3.1. Chemistry
Reagents and solvents were obtained from commercial suppliers and used as received. 1H-NMR spectra were obtained on an NMR spectrometer (Mercury, Varian, San Diego, CA, USA; 400 MHz). Electrospray ionization (ESI) mass spectra and high-resolution mass spectroscopy (HRMS) were performed with a liquid chromatograph/mass selective detector time-of-flight mass spectrometer (LC/MSD TOF, Agilent Technologies, Santa Clara, CA, USA). silica gel column chromatography was performed with silica gel 60G (Qingdao Haiyang Chemical, Qingdao, China). Purity was determined using HPLC, LC/MS and NMR spectroscopy. All of the synthesized compounds have the purity over than 95%.
Several commercial available compounds were purchased from Beijing innochem Co. Ltd. (Beijing, China). They are DX-01-01, DX-01-08, DX-01-09, DX-01-10, DX-01-11, DX-01-12, DX-01-18, DX-01-19, DX-01-20, DX-01-21, DX-01-22, DX-03-01.
3.1.1. Preparation of (R/S) 3-(5-fluoro-2-((hydroxyamino)methyl)-1H-indol-3-yl)pyrrolidine-2,5-dione (DX-02-05)
Preparation of (R/S) Ethyl 3-(2,5-dioxopyrrolidin-3-yl)-5-fluoro-1H-indole-2-carboxylate (DX-02-03)
Ethyl 5-fluoro-1H-indole-2-carboxylate (2 g, 9.7 mmol) and 1H-pyrrole-2,5-dione (1.40 g, 14.4 mmol) were added to dry 1,2-dichloroethane (50 mL), and 46.5% BF3-Et2O (3.54 g, 11.6 mmol) was added to reaction mixture at 20 °C. After the reaction mixture was stirred at 90 °C, until the starting material disappeared in thin layer chromatography (TLC), the reaction mixture was distilled in vacuo, and dichloromethane (50 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaHCO3 and NaCl (20 mL) respectively, and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 1.62 g, yield 55%. 1H-NMR (400 MHz, DMSO-d6): δ = 12.00 (s, 1H, NH-indolyl), 11.31 (s, 1H, NH), 7.49–7.45 (m, 2H, H-indolyl), 7.18 (td, J = 9.2, 2.6 Hz, 1H, H-indolyl), 4.82 (dd, J = 9.6, 6.6 Hz, 1H, CH), 4.33–4.27 (m, 2H, CH2), 3.08 (dd, J = 17.7, 9.6 Hz, 1H, CH’H’’), 2.70 (dd, J = 17.7, 6.6 Hz, 1H, CH’H’’), 1.30 (t, J = 7.1 Hz, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ =182.17, 180.15, 162.66, 160.66, 134.24, 129.01, 126.54, 119.11, 115.66, 114.82, 104.78, 62.19, 39.68, 39.37, 14.63. HRMS (ESI): m/z [M + H]+ calculated for C15H14O4N2F: 305.09321; found: 305.09290.
Preparation of (R/S) 3-(5-fluoro-2-(hydroxymethyl)-1H-indol-3-yl) pyrrolidine-2,5-dione (1)
Ethyl 3-(2,5-dioxopyrrolidin-3-yl)-5-fluoro-1H-indole-2-carboxylate (DX-02-03, 1.3 g, 4.3 mmol) were added to dry CH3OH (20 mL), LiAlH4 (0.13 g, 4.3 mmol) was added to reaction mixture gradually at 20 °C. The reaction mixture was stirred at 20 °C until the starting material disappeared in TLC. The reaction mixture was distilled in vacuo, and ethyl acetate (50 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaCl (20 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 0.97 g, yield 86%. 1H-NMR (400 MHz, CD3OD): δ = 7.30 (dd, J = 8.8, 4.4 Hz, 1H, H-phenyl), 6.93 (dd, J = 9.9, 2.4 Hz, 1H, H-phenyl), 6.89–6.83 (m, 1H, H-phenyl), 4.72 (d, J = 1.3 Hz, 2H, CH2), 4.46 (dd, J = 9.7, 5.6 Hz, 1H, CH), 3.20 (dd, J = 18.4, 9.7 Hz, 1H, CH’H’’), 2.81 (dd, J = 18.4, 5.6 Hz, 1H, CH’H’’). 13C-NMR (101 MHz, CD3OD): δ = 182.67, 180.16, 160.22, 157.85, 139.57, 133.85, 127.82, 113.46, 110.70, 103.45, 56.67, 39.93, 39.00. HRMS (ESI): m/z [M + H]+ calculated for C13H12N2O3F: 263.08265; found: 263.08160.
Preparation of (R/S) 3-(2,5-dioxopyrrolidin-3-yl)-5-fluoro-1H-indole-2-carbaldehyde (2)
3-(5-Fluoro-2-(hydroxymethyl)-1H-indol-3-yl)pyrrolidine-2,5-dione (1, 0.8 g, 3.1 mmol) was added to dry dichloridemethane (20 mL), MnO2 (0.36 g, 4.1 mmol) was added to reaction mixture gradually at 20 °C. The reaction mixture was stirred at 20 °C until the starting material disappeared in TLC. The reaction mixture was filtered, the solvent was removed under vacuum, and the product was purified as a white solid by silica gel column chromatography, 0.73g, yield 91%. 1H-NMR (400 MHz, CD3OD): δ = 10.00 (s, 1H, CHO), 7.50 (dd, J = 9.1, 4.3 Hz, 1H, H-phenyl), 7.27 (dd, J = 9.5, 2.3 Hz, 1H, H-phenyl), 7.19 (td, J = 9.1, 2.4 Hz, 1H, H-phenyl), 4.91 (dd, J = 9.7, 6.0 Hz, 1H, CH), 3.29–3.21 (m, 1H, CH’H’’), 2.84 (dd, J = 18.1, 6.0 Hz, 1H, CH’H’’). 13C-NMR (101 MHz, CD3OD): δ = 183.66, 181.23, 179.66, 160.59, 158.30, 135.75, 135.36, 127.61, 117.26, 115.66, 105.59, 39.77, 39.35. HRMS (ESI): m/z [M + H]+ calculated for C13H10N2O3F: 261.06700; found: 261.06638.
Preparation of (R/S) (E)-3-(2,5-dioxopyrrolidin-3-yl)-5-fluoro-1H-indole-2-carbaldehyde oxime (DX-02-04)
3-(2,5-Dioxopyrrolidin-3-yl)-5-fluoro-1H-indole-2-carbaldehyde (0.5 g, 1.92 mmol) was added to dry CH3OH (30 mL), then N,N-Diisopropylethylamine (0.5 g, 3.84 mmol) and hydroxylamine hydro -chloride (0.13 g, 1.92 mmol) were added respectively at 20 °C. The reaction mixture was stirred at 60 °C until the starting material disappeared in TLC. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 0.35 g, yield 66%. 1H-NMR (400 MHz, CD3OD): δ = 8.22 (s, 1H, =CH), 7.32 (dd, J = 8.8, 4.4 Hz, 1H, H-indolyl), 7.06–6.86 (m, 2H, H-indolyl), 4.63 (dd, J = 9.7, 5.9 Hz, 1H, CH), 3.19 (dd, J = 18.3, 9.7 Hz, 1H, CH’H’’), 2.81 (dd, J = 18.7, 6.2 Hz, 1H, CH’H’’). 13C-NMR (101 MHz, CD3OD): δ =181.92, 179.89, 160.31, 157.98, 141.03, 136.51, 134.81, 132.30, 113.78, 112.58, 103.95, 39.83, 38.66. HRMS (ESI): m/z [M + H]+ calculated for C13H11N3O3F: 276.07790; found: 276.07718.
Preparation of (R/S) 3-(5-fluoro-2-((hydroxyamino)methyl)-1H-indol-3-yl) pyrrolidine-2,5-dione (DX-02-05)
3-(2,5-Dioxopyrrolidin-3-yl)-5-fluoro-1H-indole-2-carbaldehyde oxime (DX-02-04, 0.3 g, 1.1 mmol) was added to CH3OH (20 mL), then NaBH3CN (0.07 g, 1.1 mmol) was added at 20 °C. 12N HCl was added to reaction mixture continuously to keep acidic. The reaction mixture was stirred at 20 °C until the starting material disappeared in TLC. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 0.13 g, yield 43%. 1H-NMR (400 MHz, CD3OD): δ = 7.30 (dd, J = 8.8, 4.5 Hz, 1H, H-indolyl), 6.93–6.88 (m, 1H, H-indolyl), 6.88–6.82 (m, 1H, H-indolyl), 4.50 (dd, J = 9.7, 5.5 Hz, 1H, CH), 4.12 (d, J = 3.6 Hz, 2H, CH2NH), 3.21 (dd, J = 18.5, 9.7 Hz, 1H, CH’H’’), 2.85 (dd, J = 18.5, 5.5 Hz, 1H, CH’H’’). 13C-NMR (101 MHz, CD3OD): δ = 160.27, 157.92, 135.20, 130.85, 130.02, 113.00, 110.85, 110.54, 104.55, 104.31, 50.48, 38.34, 30.37. HRMS (ESI): m/z [M + H]+ calculated for C13H13N3O3F: 278.09355; found: 278.09291.
