4. Materials and Methods
All UV-vis and chiroptical measurements were made with solutions prepared in spectroscopic grade solvents; MeOH, CHCl
3 (stabilized with amylenes) or CF
3CH
2OH (TFE), purchased from Acros Organics (NJ, USA). Optical rotations were recorded on a JASCO P2000 digital polarimeter (Easton, MD, USA) at the D-line of Na emission on solutions in quartz cells (0.100 dm pathlength). UV-visible spectra were recorded on a JASCO V630 dual beam spectrometer (Easton, MD, USA) in quartz cuvettes (1.00 cm or 0.100 cm pathlength). Electronic circular dichroism spectra were recorded on either a Jasco J810 spectropolarimeter (Easton, MD, USA) or an Applied PhotoPhysics Chirascan with solutions in quartz cells (0.100 or 0.500 cm). HRMS were acquired on two instruments: a Bruker micrOTOF Q II mass spectrometer (Billerica, MA, USA) using external calibration of sodium formate clusters, and an Agilent 6230 TOFMS (Santa Clara, CA, USA) with a Jetstream electrospray ionization source, calibrated by using Agilent ESL-L Low Concentration Tuning Mix (Part number G1969-85000, Agilent Technologies).
1H-NMR spectra were recorded at 500.16 MHz on a Jeol ECA 500 spectrometer, at 399.91 MHz on a Mercury 400, at 400.13 MHz on a Bruker Avance AVIII 400 MHz or at 500.19 MHz on a Bruker Avance AVIII-HD 500 MHz spectrometer (Billerica, MA, USA).
13C-NMR spectra were recorded at 125.69 MHz on a Varian VX 500 spectrometer (Palo Alto, CA, USA) equipped with an Xsens
13C{
1H} cryoprobe. Spectra were referenced to solvent (residual CHCl
3 δ 7.26 ppm; CDCl
3,
δ 77.16 ppm; residual CHD
2OD
δ 3.31 ppm; CD
3OD
δ 49.00 ppm). Dry pyridine was distilled from CaH
2 under an atmosphere of N
2, and dry CH
2Cl
2, CH
3CN were obtained by passage through commercial basic alumina cartridges under an atmosphere of Ar. Jacobsen’s catalysts [
24] (
S,
S)-(+)- and (
R,
R)-(−)-
N,
N′-
bis(3,5-
tert-butylsalicylidene)-2,2cyclohexanediaminecobalt(II), (±)-1,2-epoxybutane and (±)-1,2-epoxyhexane were obtained from Sigma-Aldrich (Milwaukee, WI, USA).
Animal material. The ascidian consisting of mixed color-morphs of Pseudodistoma opacum was collected on September 29, 2017, at a depth of 4–6 m in Whangateau Harbour entrance (36.3196° S, 174.7831° E), Northland, New Zealand and kept frozen until used. The white and beige color-morphs were collected separately from the same location on November 14, 2017 and kept frozen until used. A voucher specimen of the ascidian is held at NIWA, Wellington, New Zealand as MNP 99203. The organisms were collected under MPI/UoA special permit 549. The sample of P. cereum was collected on November 24, 2002, at a depth of 15 m from Princes Island (34.1786° S, 172.0418° E), Three Kings island group, Northland, New Zealand and kept frozen until used. A voucher specimen of P. cereum is held at NIWA as MNP7042.
Isolation and purification. The freeze-dried ascidians were macerated and cold extracted with MeOH (4 × 200 mL), and the extract filtered, concentrated in vacuo affording a green solid, which was subjected to repeated C18 reversed-phase column chromatography using a gradient of H2O (0.05% TFA) to 100% MeOH. Distaminolyne A∙TFA salt eluted with 70% MeOH/H2O as a pale yellow gum.
Distaminolyne A (NP-9-161-3,
P. opacum mixed colors;
1a): 30 mg, 0.08% dry wt;
+1.0 (
c 1.0 MeOH); The
1H NMR spectrum (CD
3OD, 500 MHz) was identical with the published data [
7]; (+)-HRESIMS
m/
z 262.2163 [M + H]
+, (calcd. for C
17H
28NO, 262.2165).
Distaminolyne A (NP-12-8-1,
P. opacum white color-morph;
1a): 27 mg, 0.2% dry wt;
+1.9 (
c 0.73, MeOH); The
1H NMR (CD
3OD, 500 MHz) spectrum was identical with the published data [
7]; (+)-HRESIMS
m/
z 262.2161 [M + H]
+, (calcd. for C
17H
28NO, 262.2165).
Distaminolyne A (NP12-9-1,
P. opacum beige color-morph;
1a): 7 mg, 0.1% dry wt;
−1.1 (
c 0.70, MeOH); The
1H NMR (CD
3OD, 500 MHz) spectrum was identical with the published data [
7]; (+)-HRESIMS
m/
z 262.2159 [M + H]
+, (calcd. for C
17H
28NO, 262.2165).
Distaminolyne A (NP-12-10-1, P. cereum; 1a): 8.5 mg, 0.04% dry wt; +1.5 (c 0.275, MeOH); 1H NMR (CD3OD, 500 MHz) δ 5.89–5.83 (1H, m), 5.12–5.08 (1H, m), 5.06–5.03 (1H, m), 3.76–3.73 (1H, m), 3.03 (1H, dd, J = 12.8, 3.0 Hz), 2.77 (1H, dd, J = 12.8, 9.5 Hz), 2.35 (2H, t, J = 6.6 Hz), 2.28–2.24 (4H, m), 1.56–1.29 (12H, m); 13C NMR (CD3OD, 125 MHz) δ 137.8, 116.3, 78.0, 77.1, 68.7, 66.7, 66.3, 46.1, 36.0, 33.7, 30.5, 30.1, 29.8, 29.5, 26.3, 19.6; (+)-HRESIMS m/z 262.2157 [M + H]+, (calcd. for C17H28NO, 262.2165).
