
Design of a Human Rhinovirus-14 3C Protease-Inducible Caspase-3

Abstract
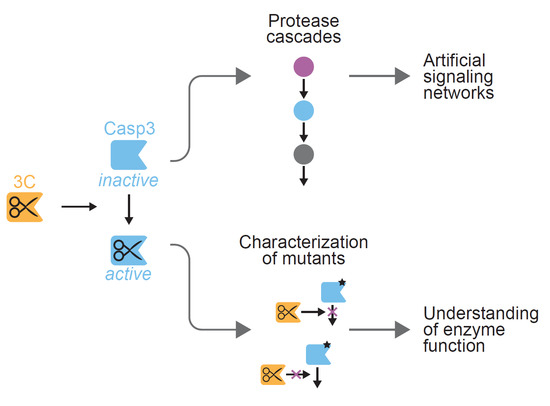
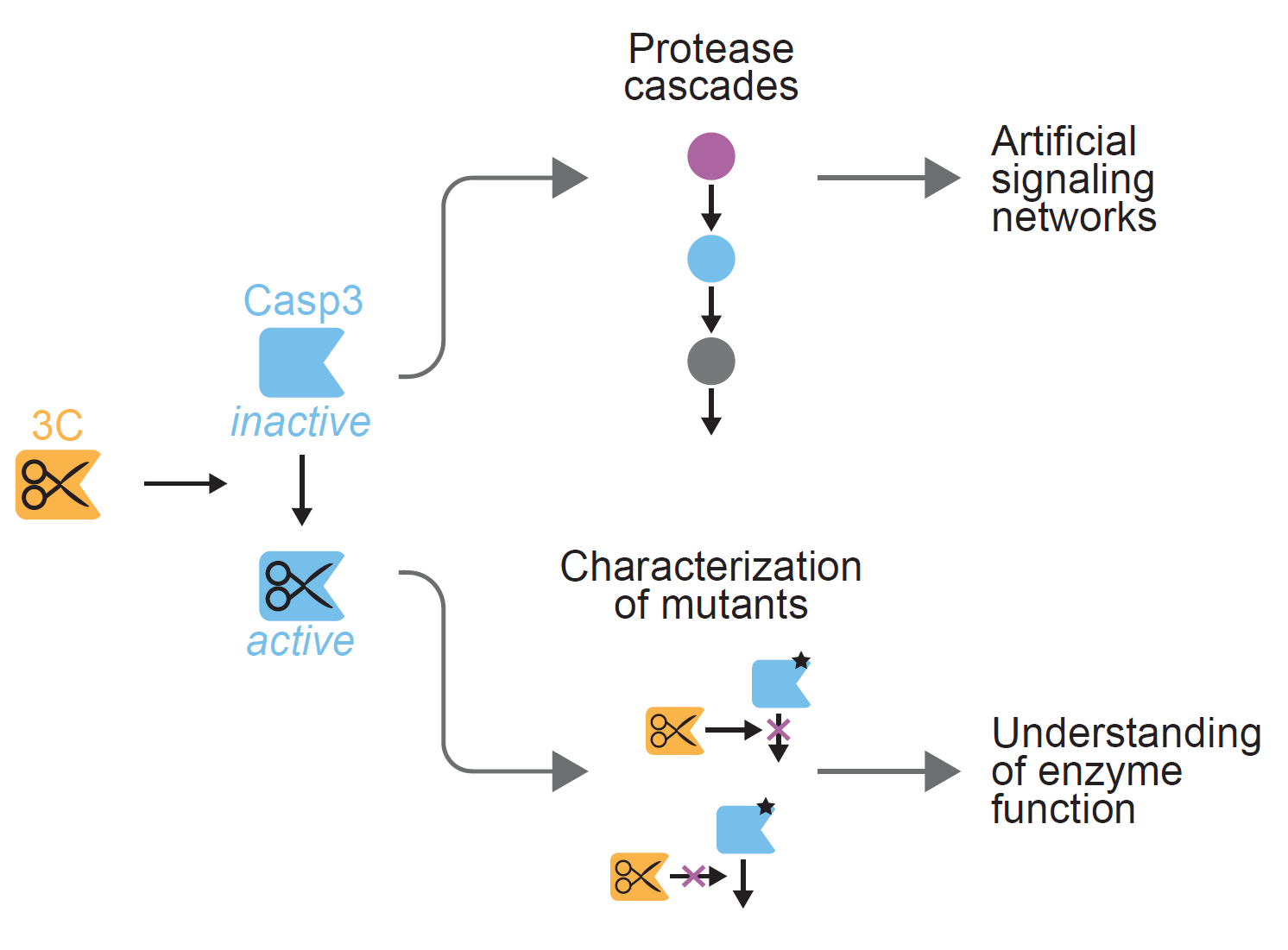
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
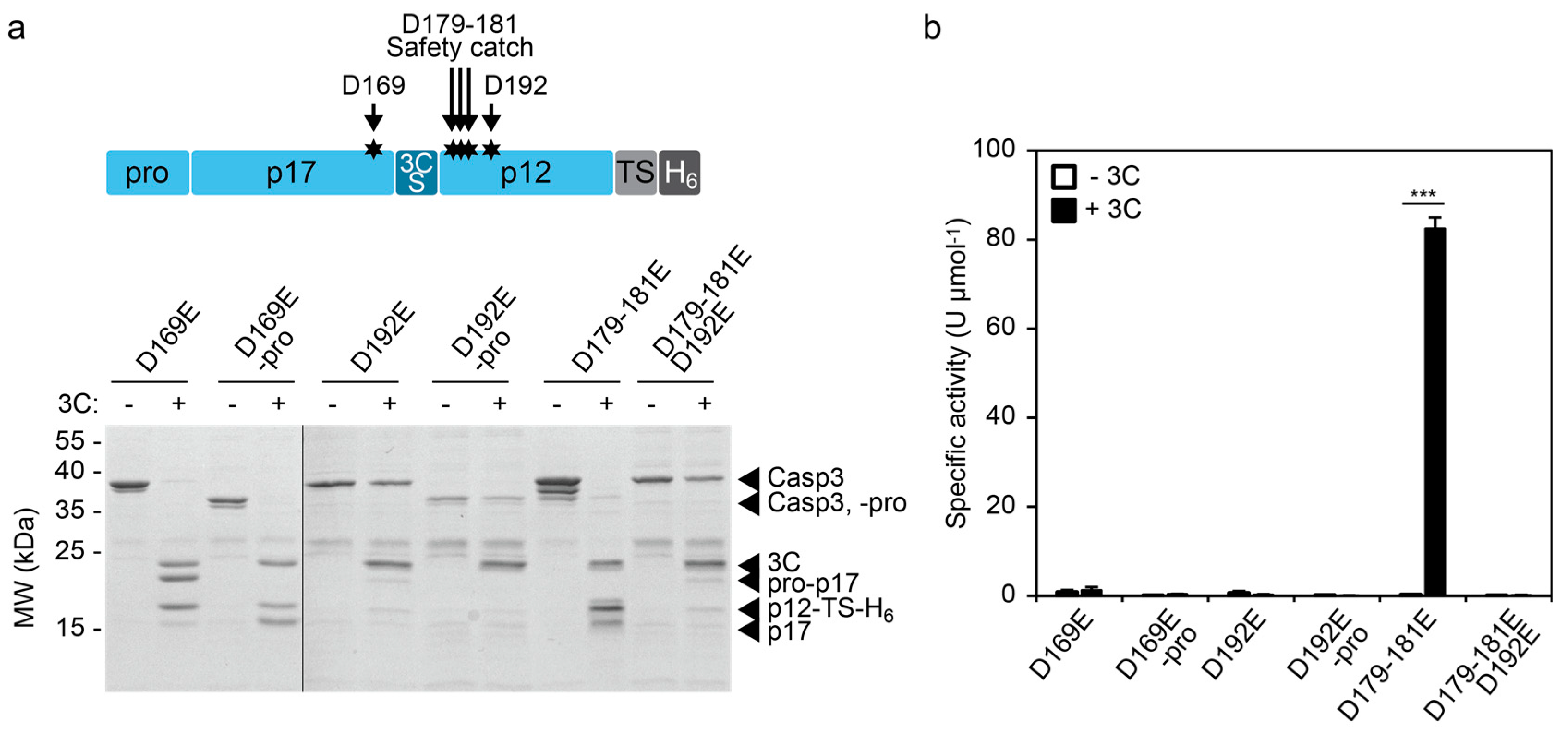
2.1. Design and Testing of an Inducible CASP3 Via Endogenous Cleavage Site Engineering
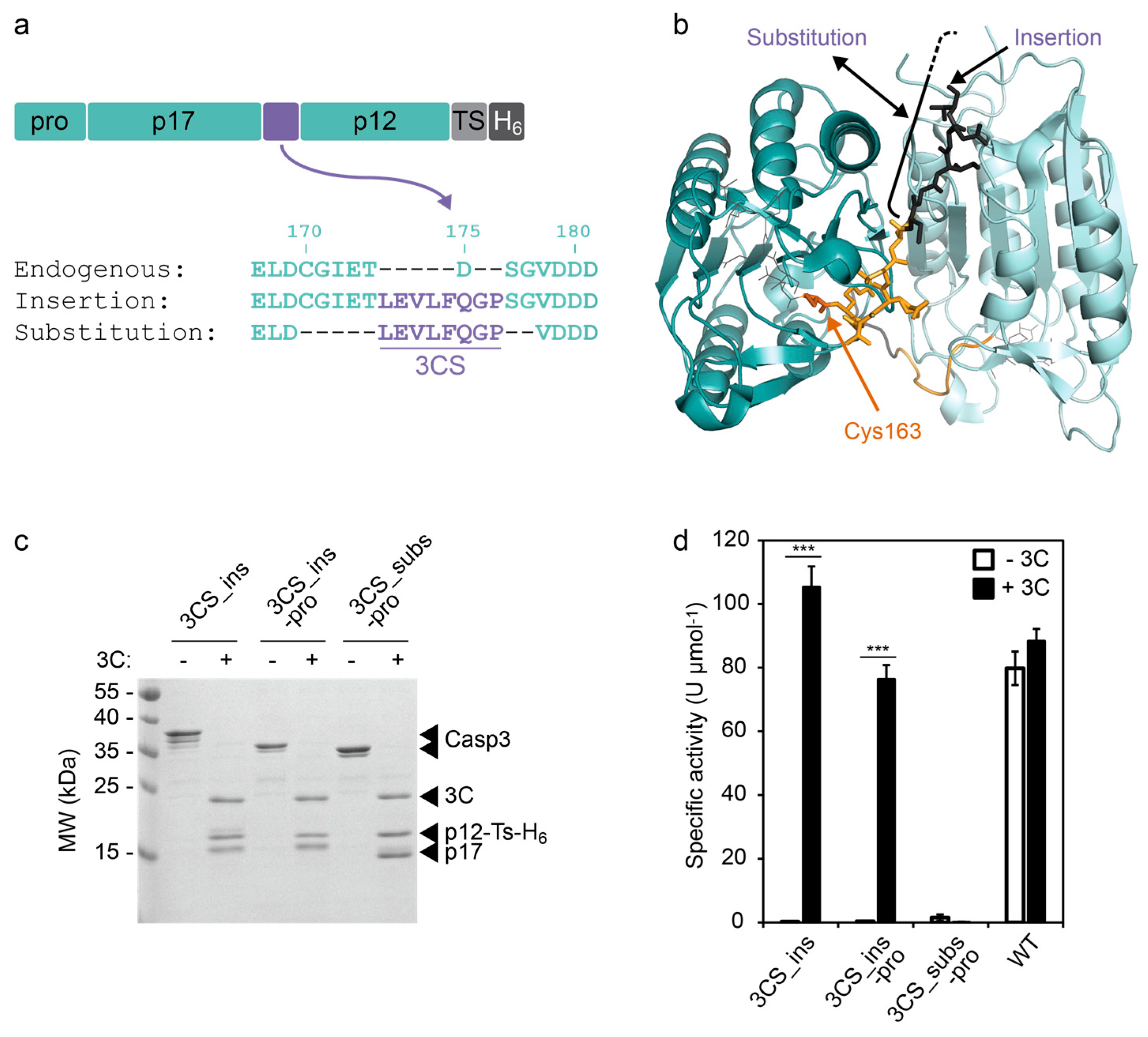
2.2. Design and Testing of an Inducible Caspase-3 by Inserting a Bulky Protein Domain
2.3. Inducible CASP3 as a Research Tool
2.3.1. Control of Proteolytic Cascades
2.3.2. Characterizing the Effect of CASP3 Mutations
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Protein Production and Purification
4.3. Cleavage and Analysis of CASP3 Constructs
4.4. Protease Cascade
4.5. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3CS_ins | CASP3 with inserted 3C protease cleavage site |
| 3CS_ins -pro | 3CS_ins with deleted prodomain |
| 3CS_subs | CASP3 with substituted cleavage site |
| 3CS_subs -pro | 3CS_subs with deleted prodomain |
| 3CS | HRV14 3C protease cleavage site |
| BSA | Bovine serum albumin |
| CASP3 | Human caspase-3 |
| CCS | CASP3 cleavage site |
| DEVD-pNA | p-nitroaniline-labeled DEVD peptide for colorimetric CASP3 assay |
| DTT | Dithiothreitol |
| E. coli | Escherichia coli |
| GrB | Granzyme B |
| GyrB | Bacterial gyrase subunit B |
| His-tag | Hexa-histidine tag |
| HRV14 | Human rhinovirus serotype 14 |
| IMAC | Immobilized metal affinity chromatography |
| IPTG | Isopropyl β-d-1-thiogalactopyranoside |
| mCherry-3CS_ins | CASP3 with incorporated mCherry and inserted 3C protease cleavage site |
| mCherry-3CS_subs | CASP3 with incorporated mCherry and substituted cleavage site |
| 2-ME | 2-mercaptoethanol |