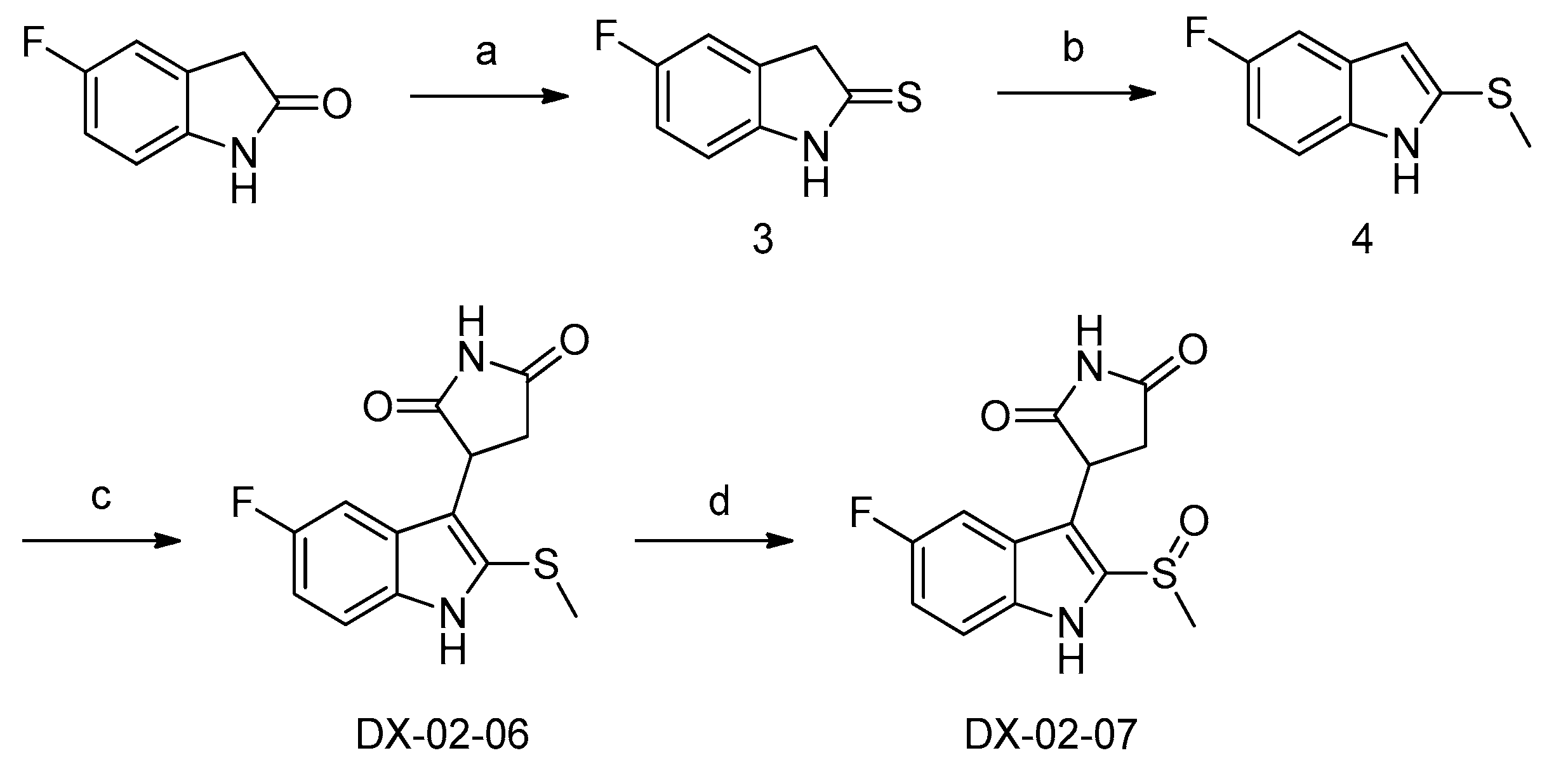
3.1.2. Preparation of (R/S) 3-(5-fluoro-2-(methylsulfinyl)-1H-indol-3-yl)pyrrolidine-2,5-dione (DX-02-07)
Preparation of 5-fluoroindoline-2-thione (3)
5-fluoroindolin-2-one (2 g, 13 mmol) and P2S5 (2.92 g, 13 mmol) were added to dry tetrahydrofuran (30 mL). After the reaction mixture was stirred at room temperature for 1 h. Until the starting material disappeared in thin layer chromatography (TLC), the NaHCO3 (3.43 g, 39 mmol) was added gradually. After stirring at room temperature for 1 h, the reaction mixture was distilled in vacuo, dichloromethane (50 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaCl (20 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was obtained as yellow crystals by re-crystallization using ethanol, 1.62 g, yield 75%. 1H-NMR (400 MHz, CDCl3): δ = 10.14 (s, 1H, NH), 7.05–6.95 (m, 1H, H-phenyl), 6.94–6.89 (m, 1H, H-phenyl), 4.08 (s, 1H, CH2). 13C-NMR (101 MHz, CDCl3)): δ = 203.39, 159.10, 140.24, 132.15, 114.87, 112.47, 110.47, 49.15. HRMS (ESI): m/z [M + H]+ calculated for C8H7NFS: 168.02777; found: 168.02773.
Preparation of 5-fluoro-2-(methylthio)-1H-indole (4)
5-fluoroindoline-2-thione (3, 1 g, 6 mmol), CH3I (0.99 g, 7 mmol) and K2CO3 (0.97 g, 7 mmol) were added to acetone (30 mL), and the reaction mixture was stirred at room temperature until the starting material disappeared in TLC. The reaction mixture was distilled in vacuo, ethyl acetate (50 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaCl (20 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 0.90 g, yield 83%. 1H-NMR (400 MHz, CDCl3): δ = 8.04 (s, 1H, NH), 7.23–7.13 (m, 2H, H-indolyl), 6.91 (s, 1H, H-indolyl), 6.49 (s, 1H, H-indolyl), 2.52 (s, 3H, CH3). 13C-NMR (101 MHz, CDCl3): δ = 159.18, 156.85, 133.57, 129.02, 110.94, 110.47, 105.31, 104.73, 18.86. HRMS (ESI): m/z [M + H]+ calculated for C9H9NFS: 182.04342; found: 182.04425.
Preparation of (R/S) 3-(5-fluoro-2-(methylthio)-1H-indol-3-yl)pyrrolidine-2,5-dione (DX-02-06)
5-Fluoro-2-(methylthio)-1H-indole (4, 0.5 g, 2.7 mmol) and 1H-pyrrole-2,5-dione (0.54 g, 5.4 mmol) were added to dry CH3CO2H (30 mL), and the reaction mixture was stirred at 120 °C until the starting material disappeared in TLC. The reaction mixture was distilled in vacuo, ethyl acetate (100 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaCl (30 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 0.47 g, yield 61%. 1H-NMR (400MHz, CD3OD): δ = 7.28 (dd, J = 8.9, 4.5 Hz, 1H, H-phenyl), 7.00 (dd, J = 9.7, 2.4 Hz, 1H, H-phenyl), 6.91 (td, J = 9.2, 2.5 Hz, 1H, H-phenyl), 4.56 (dd, J = 9.8, 5.6 Hz, 1H, CH), 3.22 (dd, J = 18.3, 9.8 Hz, 1H, CH’H’’), 2.77 (dd, J = 18.3, 5.6 Hz, 1H, CH’H’’), 2.41 (s, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ = 182.38, 180.11, 160.26, 135.05, 132.13, 127.67, 116.34, 113.18, 111.70, 103.25, 40.35, 39.10, 19.59. HRMS (ESI): m/z [M + H]+ calculated for C13H12O2N2FS: 279.05980; found: 279.05930.
Preparation of (R/S) 3-(5-fluoro-2-(methylsulfinyl)-1H-indol-3-yl)pyrrolidine-2,5-dione (DX-02-07)
3-(5-Fluoro-2-(methylthio)-1H-indol-3-yl)pyrrolidine-2,5-dione (DX-02-06, 0.2 g, 0.7 mmol) was added to dry dichloridemethane (20 mL), and 3-Chloroperoxybenzoic acid (0.15 g, 0.9 mmol) was added gradually at 0 °C. The reaction mixture was stirred at 0 °C until the starting material disappeared in TLC, and Na2S2O3 (0.11 g, 0.7 mmol) was added to the mixture. After reaction mixture was stirred for 20 min, the reaction mixture was washed twice with saturated aqueous NaCl (20 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 0.074 g, yield 35%. 1H-NMR (400MHz, DMSO-d6): δ = 12.28 (s, 1H, NH), 11.51 (s, 1H, NH), 7.51–7.48 (m, 1H, H-phenyl), 7.19–7.13 (m, 2H, H-phenyl), 4.61 (ddd, J = 26.8, 9.7, 5.8 Hz, 1H, CH), 3.18 (ddd, J = 18.0, 9.7, 2.3 Hz, 1H, CH’H’’), 2.98 (d, J = 16.7 Hz, 3H, CH3), 2.76 (ddd, J = 41.8, 18.1, 5.8 Hz, 1H, CH’H’’). 13C-NMR (101 MHz, DMSO-d6): δ = 179.74, 177.94, 158.72, 138.29, 134.14, 125.83, 114.50, 113.93, 113.53, 104.46, 41.67, 38.62, 38.11. HRMS (ESI): m/z [M + H]+ calculated for C13H12O3N2FS: 295.05472; found: 295.05508.