N,O-Dibenzoyl derivative1b: Distaminolyne A
1a (NP-12-8-1) (3.7 mg, 14 µmol) was dibenzoylated with benzoic acid according to
Method B to obtain, after RP C
18 chromatography (1:9 water/methanol) dibenzoyl derivative
1b, 2.6 mg (40%).
0 (
c 0.26, MeOH); UV (MeOH) λ
max (log ε) 203 (4.28), 228 (4.19), 267 (3.55), 282 (3.44); The
1H NMR (CD
3OD, 500 MHz) spectrum was identical with the published data [
7]; (+)-HRESIMS
m/
z 492.2500 [M + Na]
+, (calcd. for C
31H
35NNaO
3, 492.2509).
N,O-Dibenzoyl derivative1b: Distaminolyne A
1a (NP-12-9-1) (2.5 mg, 10 µmol) was dibenzoylated with benzoic acid according to
Method B to obtain, after RP C
18 chromatography (1:9 water/methanol) dibenzoyl derivative
1b, 1.8 mg (40%).
−1.1 (
c 0.18, MeOH); UV (MeOH) λ
max (log ε) 204 (4.05), 228 (3.98), 266 (3.31), 281 (3.17); The
1H NMR (CD
3OD, 400 MHz) spectrum was identical with the published data [
7]; (+)-HRESIMS
m/
z 492.2504 [M + Na]
+, (calcd. for C
31H
35NNaO
3, 492.2509).
N,O-Dibenzoyl derivative1b: Distaminolyne A
1a (NP-12-10-1) (3.5 mg, 13 µmol) was dibenzoylated with benzoic acid according to
Method B to obtain, after RP C
18 chromatography (1:9 water/methanol) dibenzoyl derivative
1b, 2.6 mg (41%).
+0.4 (
c 0.26, MeOH); UV (MeOH) λ
max (log ε) 205 (4.34), 226 (4.23), 267 (3.70), 282 (3.59); The
1H NMR (CD
3OD, 400 MHz) spectrum was identical with the published data [
7]; (+)-HRESIMS
m/
z 492.2504 [M + Na]
+, (calcd. for C
31H
35NNaO
3, 492.2509).
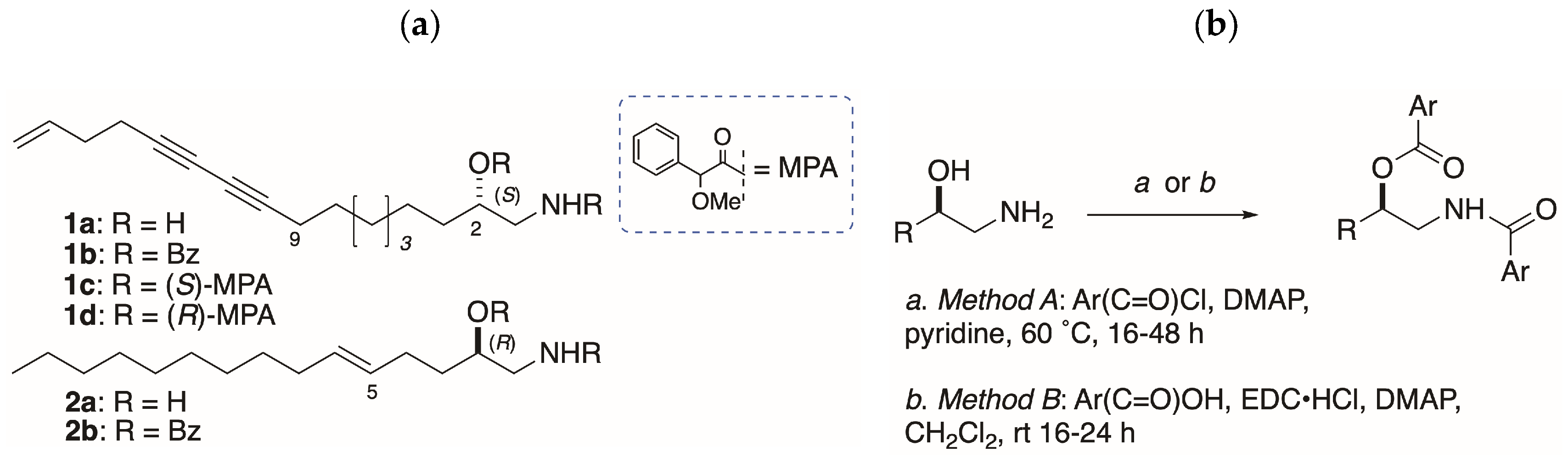
(S)-MPA derivative 1c: Distaminolyne A 1a (NP-9-161-3) (2.0 mg, 8 µmol) was treated with (S)-MPA according to Method B to yield 3.4 mg (80%) of crude product. 1H NMR (CDCl3, 500 MHz) diagnostic CαH resonances of bis-MPA: δ 4.79 (0.96H, s), 4.71 (1H, s), 4.59 (1H, s), 4.41 (0.96H, s).
(R)-MPA derivative 1d: Distaminolyne A 1a (NP-9-161-3) (2.0 mg, 8 µmol) was treated with (R)-MPA according to Method B to yield 2.4 mg (56%) of crude product. 1H NMR (CDCl3, 500 MHz) diagnostic CαH resonances of bis-MPA: δ 4.79 (0.81H, s), 4.71 (1H, s), 4.59 (1H, s), 4.41 (0.81H, s).
(S)-MPA derivative 1c: Distaminolyne A 1a (NP-12-8-1) (2.0 mg, 6 µmol) was treated with (S)-MPA according to Method B to yield 3.6 mg (84%) of crude product. 1H NMR (CDCl3, 400 MHz) diagnostic CαH resonances of bis-MPA: δ 4.79 (1H, s), 4.71 (0.93H, s), 4.59 (0.93H, s), 4.41 (1H, s).
