and function. Nutrients 2016, 8, E6. [Google Scholar] [CrossRef] [PubMed]
- Yakunin, E.; Loeb, V.; Kisos, H.; Biala, Y.; Yehuda, S.; Yaari, Y.; Selkoe, D.J.; Sharon, R. α-Synuclein neuropathology is controlled by nuclear hormone receptors and enhanced by docosahexaenoic acid in a mouse model for Parkinson's disease. Brain Pathol. 2012, 22, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Binolfi, A.; Limatola, A.; Verzini, S.; Kosten, J.; Theillet, F.X.; Rose, H.M.; Bekei, B.; Stuiver, M.; van Rossum, M.; Selenko, P. Intracellular repair of oxidation-damaged α-synuclein fails to target C-terminal modification sites. Nat. Commun. 2016, 7, 10251. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification a C:D n-X | Common Name b | Pre-Frontal Cortex FAs (%) c | Structure d |
|---|---|---|---|
| SFA | |||
| 12:0 | Lauric acid (LA) | 36 |  |
| 14:0 | Myristic acid (MA) | ||
| 16:0 | Palmitic acid (PA) | ||
| 18:0 | Stearic acid (SA) | ||
| 24:0 | Lignoceric acid (LCA) | ||
| MUFA | |||
| 18:1 n-9 | Oleic acid (OA) | 28 |  |
| 24:1 n-9 | Nervonic acid (NA) | ||
| 16:1 n-7 | Palmitoleic acid (POA) | ||
| 17:1 n-7 | Heptadecenoic acid (HA) | ||
| 18:1 n-7 | Vaccenic acid (VA) | ||
| PUFA | |||
| 22:6 n-3 | Docosahexaenoic acid (DHA) | 15–18 DHA ~ 13.5 |  |
| 18:3 n-3 | α-Linolenic acid (ALA) | ||
| 22:5 n-3 | Docosapentaenoic acid (n-3DPA) | ||
| 20:5 n-3 | Eicosapentaenoic acid (EPA) | ||
| 18:2 n-6 | Linoleic acid (LNA) | 17 AA ~ 9 | |
| 20:4 n-6 | Arachidonic acid (AA) | ||
| 22:5 n-6 | Docosapentaenoic acid (n-6DPA) | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fecchio, C.; Palazzi, L.; Polverino de Laureto, P. α-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration. Molecules 2018, 23, 1531. https://doi.org/10.3390/molecules23071531
Fecchio C, Palazzi L, Polverino de Laureto P. α-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration. Molecules. 2018; 23(7):1531. https://doi.org/10.3390/molecules23071531
Chicago/Turabian StyleFecchio, Chiara, Luana Palazzi, and Patrizia Polverino de Laureto. 2018. "α-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration" Molecules 23, no. 7: 1531. https://doi.org/10.3390/molecules23071531
APA StyleFecchio, C., Palazzi, L., & Polverino de Laureto, P. (2018). α-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration. Molecules, 23(7), 1531. https://doi.org/10.3390/molecules23071531