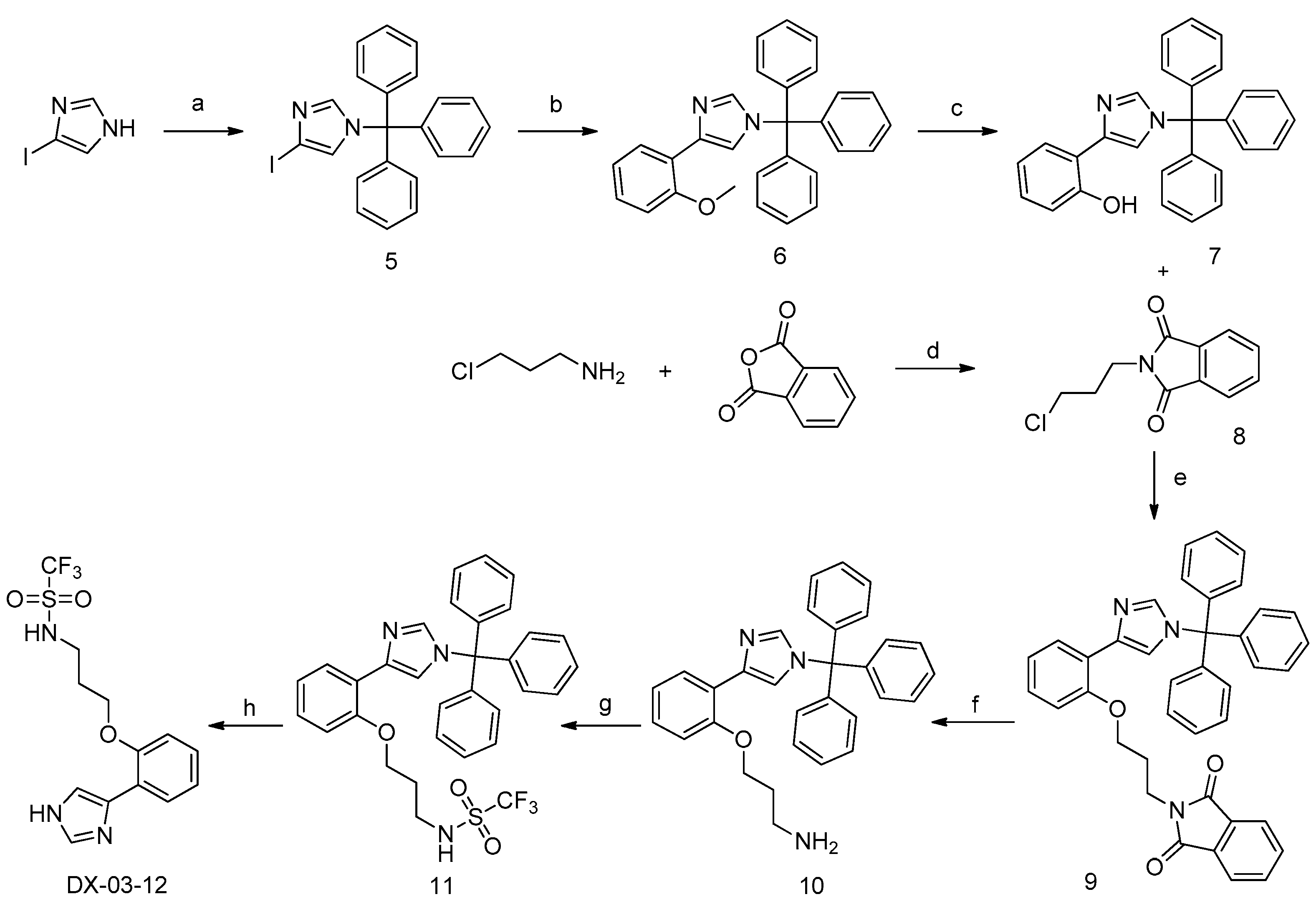
3.1.3. Preparation of N-(3-(2-(1H-imidazol-5-yl)phenoxy)propyl)-1,1,1-trifluoromethane sulfonamide (DX-03-12)
Preparation of 4-iodo-1-trityl-1H-imidazole (5)
4-Iodo-1H-imidazole (5 g, 25.8 mmol) and N,N-diisopropylethylamine (6.65 g, 51.5 mmol) were added to dry dimethylformamide (30 mL), and (chloromethanetriyl)tribenzene (7.89 g, 28.4 mmol) was added to reaction mixture gradually at 20 °C. After the reaction mixture was stirred at 20 °C until the starting material disappeared in thin layer chromatography (TLC), the reaction mixture was distilled in vacuo, ethyl acetate (200 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaCl (40 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 8.77 g, yield 78%. 1H-NMR (400 MHz, CDCl3): δ = 7.38–7.33 (m, 10H, H-phenyl, H-imidazolyl), 7.13–7.09 (m, 6H, H-phenyl), 6.92 (d, J = 1.4 Hz, 1H, H-imidazolyl). 13C-NMR (101 MHz, CDCl3): δ = 141.83, 140.56, 129.72 (6C), 128.29 (3C), 128.18 (6C), 127.91, 126.89 (3C), 75.84. HRMS (ESI): m/z [M + H]+ calculated for C22H18N2I: 437.05092; found: 437.04934.
Preparation of 4-(2-methoxyphenyl)-1-trityl-1H-imidazole (6)
4-Iodo-1-trityl-1H-imidazole (5, 2 g, 4.58 mmol), (5-chloro-2-methoxyphenyl)boronic acid (0.852 g, 4.58 mmol), K3PO4 (2.9 g, 13.7 mmol), Pd(PPh3)4 (0.74 g, 0.64 mmol) were added to dry dimethylformamide (30 mL). The reaction mixture was stirred at 100 °C under inert atmosphere, until the starting material disappeared in TLC. The reaction mixture was distilled in vacuo, and ethyl acetate (100 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaCl (20 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 1.2 g, yield 63%. 1H-NMR (400 MHz, CDCl3): δ = 7.51–7.32 (m, 12H, H-phenyl, H-imidazolyl), 7.15–7.04 (m, 8H, H-phenyl, H-imidazolyl), 6.94 (d, J = 8.1 Hz, 1H, H-phenyl), 3.99 (s, 3H, CH3). HRMS (ESI): m/z [M + H]+ calculated for C29H25N2O: 417.19614; found: 417.19647.
Preparation of 2-(1-trityl-1H-imidazol-4-yl)phenol (7)
4-(2-Methoxyphenyl)-1-trityl-1H-imidazole (6, 1.0 g, 2.4 mmol) was added to dry dichloridemethane (20 mL), BBr3 (0.71 g, 2.9 mmol) was added to reaction mixture gradually at −70 °C. The reaction mixture was stirred under inert atmosphere until the starting material disappeared in TLC. The reaction mixture was diluted with dichloridemethane (50 mL), and was washed twice with saturated aqueous NaCl (20 mL), then the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 0.63 g, yield 65%. 1H-NMR (400 MHz, DMSO-d6): δ = 11.19 (s, 1H, OH), 7.49–7.36 (m, 2H, H-phenyl), 7.33–7.10 (m, 16H, H-phenyl, H-imidazolyl), 6.84–6.70 (m, 2H, H-phenyl), 6.45 (s, 1H, H-phenyl). 13C-NMR (101 MHz, DMSO-d6): δ = 147.24, 141.47, 137.70, 136.55, 128.71 (6C), 127.80 (6C), 127.56, 127.24 (3C), 126.99 (3C), 126.10, 118.39, 117.87, 115.68, 74.61. HRMS (ESI): m/z [M + H]+ calculated for C28H23N2O: 403.18049; found: 403.18134.
Preparation of 2-(3-chloropropyl)isoindoline-1,3-dione (8)
3-Chloropropan-1-amine (3.77 g, 40.5 mmol) and isobenzofuran-1,3-dione (2 g, 13.5 mmol) were mixed without solvent, and the reaction mixture was stirred at 140 °C (molten condition), until the starting material disappeared in TLC. The reaction mixture was distilled in vacuo, ethyl acetate (100 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaCl (20 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 1.66 g, yield 55%. 1H-NMR (400 MHz, DMSO-d6): δ = 7.85–7.78 (m, 4H, H-phenyl), 3.73–3.62 (m, 4H, CH2, CH2), 2.08–2.01(m, 2H, CH2). 13C-NMR (101 MHz, DMSO-d6): δ = 167.38 (2C), 133.75 (2C), 131.19 (2C), 122.43 (2C), 42.31, 34.60, 30.39. HRMS (ESI): m/z [M + H]+ calculated for C11H11ClNO2: 224.04728; found: 224.04683.
Preparation of 2-(3-(2-(1-trityl-1H-imidazol-5-yl)phenoxy)propyl)isoindoline-1,3-dione (9)
2-(1-Trityl-1H-imidazol-4-yl)phenol (7, 0.5 g, 1.2 mmol) and KOH (0.14 g, 2.5 mmol) were added to dry dimethylformamide (20 mL), after the reaction mixture was stirred at 20 °C for 15 min, 2-(3-chloropropyl)isoindoline-1,3-dione (8, 0.294 g, 1.32 mmol) was added to reaction mixture at 20 °C. After the reaction mixture was stirred at 60 °C until the starting material disappeared in thin layer chromatography (TLC), the reaction mixture was distilled in vacuo, ethyl acetate (100 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaCl (20 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product was purified as a white solid by silica gel column chromatography, 0.5 g, yield 71%. 1H-NMR (400 MHz, DMSO-d6): δ = 8.08 (dd, J = 7.7, 1.6 Hz, 1H, H-phenyl), 7.88–7.81 (m, 4H, H-phenyl, H-imidazolyl), 7.44–7.35 (m, 10H, H-phenyl), 7.17–7.13 (m, 7H, H-phenyl, H-imidazolyl), 7.12–7.08 (m, 1H, H-phenyl), 6.97 (t, J = 7.5 Hz, 1H, H-phenyl), 6.92 (d, J = 8.2 Hz, 1H, H-phenyl), 3.93 (t, J = 6.4 Hz, 2H, CH2), 3.43 (t, J = 6.5 Hz, 2H, CH2), 1.82–1.69 (m, 2H, CH2). 13C-NMR (101 MHz, DMSO-d6): δ = 167.25, 154.09, 141.77, 137.17, 134.72, 133.82, 131.11, 128.71 (6C), 128.65 (3C), 127.69, 127.66 (6C), 127.46, 127.36 (3C), 126.71, 125.92, 122.47, 121.86, 121.01, 120.60, 119.93, 111.33, 74.14, 64.42, 33.61, 27.54. HRMS (ESI): m/z [M + H]+ calculated for C39H32N3O3: 590.24382; found: 590.24541.