(R)-MPA derivative 1d: Distaminolyne A 1a (NP-12-8-1) (2.0 mg, 6 µmol) was treated with (R)-MPA according to Method B to yield 3.8 mg (89%) of crude product. 1H NMR (CDCl3, 400 MHz) diagnostic CαH resonances of bis-MPA: δ 4.79 (0.68H, s), 4.71 (1H, s), 4.59 (1H, s), 4.41 (0.68H, s).
(S)-MPA derivative 1c: Distaminolyne A 1a (NP-12-9-1) (2.0 mg, 6 µmol) was treated with (S)-MPA according to Method B to yield 2.3 mg (54%) of crude product. 1H NMR (CDCl3, 400 MHz) diagnostic CαH resonances of bis-MPA: δ 4.79 (0.97H, s), 4.71 (1H, s), 4.59 (1H, s), 4.40 (0.97H, s).
(R)-MPA derivative 1d: Distaminolyne A 1a (NP-12-9-1) (2.0 mg, 6 µmol) was treated with (R)-MPA according to Method B to yield 3.5 mg (82%) of crude product. 1H NMR (CDCl3, 400 MHz) diagnostic CαH resonances of bis-MPA: δ 4.79 (0.51H, s), 4.71 (1H, s), 4.59 (1H, s), 4.41 (0.51H, s).
(S)-MPA derivative 1c: Distaminolyne A 1a (NP-12-10-1) (1.5 mg, 6 µmol) was treated with (S)-MPA according to Method B to yield 2.4 mg (75%) of crude product. 1H NMR (CDCl3, 500 MHz) diagnostic CαH resonances of bis-MPA: δ 4.79 (1H, s), 4.71 (0.59H, s), 4.60 (0.59H, s), 4.40 (1H, s).
(R)-MPA derivative 1d: Distaminolyne A 1a (NP-12-10-1) (1.5 mg, 6 µmol) was treated with (R)-MPA according to Method B to yield 2.8 mg (88%) of crude product. 1H NMR (CDCl3, 500 MHz) diagnostic CαH resonances of bis-MPA: δ 4.79 (0.4H, s), 4.71 (1H, s), 4.60 (1H, s), 4.40 (0.4H, s).
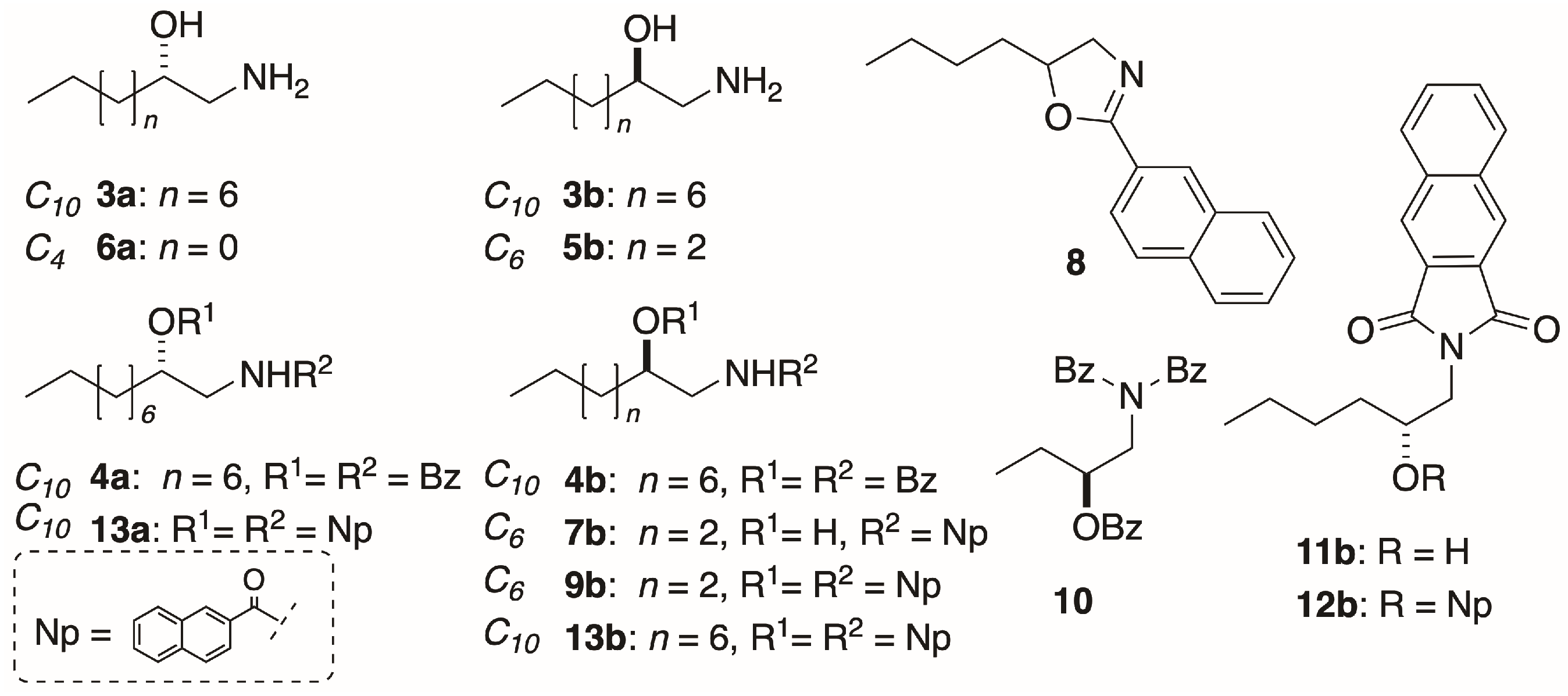
(R)-1-Amino-2-hexanol,5b: 1-Amino-2-alkanols were prepared from the corresponding optically enriched 1,2-epoxides (>97%ee, prepared by Jacobsen’s hydrolytic kinetic resolution of the corresponding racemic epoxide [
24] with (
S,
S)- or (
R,
R)-catalyst) by ammoniolysis using a variation of the published procedure for
4a,
b [
12]. To an ice-cold, stirred solution of (
R)-1,2-epoxyhexane (82.7 mg, 0.83 mmol) in EtOH (3.0 mL) contained in a 20 mL vial was added, dropwise, aqueous NH
4OH (20%
v/
v, 3.0 mL). The vial was sealed tightly and heated at 60 °C for 23 h. The cooled contents of the vial were transferred to a round bottom flask and the volatiles removed under reduced pressure. The residue was taken up in CHCl
3 and passed through plug of anhydrous Na
2SO
4 to give, after removal of solvent, amino alcohol (
R)-
5b as a straw yellow oil (210 mg, 58%). Distillation (twice, Kügelrohr, 120–30 °C/1 mm Hg) gave an analytically pure sample.