| Ni-NTA | Ni2+-nitrilotriacetic acid |
| p12 | Small CASP3 subunit |
| p17 | Large CASP3 subunit |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| TEV | Tobacco etch virus |
| TS | TEV protease cleavage site |
| WT | Wild-type |
References
- Raran-Kurussi, S.; Waugh, D.S. A dual protease approach for expression and affinity purification of recombinant proteins. Anal. Biochem. 2016, 504, 30–37. [Google Scholar] [CrossRef]
- Waugh, D.S. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr. Purif. 2011, 80, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.A.; Leong, L.E.-C.; Ng, P.W.P.; Tan, S.H.; Waller, S.; Murphy, D.; Porter, A.G. Efficient and Rapid Affinity Purification of Proteins Using Recombinant Fusion Proteases. Nat. Biotechnol. 1994, 12, 601–605. [Google Scholar] [CrossRef]
- Fink, T.; Lonzarić, J.; Praznik, A.; Plaper, T.; Merljak, E.; Leben, K.; Jerala, N.; Lebar, T.; Strmšek, Ž.; Lapenta, F.; Benčina, M.; et al. Design of fast proteolysis-based signaling and logic circuits in mammalian cells. Nat. Chem. Biol. 2019, 15, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Stein, V.; Alexandrov, K. Protease-based synthetic sensing and signal amplification. Proc. Natl. Acad. Sci. USA 2014, 111, 15934–15939. [Google Scholar] [CrossRef]
- Fernandez-Rodriguez, J.; Voigt, C.A. Post-translational control of genetic circuits using Potyvirus proteases. Nucleic Acids Res. 2016, 44, 6493–6502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.J.; Chong, L.S.; Kim, M.S.; Elowitz, M.B. Programmable protein circuits in living cells. Science 2018, 361, 1252–1258. [Google Scholar] [CrossRef]