Preparation of 3-(2-(1-trityl-1H-imidazol-4-yl)phenoxy)propan-1-amine (10)
2-(3-(2-(1-Trityl-1H-imidazol-4-yl)phenoxy)propyl)isoindoline-1,3-dione (9, 0.4 g, 0.68 mmol) and 35% hydrazine hydrate (0.12 g, 1.35 mmol) were added to CH3OH, and the reaction mixture was stirred at 50 °C under inert atmosphere. After the starting material disappeared in thin layer chromatography (TLC), the reaction mixture was distilled in vacuo, ethyl acetate (100 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaHCO3 and NaCl (20 mL) respectively, and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the unpurified product was added to the next step directly.
Preparation of 1,1,1-trifluoro-N-(3-(2-(1-trityl-1H-imidazol-4-yl)phenoxy)propyl) Methane Sulfonamide (11)
3-(2-(1-Trityl-1H-imidazol-4-yl)phenoxy)propan-1-amine (crude 10, 0.3 g, 0.65 mmol) and N,N-diisopropylethylamine (0.168 g, 1.3 mmol) were added to dry dichloridemethane (20 mL), and trifluoromethanesulfonylchloride (0.12 g, 0.71 mmol) was added to reaction mixture gradually at 20 °C. After the starting material disappeared in thin layer chromatography (TLC), the reaction mixture was distilled in vacuo, the reaction mixture was diluted with dichloridemethane (50 mL), and the organic extract was washed twice with saturated aqueous NaCl (20 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the product 11 was purified as a white solid by silica gel column chromatography, 0.257 g, yield 67%. 1H-NMR (400 MHz, CDCl3): δ = 7.80 (s, 1H, NH), 7.43–7.32 (m, 10H, H-phenyl, H-imidazolyl), 7.23–7.16 (m, 8H, H-phenyl, H-imidazol), 7.07 (d, J = 1.5 Hz, 1H, H-phenyl), 6.97–6.86 (m, 2H, H-phenyl), 4.16 (t, J = 5.2 Hz, 2H, CH2), 3.57–3.46 (m, 2H, CH2), 2.19–2.06 (m, 2H, CH2). 13C-NMR (101 MHz, CDCl3): δ= 154.95, 142.08, 139.92, 138.22, 129.77 (6C), 128.56 (3C), 128.36, 128.15 (3C), 128.09 (6C), 121.68, 120.87, 119.04, 118.47, 111.86, 75.75, 67.83, 43.24, 29.80. HRMS (ESI): m/z [M + H]+ calculated for C32H29N3O3F3S: 592.18762; found: 592.18616.
Preparation of N-(3-(2-(1H-imidazol-5-yl)phenoxy)propyl)-1,1,1-trifluoromethane Sulfonamide (DX-03-12)
1,1,1-Trifluoro-N-(3-(2-(1-trityl-1H-imidazol-4-yl)phenoxy)propyl)methanesulfonamide (11, 0.2 g, 0.34 mmol) and AcOH (0.5 mL) were added to CH3OH (10 mL), and the reaction mixture was stirred at 20 °C. After the starting material disappeared in thin layer chromatography (TLC), the reaction mixture was distilled in vacuo, the product was purified as a white solid by silica gel column chromatography, 0.101 g, yield 86%. 1H-NMR (400 MHz, DMSO-d6): δ =7.99 (d, J = 7.4 Hz, 1H, H-phenyl), 7.72 (s, 1H, H-imidazolyl), 7.49 (s, 1H, H-imidazolyl), 7.18 (t, J = 7.8 Hz, 1H, H-phenyl), 7.03 (d, J = 8.1 Hz, 1H, H-phenyl), 6.98 (t, J = 7.5 Hz, 1H, H-phenyl), 4.15 (t, J = 6.0 Hz, 2H, CH2), 3.40 (t, J = 6.8 Hz, 2H, CH2), 2.06–2.09 (m, 2H, CH2). 13C-NMR (101 MHz, DMSO-d6): δ = 154.86, 135.47, 134.49, 127.46, 127.25, 124.99, 122.69, 120.97, 118.57, 112.34, 65.09, 41.39, 30.14. HRMS (ESI): m/z [M + H]+ calculated for C13H15O3N3F3S: 350.07807; found: 350.07782.
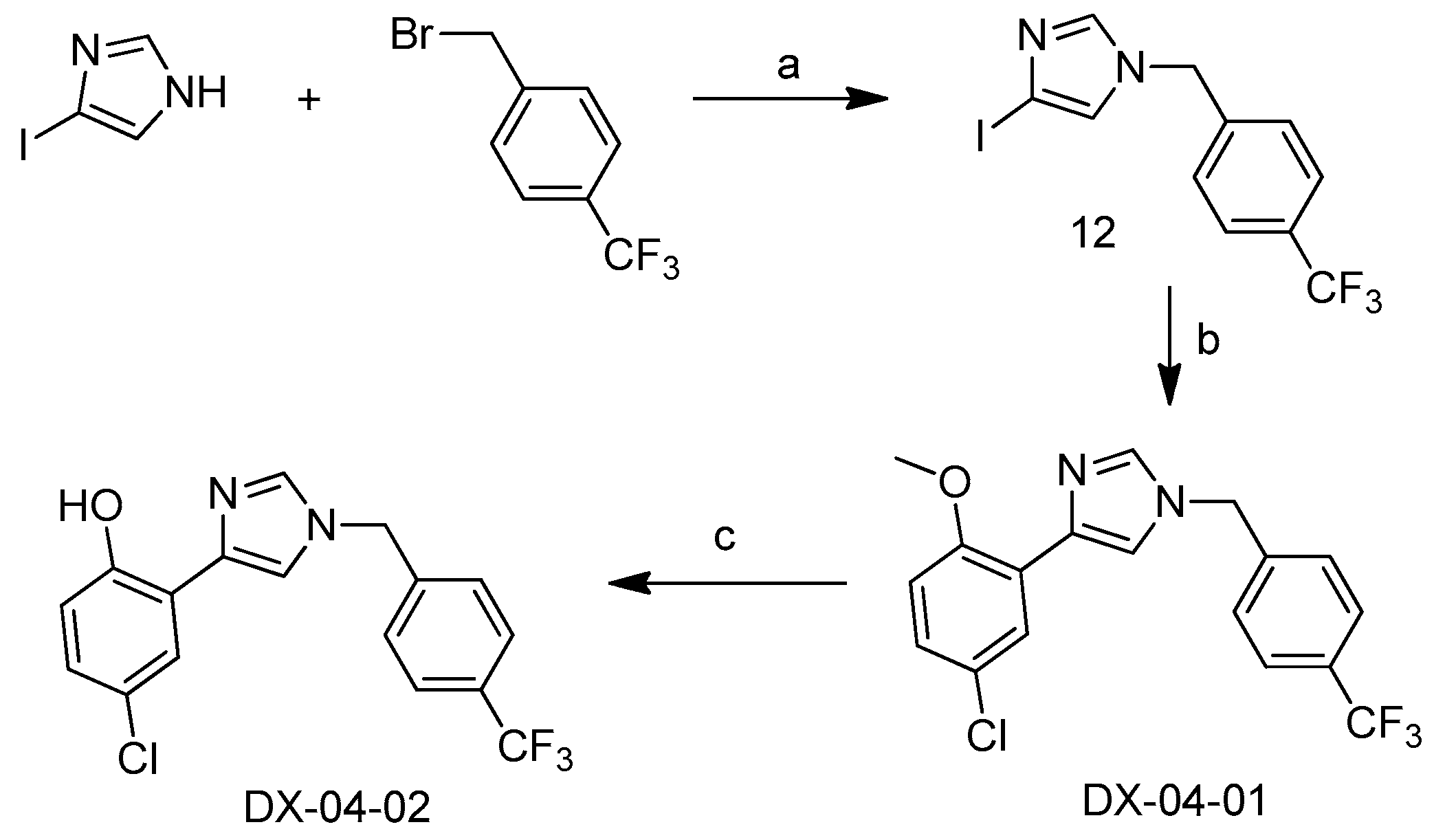
3.1.4. Preparation of 4-chloro-2-(1-(4-(trifluoromethyl)benzyl)-1H-imidazol-4-yl) Phenol (DX-04-02)
Preparation of 4-iodo-1-(4-(trifluoromethyl)benzyl)-1H-imidazole (12)
4-Iodo-1H-imidazole (1 g, 5.2 mmol) and N,N-diisopropylethylamine (1.34 g, 10.4 mmol) were added to dry dimethylformamide (20 mL), and 1-(bromomethyl)-4-(trifluoromethyl)benzene (1.48 g, 6.2 mmol) was added to reaction mixture at 20 °C. After the reaction mixture was stirred at 20 °C until the starting material disappeared in thin layer chromatography (TLC), the reaction mixture was distilled in vacuo, dichloromethane (50 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaHCO3 and NaCl (20 mL) respectively, and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, the unpurified product was added to the next step directly.