1H and
13C NMR and MS data for (
R)-
5b matched the literature values [
25].
(S)-1-Amino-2-butanol,6a: The title compound was prepared from (
S)-1,2-epoxybutane according to the above-described procedure for
5b, with final purification by distillation (Kügelrohr, 130 °C/1 mm Hg) have an analytically pure sample. Colorless oil,
+17 (
c 1.0, CHCl
3), lit. +7.3 (
c 1.0, CHCl
3) [
26];
1H NMR and
13C NMR matched the literature values [
26].
Acylation, Method A: A solution of 1-amino-2-alkanol (1-AA, 1 equiv), freshly distilled benzoyl chloride (4 equiv) and DMAP (1 crystal) in pyridine (1-AA concentration ~0.1 M) was heated at 60 °C under an atmosphere of N2 for 24–48 h. The volatiles were removed under high vacuum and the residue purified by elution (CH2Cl2) through a plug of basic alumina, followed by flash chromatography (silica or RP C18) followed by HPLC (silica or RP C18) if required.
Acylation, Method B: A solution of benzoic or 2-naphthoic acid (~1 mmol, 5 equiv), EDC∙HCl (5.5 equiv) and DMAP in CH2Cl2 was stirred at 0 °C for 20 min under N2. A solution of 1-AA in CH2Cl2 was (final concentration of 1-AA ~0.01 M) and the mixture warmed to rt over 24 h. Additional CH2Cl2 (20 mL) was added and the mixture washed successively with equal volumes of 10% HCl, water, NaHCO3 (satd.) and H2O. After drying (Na2SO4), the volatiles were removed and the residue purified by flash chromatography (silica), followed by HPLC (silica or RP C18) if required.
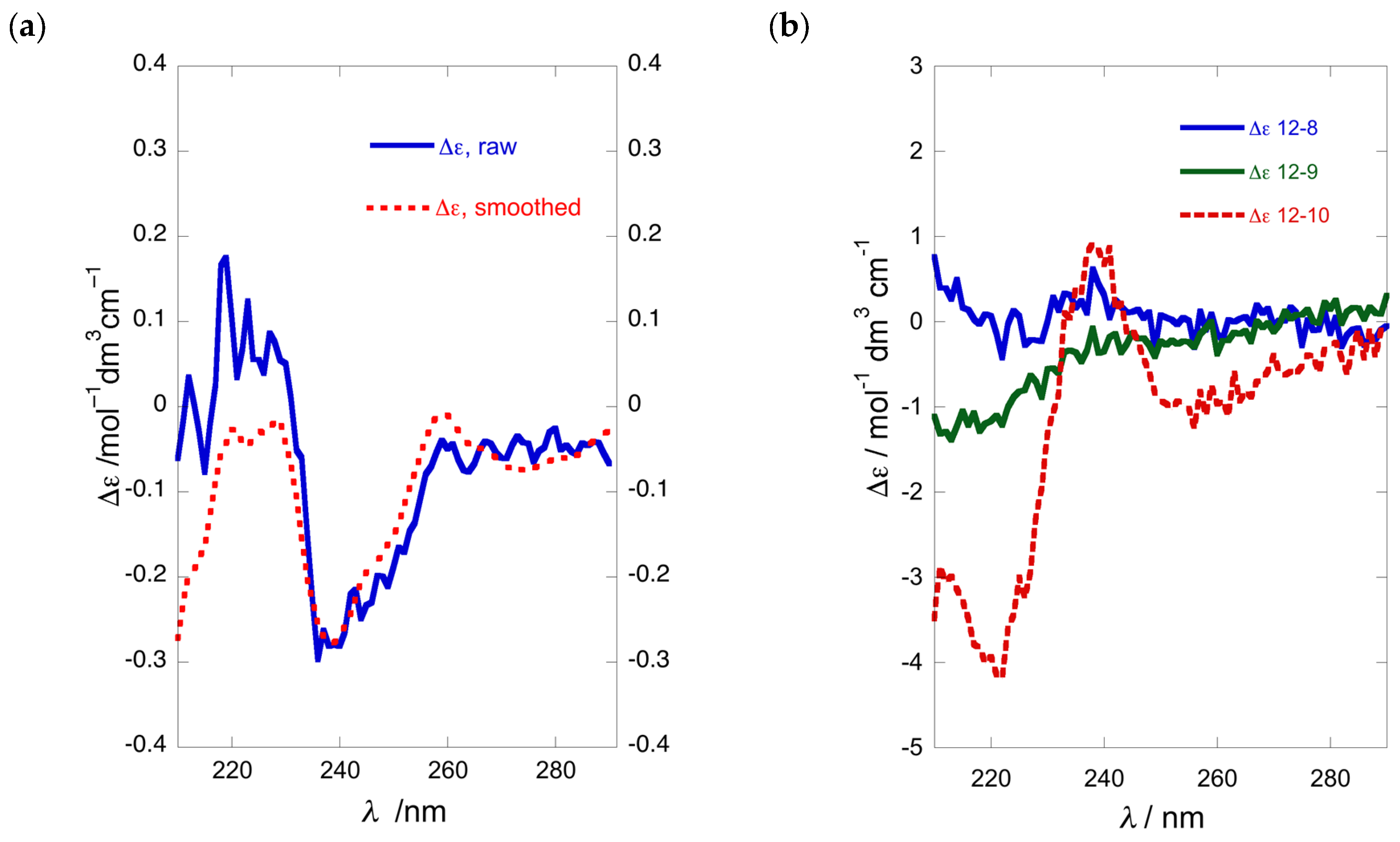
Acylation, Method C: A standard solution was prepared by dissolving EDC∙HCl (18.6 mg, 97 µmol), DMAP (11.2 mg, 97 µmol) and 2-naphthoic acid (16.3 mg, 95 µmol) in 1,2-dichloroethane (DCE, 820 µL; final concentration 0.13 M in 2-naphthoic acid). A solution of 1-AA in DCE (6.1 µg, 7.5 µL, 0.35 µmol) and an aliquot