- Gray, D.C.; Mahrus, S.; Wells, J.A. Activation of Specific Apoptotic Caspases with an Engineered Small-Molecule-Activated Protease. Cell 2010, 142, 637–646. [Google Scholar] [CrossRef]
- Smart, A.D.; Pache, R.A.; Thomsen, N.D.; Kortemme, T.; Davis, G.W.; Wells, J.A. Engineering a light-activated caspase-3 for precise ablation of neurons in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E8174–E8183. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.; Chen, X.; Pham, E.; Wong, S.; Truong, K. Engineering a Photoactivated Caspase-7 for Rapid Induction of Apoptosis. ACS Synth. Biol. 2012, 1, 75–82. [Google Scholar] [CrossRef]
- Kolar, K.; Knobloch, C.; Stork, H.; Žnidarič, M.; Weber, W. OptoBase: A Web Platform for Molecular Optogenetics. ACS Synth. Biol. 2018, 7, 1825–1828. [Google Scholar] [CrossRef]
- Schwarz, K.A.; Daringer, N.M.; Dolberg, T.B.; Leonard, J.N. Rewiring human cellular input–output using modular extracellular sensors. Nat. Chem. Biol. 2017, 13, 202–209. [Google Scholar] [CrossRef]
- Antalis, T.M.; Shea-Donohue, T.; Vogel, S.N.; Sears, C.; Fasano, A. Mechanisms of Disease: Protease functions in intestinal mucosal pathobiology. Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 393–402. [Google Scholar] [CrossRef]
- Turk, B.; Turk, D.; Turk, V. Protease signalling: the cutting edge. EMBO J. 2012, 31, 1630–1643. [Google Scholar] [CrossRef] [Green Version]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019. [Google Scholar] [CrossRef]
- Vocero-Akbani, A.M.; Heyden, N.V.; Lissy, N.A.; Ratner, L.; Dowdy, S.F. Killing HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein. Nat. Med. 1999, 5, 29–33. [Google Scholar] [CrossRef]