Preparation of 4-(5-chloro-2-methoxyphenyl)-1-(4-(trifluoromethyl)benzyl)-1H-imidazole (DX-04-01)
4-Iodo-1-(4-(trifluoromethyl)benzyl)-1H-imidazole (crude 12, 0.5 g, 1.4 mmol), (5-chloro-2-methoxyphenyl)boronic acid (0.27 g, 1.4 mmol), K3PO4 (0.9 g, 4.3 mmol), Pd(PPh3)4 (0.23 g, 0.2 mmol) were added to dry dimethylformamide (10 mL). The reaction mixture was stirred at 100 °C under inert atmosphere, until the starting material disappeared in TLC. The reaction mixture was distilled in vacuo, ethyl acetate (50 mL) was added to the mixture. The organic extract was washed twice with saturated aqueous NaCl (20 mL), and the organic extract was dried over Na2SO4. After solvent was removed under vacuum, DX-04-01 was purified as a white solid by silica gel column chromatography, 0.37 g, yield 72%. 1H-NMR (400 MHz, CD3OD): δ = 7.94 (d, J = 2.5 Hz, 1H, H-phenyl), 7.81 (s, 1H, H-imidazolyl), 7.65–7.63 (m, 3H, H-imidazolyl, H-phenyl), 7.40 (d, J = 8.0 Hz, 2H, H-phenyl), 7.15 (dd, J = 8.7, 2.5 Hz, 1H, H-phenyl), 6.96 (d, J = 8.8 Hz, 1H, H-phenyl), 5.34 (s, 2H, CH2), 3.86 (s, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ = 156.09, 142.76, 138.77, 137.13, 131.46, 129.05 (2C), 128.22, 127.44, 126.92 (2C), 124.85, 124.18, 121.78, 113.52, 112.48, 56.17, 51.22. HRMS (ESI): m/z [M + H]+ calculated forC18H15ON2ClF3: 367.08195; found: 367.08374.
Preparation of 4-chloro-2-(1-(4-(trifluoromethyl)benzyl)-1H-imidazol-4-yl)phenol (DX-04-02)
4-(5-Chloro-2-methoxyphenyl)-1-(4-(trifluoromethyl)benzyl)-1H-imidazole (DX-04-01, 0.3 g, 0.82 mmol) was added to dry dichloridemethane (20 mL), BBr3 (0.24 g, 0.98 mmol) was added to reaction mixture gradually at −70 °C. The reaction mixture was stirred under inert atmosphere until the starting material disappeared in TLC. The reaction mixture was diluted with dichloridemethane (50 mL), and was washed twice with saturated aqueous NaCl (20 mL), then the organic extract was dried over Na2SO4. After solvent was removed under vacuum, DX-04-02 was purified as a white solid by silica gel column chromatography, 0.15 g, yield 53%. 1H-NMR (400 MHz, CD3OD): δ = 7.85 (s, 1H, H-imidazolyl), 7.70–7.65 (m, 3H, H-imidazolyl, H-phenyl), 7.63 (d, J = 2.6 Hz, 1H, H-phenyl), 7.45 (d, J = 8.1 Hz, 2H, H-phenyl), 7.02 (dd, J = 8.7, 2.6 Hz, 1H, H-phenyl), 6.80 (d, J = 8.7 Hz, 1H, H-phenyl), 5.37 (s, 2H, CH2). 13C-NMR (101 MHz, CD3OD): δ = 155.21, 142.66, 140.49, 137.67, 129.87, 129.60, 129.12 (2C), 128.42, 126.89 (2C), 126.06, 125.01, 121.01, 118.79, 118.28, 51.17. HRMS (ESI): m/z [M + H]+ calculated forC17H13ON2ClF3: 353.06630; found: 353.06635.
3.1.5. Preparation of 4-methyl-1H-indole-3-carboxylic Acid (DX-01-02)
Methyl 4-methyl-1H-indole-3-carboxylate (200 mg, 1.06 mmol) used hydrolysis reaction (2 N NaOH water solution) to afford DX-01-02 (white solid, 151 mg, yield 82%). 1H-NMR (400 MHz, CD3OD): δ = 8.18 (dd, J = 3.9, 1.9 Hz, 1H, H-indolyl), 7.30 (d, J = 8.0 Hz, 1H, H-indolyl), 7.17 (t, J = 7.7 Hz, 1H, H-indolyl), 7.01 (dd, J = 7.3, 0.7 Hz, 1H, H-indolyl), 2.77 (s, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ = 139.17, 133.89, 126.39, 125.96, 125.68, 120.61, 117.71, 112.19, 111.02, 23.30. HRMS (ESI): m/z [M + H]+ calculated for C10H10NO2: 176.07061; found: 176.07129.
3.1.6. Preparation of 5-methyl-1H-indole-3-carboxylic Acid (DX-01-03)
Methyl 5-methyl-1H-indole-3-carboxylate (200 mg, 1.06 mmol) used hydrolysis reaction (2 N NaOH water solution) to afford DX-01-03 (white solid, 150 mg, yield 81%). 1H-NMR (400 MHz, CD3OD): δ = 8.15 (d, J = 1.8 Hz, 1H, H-indolyl), 8.07 (s, 1H, H-indolyl), 7.39 (d, J = 8.3 Hz, 1H, H-indolyl), 7.15 (dd, J = 8.3, 1.1 Hz, 1H, H-indolyl), 2.46 (s, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ = 137.41, 134.35, 127.80, 127.00, 122.51, 120.09, 117.20, 113.08, 110.78, 21.77. HRMS (ESI): m/z [M + H]+ calculated for C10H10NO2: 176.07061; found: 176.07094.
3.1.7. Preparation of 5-nitro-1H-indole-3-carboxylic Acid (DX-01-04)
Methyl 5-nitro-1H-indole-3-carboxylate (200 mg, 0.91 mmol) used hydrolysis reaction (2 N NaOH water solution) to afford DX-01-04 (white solid, 172 mg, yield 92%). 1H-NMR (400 MHz, CD3OD): δ = 8.99 (d, J = 2.2 Hz, 1H, H-indolyl), 8.14 (s, 1H, H-indolyl), 8.12 (dd, J = 9.0, 2.3 Hz, 1H, H-indolyl), 7.58 (d, J = 9.0 Hz, 1H, H-indolyl). 13C-NMR (101 MHz, CD3OD): δ = 167.81, 144.32, 141.16, 136.51, 127.01, 118.93, 118.80, 113.45, 111.10, 49.00. HRMS (ESI): m/z [M + H]+ calculated for C9H7N2O4: 207.04003; found: 207.03940.
3.1.8. Preparation of 6-nitro-1H-indole-3-carboxylic Acid (DX-01-05)
Methyl 6-nitro-1H-indole-3-carboxylate (200 mg, 0.91 mmol) used hydrolysis reaction (2 N NaOH water solution) to afford DX-01-05 (white solid, 168 mg, yield 90%). 1H-NMR (400 MHz, CD3OD): δ = 8.38 (d, J = 1.5 Hz, 1H, H-indolyl), 8.22 (s, 1H, H-indolyl), 8.18 (d, J = 8.9 Hz, 1H, H-indolyl), 8.05 (dd, J = 8.8, 1.7 Hz, 1H, H-indolyl). 13C-NMR (101 MHz, CD3OD): δ = 167.87, 144.97, 138.31, 136.78, 132.29, 122.19, 117.33, 109.89, 109.69. HRMS (ESI): m/z [M + H]+ calculated for C9H7N2O4: 207.04003; found: 207.03949.
3.1.9. Preparation of 5-methoxy-1H-indole-3-carboxylic Acid (DX-01-06)
Methyl 5-methoxy-1H-indole-3-carboxylate (200 mg, 0.97 mmol) used hydrolysis reaction (2 N NaOH water solution) to afford DX-01-06 (white solid, 140 mg, yield 75%). 1H-NMR (400 MHz, CD3OD): δ = 7.88 (s, 1H, H-indolyl), 7.57 (d, J = 2.4 Hz, 1H, H-indolyl), 7.31 (d, J = 8.8 Hz, 1H, H-indolyl), 6.83 (dd, J = 8.8, 2.5 Hz, 1H, H-indolyl), 3.83 (s, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ = 156.99, 133.58, 128.36, 115.37, 114.23, 113.88, 113.61, 104.51, 103.64, 56.06. HRMS (ESI): m/z [M + H]+ calculated for C10H10NO3: 192.06552; found: 192.06645.
3.1.10. Preparation of 7-bromo-1H-indole-3-carboxylic Acid (DX-01-07)
Methyl 7-bromo-1H-indole-3-carboxylate (200 mg, 0.79 mmol) used hydrolysis reaction (2 N NaOH water solution) to afford DX-01-07 (white solid, 160 mg, yield 85%). 1H-NMR (400 MHz, CD3OD): δ = 8.06 (dd, J = 8.0, 0.9 Hz, 1H, H-indolyl), 7.98 (s, 1H, H-indolyl), 7.36 (dd, J = 7.6, 0.7 Hz, 1H, H-indolyl), 7.08 (t, J = 7.8 Hz, 1H, H-indolyl). 13C-NMR (101 MHz, CD3OD): δ = 168.56, 138.29, 134.03, 129.07, 126.24, 123.59, 121.46, 117.02, 106.02. HRMS (ESI): m/z [M + H]+ calculated for C9H7NO2Br: 239.96547; found: 239.96452.