of the standard solution (7.5 µL, 0.98 µmol, 2.8 equiv.) were transferred into a melting point capillary using a gas tight syringe which was then flame sealed. The capillary tube was placed into a melting point apparatus set at 68 °C and left for 60 min, after which the liquid contents of the tube were removed with a narrow capillary and applied directly onto a glass backed TLC plate (6 × 6 cm, silica, 250 µm, prewashed by 2× development in 1:1 EtOAc/n-hexane) in replicate lanes (n = 6). After development of the TLC plate in 1:4 EtOAc/n-hexane, the fluorescent spots corresponding to the N,O-dinaphthoyl-1-AA product (for (R)-12b, Rf 0.20) were scraped from the plate into a 6 mL vial containing a magnetic stir bar, and the mixture extracted with TFE (1.0 mL) by vigorous stirring for 30 min. The vial was centrifuged and measurements of the UV-vis and ECD spectra were carried out directly on the supernatant (30–40% yield based on absorbance at λmax 231 nm).
N,O-Dibenzoyl-1-amino-2-decanol (R)-4b: (
R)-1-Amino-2-decanol (
3b) (2.5 mg, 14 µmol) was dibenzoylated with benzoic acid according to
Method B to obtain, after ‘pencil column’ chromatography (silica, 1:3 EtOAc/
n-hexane) dibenzoyl derivative (
R)-
4b (7.3 mg, quant).
−24.1 (
c 1.48, MeOH). lit −26.2 (
c 1.78, MeOH) [
12]. The
1H-NMR spectrum (CDCl
3) was identical with the published data [
12].
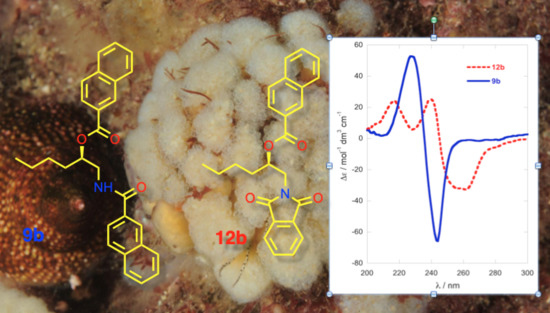
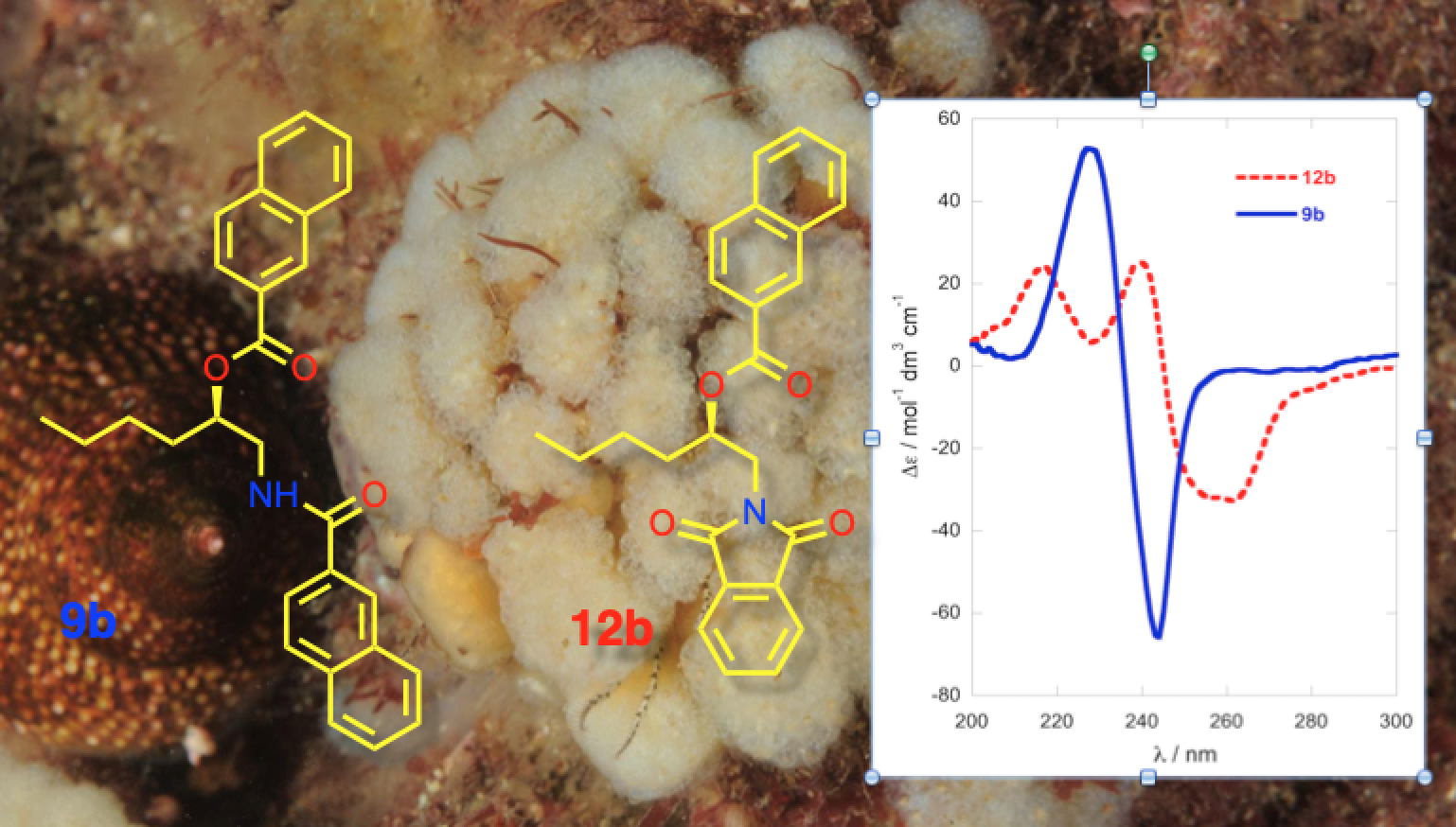
Naphthoylation of Amino Alcohol (R)-5b: 2-Naphthoyl chloride (304 mg, 1.60 mmol, 2.5 equiv), freshly prepared from 2-naphthoic acid (SOCl2, reflux), was added to a stirred solution of 1-AA (R)-5b (75.3 mg, 0.64 mmol) and DMAP (1 crystal) in pyridine (5.0 mL) under N2, and the mixture heated at 60 °C for 96 h. The volatiles removed under high vacuum and the pale pink