- Lavrik, I.N.; Golks, A.; Krammer, P.H. Caspases: pharmacological manipulation of cell death. J. Clin. Invest. 2005, 115, 2665–2672. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Bayly, C.I.; Gareau, Y.; Houtzager, V.M.; Kargman, S.; Keen, S.L.C.; Rowland, K.; Seiden, I.M.; Thornberry, N.A.; Nicholson, D.W. Maintenance of caspase-3 proenzyme dormancy by an intrinsic “safety catch” regulatory tripeptide. Proc. Natl. Acad. Sci. USA 2001, 98, 6132–6137. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Lee, Y.; Jeong, Y.-J.; Park, K.; Jang, M.; Park, S.; Bae, K.-H.; Kim, M.; Chung, S.J. Large-scale preparation of active caspase-3 in E. coli by designing its thrombin-activatable precursors. BMC Biotechnol. 2008, 8, 92. [Google Scholar] [CrossRef]
- Ponder, K.G.; Boise, L.H. The prodomain of caspase-3 regulates its own removal and caspase activation. Cell Death Discov. 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Chumanevich, A.A.; Agniswamy, J.; Fang, B.; Harrison, R.W.; Weber, I.T. Structural basis for executioner caspase recognition of P5 position in substrates. Apoptosis 2008, 13, 1291–1302. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System; Version 1.3; Schrödinger, LLC: New York, NY, USA, 2010.
- Wagner, H.J.; Engesser, R.; Ermes, K.; Geraths, C.; Timmer, J.; Weber, W. Synthetic biology-inspired design of signal-amplifying materials systems. Mater. Today 2019, 22, 25–34. [Google Scholar] [CrossRef]
- Wagner, H.J.; Kemmer, S.; Engesser, R.; Timmer, J.; Weber, W. Biofunctionalized Materials Featuring Feedforward and Feedback Circuits Exemplified by the Detection of Botulinum Toxin A. Adv. Sci. 2019, 6, 1801320. [Google Scholar] [CrossRef]
- Beyer, H.M.; Engesser, R.; Hörner, M.; Koschmieder, J.; Beyer, P.; Timmer, J.; Zurbriggen, M.D.; Weber, W. Synthetic Biology Makes Polymer Materials Count. Adv. Mater. 2018, 30, 1800472. [Google Scholar] [CrossRef] [PubMed]