3.1.11. Preparation of (E) 1H-indole-3-carbaldehyde Oxime (DX-01-13)
1H-indole-3-carbaldehyde (200 mg, 1.38 mmol) following the similar procedure described for the preparation of DX-02-04 afforded DX-01-13 (white solid, 158 mg, yield 72%). 1H-NMR (400 MHz, DMSO-d6): δ = 11.56 (s, 1H, OH), 11.17 (s, 1H, NH), 8.26 (s, 1H, =CH), 8.22 (d, J = 1.3 Hz, 1H, H-indolyl), 7.97 (d, J = 7.9 Hz, 1H, H-indolyl), 7.44 (d, J = 8.1 Hz, 1H, H-indolyl), 7.16 (t, J = 7.5 Hz, 2H, H-indolyl). 13C-NMR (101 MHz, CD3OD): δ = 146.89, 138.66, 132.19, 127.98, 123.30, 121.40, 118.79, 112.59, 107.56. HRMS (ESI): m/z [M + H]+ calculated for C9H9ON2: 161.07094; found: 161.07076.
3.1.12. Preparation of N-((1H-indol-3-yl)methyl)hydroxylamine (DX-01-14)
DX-01-13 (100 mg, 0.62 mmol) following the similar procedure described for the preparation of DX-02-05 afforded DX-01-14 (white solid, 70 mg, yield 69%). 1H-NMR (400 MHz, DMSO-d6): δ = 10.87 (s, 1H, NH-indolyl), 7.58 (d, J = 7.8 Hz, 1H, H-indolyl), 7.34 (d, J = 8.1 Hz, 1H, H-indolyl), 7.29 (br, 1H, NH), 7.24 (s, 1H, H-indolyl), 7.06 (t, J = 8.0 Hz, 1H, H-indolyl), 6.99–6.95 (m, 1H, H-indolyl), 5.66 (br, 1H, OH), 4.04 (s, 2H, CH2). 13C-NMR (101 MHz, CD3OD): δ = 138.02, 128.61, 125.94, 122.58, 120.11, 119.41, 112.28, 110.24, 55.80. HRMS (ESI): m/z [M + H]+ calculated for C9H11ON2: 163.08659; found: 163.08611.
3.1.13. Preparation of N-((5-(o-tolyl)-1H-indol-3-yl)methyl)hydroxylamine (DX-01-15)
5-Bromo-1H-indole-3-carbaldehyde (300 mg, 1.34 mmol) and o-tolylboronic acid (183 mg, 1.34 mmol) following the similar procedure described for the preparation of DX-03-12 and DX-02-05 afforded DX-01-15 (white solid, 145 mg, yield 43% from 5-bromo-1H-indole-3-carbaldehyde). 1H-NMR (400 MHz, CD3OD): δ = 7.57–7.39 (m, 3H, H-phenyl, H-indolyl), 7.26–7.04 (m, 5H, H-phenyl, H-indolyl), 5.13 (s, 2H, CH2), 2.24 (s, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ = 144.63, 137.29, 136.60, 135.38, 131.19, 131.12, 128.56, 127.79, 126.62, 125.69, 124.87, 119.47, 112.33, 107.61, 62.12, 29.31. HRMS (ESI): m/z [M + H]+ calculated for C16H17N2O: 253.13354; found: 253.13471.
3.1.14. Preparation of (E)-1H-indole-2-carbaldehyde Oxime (DX-01-16)
1H-indole-2-carbaldehyde (200 mg, 1.38 mmol) following the similar procedure described for the preparation of DX-02-04 afforded DX-01-16 (white solid, 117 mg, yield 53%). 1H-NMR (400 MHz, DMSO-d6): δ = 11.29 (s, 1H, OH), 11.17 (s, 1H, NH), 8.14 (s, 1H, =CH), 7.52 (d, J = 8.0 Hz, 1H, H-indolyl), 7.37 (d, J = 8.2 Hz, 1H, H-indolyl), 7.12 (t, J = 7.6 Hz, 1H, H-indolyl), 6.98 (t, J = 7.5 Hz, 1H, H-indolyl), 6.56 (s, 1H, H-indolyl). 13C-NMR (101 MHz, CD3OD): δ = 142.67, 138.93, 132.77, 129.48, 124.71, 122.20, 120.89, 112.84, 108.61. HRMS (ESI): m/z [M + H]+ calculated for C9H9ON2: 161.07094; found: 161.07077.
3.1.15. Preparation of N-((1H-indol-2-yl)methyl)hydroxylamine (DX-01-17)
DX-01-16 (60 mg, 0.37 mmol) following the similar procedure described for the preparation of DX-02-05 afforded DX-01-17 (white solid, 39 mg, yield 65%). 1H-NMR (400 MHz, DMSO-d6): δ = 10.88 (s, 1H, NH-indolyl), 7.46–7.39 (m, 2H, H-indolyl, NH), 7.32 (d, J = 8.3 Hz, 1H, H-indolyl), 7.03–6.97 (m, 1H, H-indolyl), 6.93 (t, J = 8.1 Hz, 1H), 6.28 (s, 1H, H-indolyl), 6.17 (br, 1H, OH), 4.00 (s, 2H, CH2). 13C-NMR (101 MHz, CD3OD): δ = 137.98, 136.56, 122.09, 120.86, 120.84, 120.01, 111.84, 101.86, 51.98. HRMS (ESI): m/z [M + H]+ calculated for C9H11ON2: 163.08659; found: 163.08636.
3.1.16. Preparation of (E)-3-((1H-indol-3-yl)methylene)pyrrolidine-2,5-dione (DX-02-01)
1H-indole-3-carbaldehyde (500 mg, 3.45 mmol) and 1H-pyrrole-2,5-dione (401 mg, 4.14 mmol) following the similar procedure described for the preparation of DX-02-04 afforded DX-02-01 (white solid, 398 mg, yield 51% from 1H-indole-3-carbaldehyde). 1H-NMR (400 MHz, DMSO-d6): δ = 11.94 (s, 1H, NH-indolyl), 11.18 (s, 1H, NH), 7.81 (dd, J = 12.1, 5.1 Hz, 2H, H-indolyl, =CH), 7.70 (t, J = 2.1 Hz, 1H, H-indolyl), 7.54–7.41 (m, 1H, H-indolyl), 7.21–7.16 (m, 2H, H-indolyl), 3.52 (d, J = 2.2 Hz, 2H). HRMS (ESI): m/z [M + H]+ calculated for C13H11O2N2: 227.08150; found: 227.08134.
3.1.17. Preparation of (R/S) 3-((1H-indol-3-yl)methyl)pyrrolidine-2,5-dione (DX-02-02)
DX-02-01 (200 mg, 3.45 mmol) following the similar procedure described for the preparation of DX-02-04 afforded DX-02-02 (white solid, 65 mg, yield 32%). 1H-NMR (400 MHz, DMSO-d6): δ = 11.06 (s, 1H, NH-indolyl), 10.91 (s, 1H, NH), 7.51 (d, J = 7.8 Hz, 1H, H-indolyl), 7.33 (d, J = 8.0 Hz, 1H, H-indolyl), 7.16 (s, 1H, H-indolyl), 7.06 (t, J = 7.5 Hz, 1H, H-indolyl), 6.97 (t, J = 7.5 Hz, 1H, H-indolyl), 3.20–3.12 (m, 1H, CH’H’’), 2.95 (dd, J = 14.0, 8.5 Hz, 1H, CH’H’’), 2.70–2.61 (m, 2H, H-pyrrolidine-2,5-dione), 2.34 (dd, J = 18.0, 3.5 Hz, 1H, H-pyrrolidine-2,5-dione). HRMS (ESI): m/z [M + H]+ calculated for C13H13N2O2: 229.09715; found: 229.09663
3.1.18. Preparation of (E) 2-(1H-imidazol-4-yl)benzaldehyde Oxime (DX-03-02)
Compound 5 (400 mg, 0.92 mmol) and (2-formylphenyl)boronic acid (138 mg, 0.92 mmol) following the similar procedure described for the preparation of DX-03-12 and DX-02-04 afforded DX-03-02 (white solid, 98 mg, yield 57% from (2-formylphenyl)boronic acid). 1H-NMR (400 MHz, CD3OD): δ = 8.32 (s, 1H, =CH), 7.85–7.84 (m, 1H, H-phenyl), 7.79 (s, 1H, H-imidazolyl), 7.52 (d, J = 7.7 Hz, 1H, H-phenyl), 7.42–7.38 (m, 1H, H-phenyl), 7.34–7.30 (m, 1H, H-phenyl), 7.12 (s, 1H, H-imidazolyl). 13C-NMR (100 MHz, DMSO-d6): δ = 148.70, 136.36, 133.83, 130.10, 129.50, 129.27, 128.72, 128.34, 127.09, 126.20. HRMS (ESI): m/z [M + H]+ calculated for C10H10ON3: 188.08184; found: 188.08191.
3.1.19. Preparation of N-(2-(1H-imidazol-4-yl)benzyl)hydroxylamine (DX-03-03)
Compound DX-03-02 (50 mg, 0.27 mmol) following the similar procedure described for the preparation of DX-02-04 afforded DX-03-03 (white solid, 32 mg, yield 63%). 1H-NMR (400 MHz, CD3OD): δ = 7.75 (s, 1H, H-imidazolyl), 7.53 (d, J = 1.7 Hz, 1H, H-phenyl), 7.47–7.44 (m, 1H, H-phenyl), 7.35–7.27 (m, 3H, H-imidazolyl, H-phenyl), 4.04 (s, 2H). HRMS (ESI): m/z [M + H]+ calculated for C10H12ON3: 190.09749; found: 190.09737.