semi-crystalline residue (220.4 mg) was eluted through a plug of basic alumina with CH2Cl2. The residue obtained by removal of solvent was separated by flash chromatograph (2:98 MeOH/CH2Cl2). The UV-absorbing fractions (TLC) were combined and, after removal of solvent, the residue was triturated with warm diethyl ether to give white crystals of mono-naphthamide (R)-7b (29.5 mg, 18%). The supernatant was concentrated and the residue (4.3 mg) separated, first on a ‘pencil column’ (1% MeOH/CH2Cl2) followed by HPLC (silica, 5 µ, 40:60 EtOAc/n-hexane, 3.0 mL∙min−1) to give dinaphthoyl derivative (R)-9b (1.2 mg, 0.5%) and oxazoline 8 (0.6 mg, 0.4%). With Method B, (R)-5b (3.2 mg, 19 µmol) was converted into (R)-9b (4.2 mg, 54% yield) after purification by pipette column (silica, 20% EtOAc-hexane).
Mono-Naphthamide7b: Recrystallized from CDCl3, m.p. 135 °C; −5.7 (c 0.98, MeOH); 1H NMR (CDCl3, 500 MHz) δ 8.29 (s, 1H, H-1′), 7.88–7.81 (m, 4H), 6.91 (bt, 1H, NH), 3.85 (m, 1H, H-2), 3.76 (ddd, 1H, J = 13.5, 7.0, 3.0 Hz, H-1a), 3.45 (ddd, 1H, J = 13.5, 8.0, 5.0 Hz, H-1b), 2.84 (bs, 1H, OH), 1.53 (m, 2H), 1.44 (m, 1H), 1.34 (3H), 0.90 (t, 3H, J = 7.0 Hz, H3-6). 13C NMR (CDCl3, * = interchangeable) 168.6 (C, C=O), 134.9 (C, C-2′*), 132.7 (C, C-4a*), 131.5 (C, C8a*), 129.0 (CH), 128.6 (CH), 127.85 (CH), 127.82 (CH), 127.7 (CH), 126.9 (CH), 123.7 (CH), 71.6 (CH, C-2), 46.4 (CH2, C-1), 35.0 (CH2, C-3), 27.8 (CH2, C-4), 22.8 (CH2, C-5), 14.2 (CH2, C-6); ESI TOF HRMS m/z 294.1468 [M + Na]+ calcd. for C17H21NO2Na+ 294.1465.
Oxazoline8: +2.6 (c 0.50, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 8.43 (bd, 1H, J = 1.5 Hz, H-1′), 8.03 (dd, 1H, J = 8.5, 1.5 Hz, H-3′), 7.92 (d, 1H, J = 7.5 Hz, H-8′), 7.87–7.86 (m, 2H), 7.57-7.7.50 (m, 2H); ESI TOF HRMS m/z 254.1539 [M+H]+, calcd. for C17H20NO+ 254.1539.