- Feeney, B.; Pop, C.; Swartz, P.; Mattos, C.; Clark, A.C. Role of Loop Bundle Hydrogen Bonds in the Maturation and Activity of (Pro)caspase-3. Biochemistry 2006, 45, 13249–13263. [Google Scholar] [CrossRef] [PubMed]
- Casciola-Rosen, L.; Garcia-Calvo, M.; Bull, H.G.; Becker, J.W.; Hines, T.; Thornberry, N.A.; Rosen, A. Mouse and Human Granzyme B Have Distinct Tetrapeptide Specificities and Abilities to Recruit the Bid Pathway. J. Biol. Chem. 2007, 282, 4545–4552. [Google Scholar] [CrossRef] [PubMed]
- Stein, V.; Nabi, M.; Alexandrov, K. Ultrasensitive Scaffold-Dependent Protease Sensors with Large Dynamic Range. ACS Synth. Biol. 2017, 6, 1337–1342. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Wagner, H.J.; Engesser, R.; Ermes, K.; Geraths, C.; Timmer, J.; Weber, W. Characterization of the synthetic biology-inspired implementation of a materials-based positive feedback loop. Data Brief 2018, 19, 665–677. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, H.J.; Weber, W. Design of a Human Rhinovirus-14 3C Protease-Inducible Caspase-3. Molecules 2019, 24, 1945. https://doi.org/10.3390/molecules24101945
Wagner HJ, Weber W. Design of a Human Rhinovirus-14 3C Protease-Inducible Caspase-3. Molecules. 2019; 24(10):1945. https://doi.org/10.3390/molecules24101945
Chicago/Turabian StyleWagner, Hanna J., and Wilfried Weber. 2019. "Design of a Human Rhinovirus-14 3C Protease-Inducible Caspase-3" Molecules 24, no. 10: 1945. https://doi.org/10.3390/molecules24101945
APA StyleWagner, H. J., & Weber, W. (2019). Design of a Human Rhinovirus-14 3C Protease-Inducible Caspase-3. Molecules, 24(10), 1945. https://doi.org/10.3390/molecules24101945