3.1.20. Preparation of (E) 2-(2H-tetrazol-5-yl)benzaldehyde Oxime (DX-03-04)
2-(2H-tetrazol-5-yl)benzaldehyde (200 mg, 1.15 mmol) following the similar procedure described for the preparation of DX-03-12 and DX-02-04 afforded DX-03-04 (white solid, 78 mg, yield 36% from 2-(2H-tetrazol-5-yl)benzaldehyde). 1H-NMR (400 MHz, DMSO-d6): δ = 11.23 (s, 1H, OH), 11.07 (s, 1H, H-tetrazolyl), 8.29 (s, 1H, =CH), 7.73–7.56 (m, 4H, H-phenyl). 13C-NMR (101 MHz, DMSO-d6): δ = 156.17, 137.81, 132.14, 130.64, 129.27, 127.64, 116.84, 110.69. HRMS (ESI): m/z [M + H]+ calculated for C8H8N5O: 190.07234; found: 190.07158.
3.1.21. Preparation of (E) 5-chloro-2-(1H-imidazol-4-yl)benzaldehyde Oxime (DX-03-05)
Compound 5 (300 mg, 0.69 mmol) and (4-chloro-2-formylphenyl)boronic acid (127 mg, 0.69 mmol) following the similar procedure described for the preparation of DX-03-12 and DX-02-04 afforded DX-03-05 (white solid, 70 mg, yield 46% from (4-chloro-2-formylphenyl)boronic acid). 1H-NMR (400 MHz, DMSO-d6): δ = 12.38 (s, 1H, OH), 11.32 (s, 1H, NH-imidazolyl), 8.77 (s, 1H, =CH), 7.80 (s, 1H, H-imidazolyl), 7.74 (s, 1H, H-imidazolyl), 7.63 (d, J = 2.4 Hz, 1H, H-phenyl), 7.44–7.42 (m, 2H, H-phenyl). 13C-NMR (101 MHz, DMSO-d6): δ = 147.77, 138.56, 136.60, 133.12, 131.72, 131.48, 131.05, 129.25, 125.42, 116.28. HRMS (ESI): m/z [M + H]+ calculated for C10H9ClN3O: 222.04287; found: 222.04330.
3.1.22. Preparation of (E) 2-fluoro-6-(1H-imidazol-4-yl)benzaldehyde Oxime (DX-03-06)
Compound 5 (300 mg, 0.69 mmol) and (3-fluoro-2-formylphenyl)boronic acid (116 mg, 0.69 mmol) following the similar procedure described for the preparation of DX-03-12 and DX-02-04 afforded DX-03-06 (white solid, 75 mg, yield 53% from (3-fluoro-2-formylphenyl)boronic acid). 1H-NMR (400 MHz, DMSO-d6): δ = 12.37 (s, 1H, OH), 11.24 (s, 1H, NH-imidazolyl), 8.37 (s, 1H, =CH), 7.76 (s, 1H, H-imidazolyl), 7.52 (s, 1H, H-imidazolyl), 7.36–7.13 (m, 3H, H-phenyl). 13C-NMR (101 MHz, DMSO-d6): δ = 161.48, 159.00, 144.68, 136.29, 130.27, 124.36, 122.95, 117.89, 116.35, 113.98. HRMS (ESI): m/z [M + H]+ calculated for C10H9FN3O: 206.07242; found: 206.07250.
3.1.23. Preparation of (E) 2-(1H-imidazol-4-yl)-5-methoxybenzaldehyde Oxime (DX-03-07)
Compound 5 (300 mg, 0.69 mmol) and (2-formyl-4-methoxyphenyl)boronic acid (124 mg, 0.69 mmol) following the similar procedure described for the preparation of DX-03-12 and DX-02-04 afforded DX-03-07 (white solid, 113 mg, yield 76% from (2-formyl-4-methoxyphenyl)boronic acid). 1H-NMR (400 MHz, CD3OD): δ = 8.73 (s, 1H, H-imidazolyl), 7.78 (s, 1H, H-imidazolyl), 7.52–7.38 (m, 3H, H-phenyl), 7.15 (s, 1H, H-phenyl), 7.10–6.96 (m, 4H, H-phenyl), 3.85 (s, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ = 141.55, 137.87, 133.07, 132.61, 121.44, 118.60, 117.79, 116.37, 113.90, 110.95, 55.90. HRMS (ESI): m/z [M + H]+ calculated for C11H12N3O2: 218.09240; found: 218.09206.
3.1.24. Preparation of (E) 4-(1H-imidazol-4-yl)-2′-(trifluoromethyl)-[1,1′-biphenyl]-3-carbaldehyde Oxime (DX-03-08)
Compound 5 (300 mg, 0.69 mmol) and (3-formyl-2′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl) boronic acid (202 mg, 0.69 mmol) following the similar procedure described for the preparation of DX-03-12 and DX-02-04 afforded DX-03-08 (white solid, 82mg, yield 36% from (3-formyl-2′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)boronic acid). 1H-NMR (400 MHz, DMSO-d6): δ = 12.38 (s, 1H, OH), 11.14 (s, 1H, NH-imidazolyl), 8.85(s, 1H, H-imidazolyl), 7.86–7.79 (m, 2H, H-imidazolyl, H-phenyl), 7.73–7.63 (m, 4H, H-phenyl), 7.46 (br, 2H, H-phenyl), 7.33 (s, 1H, H-phenyl). 13C-NMR (101 MHz, DMSO-d6): δ = 148.54, 140.58, 137.83, 136.56, 132.89, 132.57, 130.11, 129.84, 129.53, 128.94, 128.64, 126.58, 126.32, 126.23, 126.04, 123.22, 116.04. HRMS (ESI): m/z [M + H]+ calculated for C17H13F3N3O: 332.10052; found: 332.10123.
3.1.25. Preparation of (E) 3-(1H-imidazol-4-yl)-[1,1′-biphenyl]-4-carbaldehyde Oxime (DX-03-09)
Compound 5 (300 mg, 0.69 mmol) and (4-formyl-[1,1′-biphenyl]-3-yl)boronic acid (156 mg, 0.69 mmol) following the similar procedure described for the preparation of DX-03-12 and DX-02-04 afforded DX-03-09 (white solid, 81 mg, yield 45% from (4-formyl-[1,1′-biphenyl]-3-yl)boronic acid). 1H-NMR (400 MHz, DMSO-d6): δ = 11.01 (s, 1H, OH), 10.34 (s, 1H, NH-imidazolyl), 8.55 (s, 1H, =CH), 8.39 (s, 1H, H-imidazolyl), 8.12 (s, 1H, H-imidazolyl), 7.58 (br, 1H, H-phenyl), 7.48–7.22 (m, 2H, H-phenyl), 7.11–7.02 (m, 2H, H-phenyl), 6.87–6,81 (m, 2H, H-phenyl), 6.50 (s, 1H, H-phenyl). HRMS (ESI): m/z [M + H]+ calculated for C16H14N3O: 264.11314; found: 264.11346.
3.1.26. Preparation of 4-(2-((4-(trifluoromethyl)benzyl)oxy)phenyl)-1,2,3-thiadiazole (DX-03-10)
2-(1,2,3-Thiadiazol-4-yl)phenol (100 mg, 0.56 mmol) and 1-(bromomethyl)-4-(trifluoromethyl)benzene (134 mg, 0.56 mmol) following the similar procedure described for the preparation of 12 afforded DX-03-10 (white solid, 128 mg, yield 68%). 1H-NMR (400 MHz, DMSO-d6): δ = 9.39 (s, 1H, H-thiadiazolyl), 8.26 (d, J = 7.5 Hz, 1H, H-phenyl), 7.76 (d, J = 8.2 Hz, 2H, H-phenyl), 7.70 (d, J = 8.5 Hz, 2H, H-phenyl), 7.48–7.43 (m, 1H, H-phenyl), 7.31 (d, J = 7.7 Hz, 1H, H-phenyl), 7.17 (t, J = 7.5 Hz, 1H, H-phenyl), 5.44 (s, 2H, CH2). HRMS (ESI): m/z [M + H]+ calculated for C16H12F3N2OS: 337.06169; found: 337.06116.
3.1.27. Preparation of N-(3-(2-(1H-imidazol-5-yl)phenoxy)propyl)-aminesulfonamide (DX-03-11)
Compound 9 (200 mg, 0.34 mmol) following the similar procedure described for the preparation of DX-03-12 afforded DX-03-11 (white solid, 45 mg, yield 45% from compound 9). 1H-NMR (400 MHz, CD3OD): δ = 8.67 (s, 1H, NH-imidazolyl), 8.08 (s, 1H, H-imidazolyl), 7.83 (s, 1H, H-imidazolyl), 7.69 (d, J = 7.7 Hz, 1H, H-phenyl), 7.40 (t, J = 7.1 Hz, 1H, H-phenyl), 7.16 (d, J = 8.2 Hz, 1H, H-phenyl), 7.08 (t, J = 7.4 Hz, 1H, H-phenyl), 4.20 (t, J = 6.0 Hz, 2H, CH2), 3.44 (t, J = 6.9 Hz, 2H, CH2), 2.11–2.03 (m, 2H, CH2). HRMS (ESI): m/z [M + H]+ calculated for C12H17O3N4S: 297.10159; found: 297.10115.