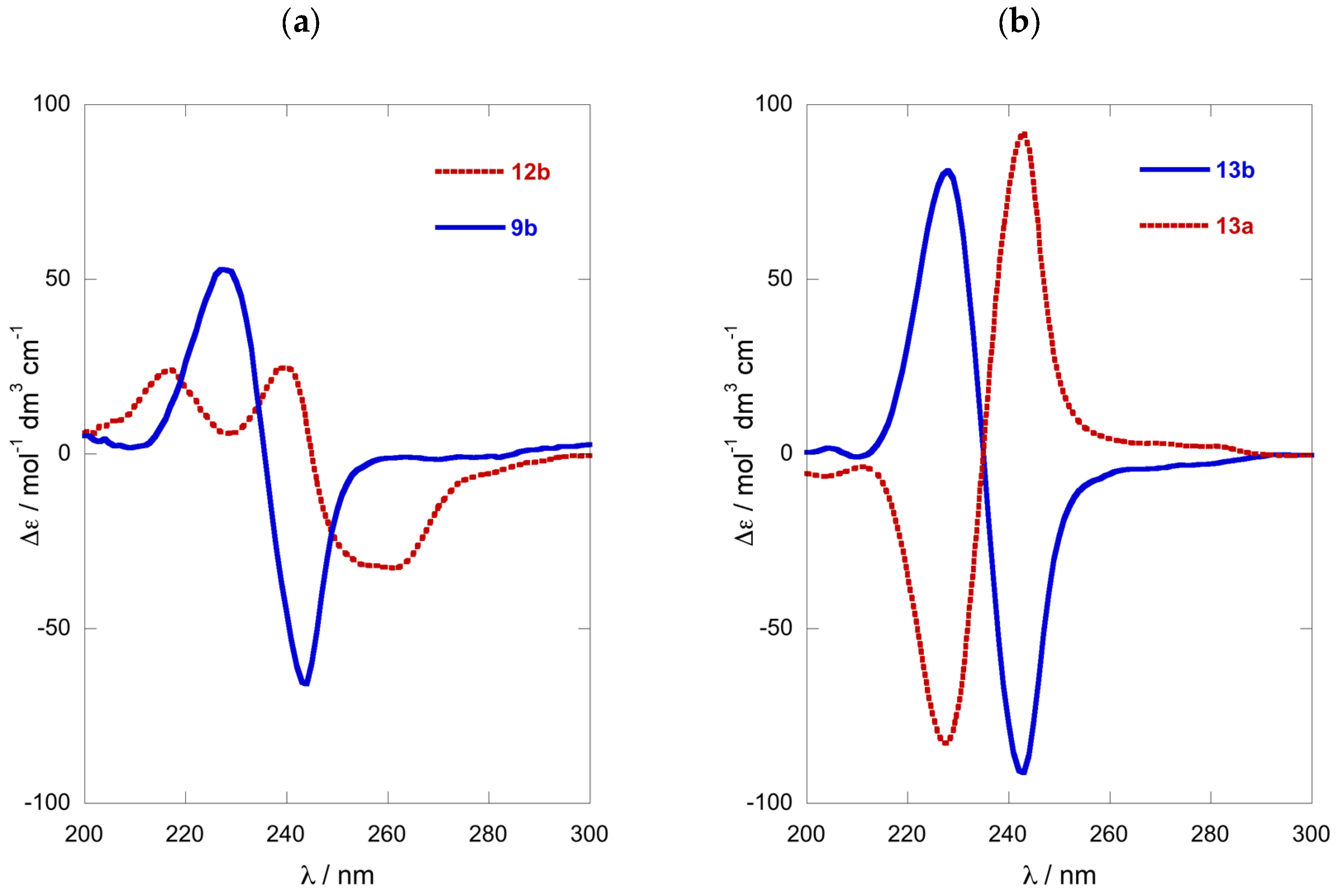
N,O-di-(2-Naphthoyl)-1-amino-2-hexanol (R)-9b: Colorless oil, −67 (c 0.8, TFE); ECD (TFE) δ 228 (∆ε +53), 236 (0), 244 (−66); 1H NMR (CDCl3, 500 MHz) δ 8.65 (s, 1H, H-1′), 8.27 (s, 1H, H-1″), 8.09 (d, 1H, J = 8.5 Hz, H-3′), 7.97 (d, 1H, J = 8.0 Hz, H-3″), 7.91 (m, 6H), 7.79 (d, 1H, J = 8.5 Hz), 7.62-7.50 (m, 4H), 6.92 (bt, 1H, J = 5.0 Hz, NH), 5.42 (m, 1H, H-2), 3.91 (ddd, 1H, J = 14.5, 5.0, 3.5 Hz, H-1a), 3.85 (ddd, 1H, J = 14.5, 9.0, 5.0 Hz, H-1b), 1.92 (m, 1H, H-3a), 1.84 (m, 1H, H-3b), 1.5 (m, 2H, H2-4), 1.42 (m, 2H, H2-5), 0.94 (t, 3H, J = 7.3 Hz, H3-6); ESI TOF HRMS m/z 426.2061 [M + H]+ calcd. for C28H28NO3+ 426.2064.
Tribenzoyl-1-amino-butanol (S)-10a: A solution of 1-AA (S)-6a (66.7 mg, 0.75 mmol) in dry pyridine (2.0 mg) and DMAP (1 crystal) was treated with freshly distilled benzoyl chloride (271 µL, 2.99 mmol) and the mixture heated at 60 °C under at atmosphere of N2 with stirring for 48 h. The volatiles were removed under high vacuum and the semi-crystalline residue (1.28 g) taken up in CH2Cl2 and filtered through a plug of basic alumina. The eluate was concentrated and the residue separated on a ‘pencil column’ (silica, elution with 1:4 EtOAc/n-hexane) to give a non-polar, clear oil (67 mg) that was further purified by HPLC (silica, 10 × 250 mm, 5 µ, 1:4 EtOAc/n-hexane) to give tribenzoyl derivative (S)-10b (14.9 mg, 5%). +44.6 (c 2.92, MeOH); 1H NMR (CDCl3, 500 MHz) δ 7.81 (d, 2H, J = 7.5 Hz, H-2″,6″), 7.39 (t, 1H, J = 7.4 Hz, H-4″), 7.33 (d, 4H, J = 7.5 Hz, H-2′,6′), 7.23 (t, 2H, J = 7.5 Hz, H-3″,5″), 7.12 (t, 2H, J = 7.5 Hz, H-4′), 7.02 (t, 4H, J = 7.5 Hz, H-3′,5′), 5.65 (m, 1H, H-2), 4.53 (dd, 1H, J = 14.0, 9.1 Hz, H-1a), 4.25 (dd, 1H, J = 14.1, 2.8 Hz, H-1b), 1.87 (m, 2H, H2-3), 1.06 (t, 3H, J = 7.5 Hz, H3-6). 13C NMR (CDCl3) δ (C, N(C=O)2), 166.4 (C, O(C=O)), 136.3 (C, C2-1′), 132.8 (CH, C-4″), 131.9 (C, C2-4′), 130.0 (CH, C-1″), 129.6 (CH, C-2′,6′), 129.0 (CH, C4-2′,6′), 128.16 (CH, C2-3′), 128.15 (CH, C-3″), 75.0 (CH, C-2), 49.9 (CH2, C-1), 25.8 (CH2, C-3), 9.6 (CH3, C-4); HRMS m/z 424.1520 [M + Na]+, calcd. for C25H23NO4Na+ 424.1519.
Naphthimide (R)-11b: A solution of the 1-AA (R)-5b (3.5 mg, 30.3 µmol) in dry pyridine (0.175 mL) was added to a stirred solution of freshly sublimed 2,3-naphthalenedicarboxylic acid anhydride (9.0 mg, 45.4 µmol, 1.5 equiv.) in dry pyridine (0.325 mL) and the mixture heated to 110 °C under N2 for 48 h. The volatiles were removed under a stream of N2. The residue was divided in half, and each applied to duplicate preparative TLC plates (silica, 200 × 200 × 0.25 mm). The plates were developed, twice (2:3 EtOAc/n-hexane) and the silica corresponding to the major fluorescent bands (TLC, Rf = 0.52, 1:1 EtOAc/n-hexane) were scrapped from the plates and extracted with EtOAc. The extracts were combined, filtered and the solvent removed from the filtrate to deliver (R)-11b (6.9 mg, 74%) as a colorless powder. +7.0 (c 0.64, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 8.35 (s, 2H, H-1′,4′), 8.05 (m, 2H, H-5′,6′), 7.71 (m, 2H, H-7′,8′), 3.94 (m, 1H, H-2), 3.88 (dd, 1H, J = 14.0, 3.5 Hz, H-1a), 3.82 (dd, 1H, J = 14.0, 8.0 Hz, H-1b), 2.46 (bs, 1H, OH), 1.58–1.48 (m, 2H, H2-3), 1.42–1.33 (m, 4H, H2-4, H2-5), 0.92 (t, 3H, J = 7.0 Hz, H3-6); ESI TOF HRMS m/z 320.1253 [M + Na]+, calcd. for C18H19NO3Na+ 320.1257.
O-Naphthoyl-naphthimide (R)-12b: An ice-cold solution of 2-naphthoic acid (4.0 mg, 32 µmol, 5.0 equiv.), DMAP (4.0 mg, 33 µmol, 5.1 equiv.) and EDC∙HCl (6.3 mg, 33 µmol, 5.1 equiv.) in CH2Cl2 (0.5 mL) was stirred for 30 min under an atmosphere of N2, then treated with a solution of naphthimide (R)-11b (2.2 mg, 6.4 µmol) in CH2Cl2 (0.3 mL). The solution was allowed to warm to 23 °C and stirred for 64 h, then diluted with additional CH2Cl2 (15 mL) and washed sequentially with 0.2M HCl, NaHCO3 (satd.) and water. After drying the solution (Na2SO4), the volatiles were removed and the residue (4.5 mg) purified by ‘pencil column’ chromatography (silica, 1:4 EtOAc/n-hexane) to give pure (R)-12b as a colorless oil (2.2 mg, 72%, Rf 0.20, 1:4 EtOAc/n-hexane). −90 (c 0.44, TFE); ECD (TFE) δ 216 (∆ε +24.7), 240 (+25.8), 245 (0), 259 (−31.8); 1H NMR (CDCl3, 500 MHz, * = interchangeable) δ 8.55 (d, 1H, J = 1.4 Hz, H-1″), 8.29 (s, 2H, H2-1′,4′), 8.00 (m, 2H, H2-5″,8″), 7.98 (dd, 1H, J = 8.3, 1.4 Hz, H-3″), 7.94 (d, 1H, J = 8.0 Hz), 7.85 (d, 1H, J = 7.0 Hz), 7.83 (d, 1H, J = 8.5 Hz), 7.85 (dd, 1H, J = 7.7, 0.8 Hz), 7.83 (d, J = 8.3 Hz), 7.66 (m, 2H, H-6′,7′), 7.57 (td, 1H, J = 7.5, 1.5 Hz, H-6″*), 7.52 (td, 1H, J = 7.5, 1.0 Hz, H-7″*), 5.52 (m, 1H, H-2), 4.16 (dd, 1H, J = 14.0, 8.5 Hz, H-1a), 4.02 (dd, 1H, J = 14.0, 3.0 Hz, H-1b), 1.85 (m, 2H, H2-3), 1.49 (m, 2H, H2-4), 1.40 (m, 2H, H2-5), 0.92 (t, 3H, J = 7.3 Hz, H3-6); ESI TOF HRMS m/z 452.1847 [M + H]+, calcd. for C29H26NO4+ 452.1856.
N,O-di-(2-Naphthoyl)-1-amino-2-decanol (S)-13a: A sample of the 1-AA (
S)-
3a (4.5 mg, 16 µmol) was acylated with 2-naphthoic acid (5 equiv.) using
Method B to give, after standard workup and purification by pipette column (silica, 1:5
n-hexane/CH
2Cl
2),
13a (3.5 mg, 46%).
+94 (
c, 0.16, CHCl
3)), which displayed
1H and
13C NMR spectra identical with those of (
R)-
13b [
12]. ESI TOF HRMS
m/
z 504.2501 [M + Na]
+, calcd. for C
32H
35NaNO
3+ 504.2509.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}