3.1.28. Preparation of 1-(3-(2-(1H-imidazol-5-yl)phenoxy)propyl)-3-(4-(trifluoromethyl) phenyl)urea (DX-03-13)
Compound 9 (200 mg, 0.34 mmol) following the similar procedure described for the preparation of DX-03-12 afforded DX-03-13 (white solid, 77 mg, yield 56% from compound 9). 1H-NMR (400 MHz, DMSO-d6): δ = 12.24 (s, 1H, NH-imidazolyl), 8.90 (s, 1H, H-imidazolyl), 8.01 (d, J = 7.4 Hz, 1H, H-phenyl), 7.75 (s, 1H, H-imidazolyl), 7.61–7.54 (m, 5H, H-phenyl), 7.21–7.15 (m, 1H, H-phenyl), 7.06 (d, J = 7.8 Hz, 1H, H-phenyl), 7.02–6.94 (m, 1H, H-phenyl), 6.48 (t, J = 5.7 Hz, 1H, H-phenyl), 4.14 (t, J = 6.1 Hz, 2H, CH2), 3.38–3.32 (m, 2H, CH2), 2.08–1.97 (m, 2H, CH2). 13C-NMR (101 MHz, DMSO-d6): δ = 155.42, 155.08, 144.71, 135.42, 129.17, 127.38, 127.13, 126.38 (2C), 123.78, 122.81, 121.53, 121.21, 120.86, 117.71 (2C), 112.33, 65.82, 36.89, 29.98. HRMS (ESI): m/z [M + H]+ calculated for C20H20O2N4F3: 405.15329; found: 405.15302.
3.1.29. Preparation of 1-(3-bromo-4-fluorobenzyl)-4-(5-chloro-2-methoxyphenyl)-1H-imidazole (DX-04-03)
2-Bromo-4-(bromomethyl)-1-fluorobenzene (500 mg, 1.88 mmol) following the similar procedure described for the preparation of DX-04-02 afforded DX-04-03 (white solid, 480 mg, yield 65% from 2-bromo-4-(bromomethyl)-1-fluorobenzene). 1H-NMR (400 MHz, CD3OD): δ = 7.95 (d, J = 2.7 Hz, 1H, H-phenyl), 7.78 (d, J = 1.3 Hz, 1H, H-imidazolyl), 7.64 (d, J = 1.3 Hz, 1H, H-imidazolyl), 7.57 (dd, J = 6.5, 2.2 Hz, 1H, H-phenyl), 7.30–7.24 (m, 1H, H-phenyl), 7.20 (d, J = 8.5 Hz, 1H, H-phenyl), 7.16 (dd, J = 8.8, 2.7 Hz, 1H, H-phenyl), 6.98 (d, J = 8.8 Hz, 1H, H-phenyl), 5.22 (s, 2H, CH2), 3.88 (s, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ = 161.35, 158.96, 156.10, 138.33, 137.40, 133.91, 129.70, 128.04, 127.36, 126.81, 125.19, 121.53, 117.84, 113.47, 110.01, 56.13, 50.30. HRMS (ESI): m/z [M + H]+ calculated for C17H14ON2BrClF: 394.99566; found: 394.99728.
3.1.30. Preparation of 2-(1-(3-bromo-4-fluorobenzyl)-1H-imidazol-4-yl)-4-chlorophenol (DX-04-04)
DX-04-03 (200 mg, 0.51 mmol) following the similar procedure described for the preparation of DX-04-02 afforded DX-04-04 (white solid, 116 mg, yield 60%). 1H-NMR (400 MHz, DMSO-d6): δ = 9.12 (s, 1H, H-imidazolyl), 8.40 (s, 1H, H-imidazolyl), 8.01 (s, 1H, H-imidazolyl), 7.89 (dd, J = 6.5, 1.7 Hz, 1H, H-phenyl), 7.56–7.49 (m, 1H, H-phenyl), 7.45 (t, J = 8.6 Hz, 1H, H-phenyl), 7.34 (dd, J = 8.8, 2.5 Hz, 1H, H-phenyl), 7.00 (d, J = 8.8 Hz, 1H, H-phenyl), 5.42 (s, 2H, CH2). 13C-NMR (101 MHz, CD3OD): δ = 161.85, 159.35, 154.93, 136.62, 134.79, 130.61, 130.34, 126.90, 125.53, 119.70, 118.77, 118.32, 118.09, 117.92, 110.27, 51.71. HRMS (ESI): m/z [M + H]+ calculated for C16H12ON2BrClF: 380.98001; found: 380.98065.
3.1.31. Preparation of 4-chloro-2-(1-(4-(trifluoromethyl)benzyl)-1H-1,2,3-triazol-4-yl)phenol (DX-04-05)
DX-04-06 (300 mg, 0.81 mmol) following the similar procedure described for the preparation of DX-04-02 afforded DX-04-05 (white solid, 176 mg, yield 61%). 1H-NMR (400 MHz, DMSO-d6): δ = 10.49 (s, 1H, OH), 7.85 (s, 1H, H-triazolyl), 7.64 (d, J = 8.1 Hz, 2H, H-phenyl), 7.34 (dd, J = 8.8, 2.7 Hz, 1H, H-phenyl), 7.20 (d, J = 8.1 Hz, 2H, H-phenyl), 7.16 (d, J = 2.7 Hz, 1H, H-phenyl), 6.98 (d, J = 8.8 Hz, 1H, H-phenyl), 5.62 (s, 2H, CH2). 13C-NMR (101 MHz, CD3OD): δ = 155.39, 140.99, 136.49, 134.75, 132.46, 131.76, 131.11, 129.36 (2C), 126.49 (2C), 125.38, 124.09, 118.30, 116.15, 53.27. HRMS (ESI): m/z [M + H]+ calculated for C16H12ON3ClF3: 354.06155; found: 354.06015.
3.1.32. Preparation of 4-(5-chloro-2-methoxyphenyl)-1-(4-(trifluoromethyl)benzyl)-1H-1,2,3-triazole (DX-04-06)
4-chloro-2-ethynyl-1-methoxybenzene (500 mg, 3.01 mmol) and 1-(bromomethyl)-4-(trifluoromethyl)benzene (717 mg, 3.01 mmol) following the similar procedure described for the preparation of DX-04-02 afforded DX-04-06 (white solid, 675 mg, yield 61% from 4-chloro-2-ethynyl-1-methoxybenzene). 1H-NMR (400 MHz, CD3OD): δ = 7.71 (s, 1H, H-triazolyl), 7.50 (d, J = 8.1 Hz, 2H, H-phenyl), 7.40 (dd, J = 8.9, 2.7 Hz, 1H, H-phenyl), 7.14 (d, J = 8.1 Hz, 2H, H-phenyl), 7.09 (d, J = 2.6 Hz, 1H, H-phenyl), 7.03 (d, J = 8.9 Hz, 1H, H-phenyl), 5.55 (s, 2H, CH2), 3.64 (s, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ = 154.74, 143.61, 131.20, 130.91, 129.93 (2C), 129.81, 129.39, 127.59, 127.02 (2C), 125.98, 125.64, 118.63, 117.43, 55.31, 55.04. HRMS (ESI): m/z [M + H]+ calculated for C17H14ON3ClF3: 368.07720; found: 368.07599.
3.1.33. Preparation of 4-chloro-2-(5-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)phenol (DX-04-07)
DX-04-08 (200 mg, 0.57 mmol) following the similar procedure described for the preparation of DX-04-02 afforded DX-04-07 (white solid, 144 mg, yield 75%). 1H-NMR (400 MHz, DMSO-d6): δ = 15.28 (s, 1H, H-triazolyl), 9.91 (s, 1H, NH-triazolyl), 7.75–7.68 (m, 4H, H-phenyl), 7.37 (br, 2H, H-phenyl), 6.94 (s, 1H, H-phenyl). 13C-NMR (101 MHz, CD3OD): δ = 155.58, 142.95, 137.77, 136.14, 131.62, 131.27, 130.86, 128.63 (2C), 126.43 (2C), 125.31, 124.28, 119.14, 118.53. HRMS (ESI): m/z [M + H]+ calculated for C15H10ON3ClF3: 340.04590; found: 340.04453.
3.1.34. Preparation of 4-(5-chloro-2-methoxyphenyl)-5-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazole (DX-04-08)
4-chloro-2-ethynyl-1-methoxybenzene (300 mg, 1.81 mmol) and 1-iodo-4-(trifluoromethyl)benzene (491 mg, 1.81 mmol) following the similar procedure described for the preparation of DX-04-02 afforded DX-04-08 (white solid, 338 mg, yield 53% from 4-chloro-2-ethynyl-1-methoxybenzene). 1H-NMR (400 MHz, DMSO-d6): δ = 15.41 (s, 1H, NH-triazolyl), 7.74 (d, J = 8.1 Hz, 2H, H-phenyl), 7.68–7.59 (m, 2H, H-phenyl), 7.54 (d, J = 8.9 Hz, 1H, H-phenyl), 7.48 (d, J = 2.7 Hz, 1H, H-phenyl), 7.15 (s, 1H, H-phenyl), 3.42 (s, 3H, CH3). 13C-NMR (101 MHz, CD3OD): δ = 157.20, 136.42, 131.71, 131.59, 131.14, 129.65, 128.37 (2C), 126.96, 126.75, 126.40 (2C), 124.27, 121.57, 114.17, 55.99. HRMS (ESI): m/z [M + H]+ calculated for C16H12ON3ClF3: 354.06155; found: 354.06079.


 ,
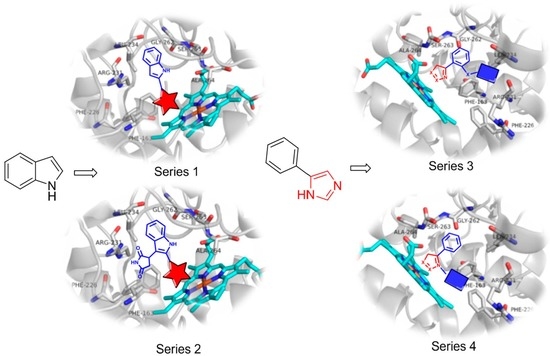
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

















































