
Three Structural Features of Functional Food Components and Herbal Medicine with Amyloid β42 Anti-Aggregation Properties



Abstract
:1. Introduction
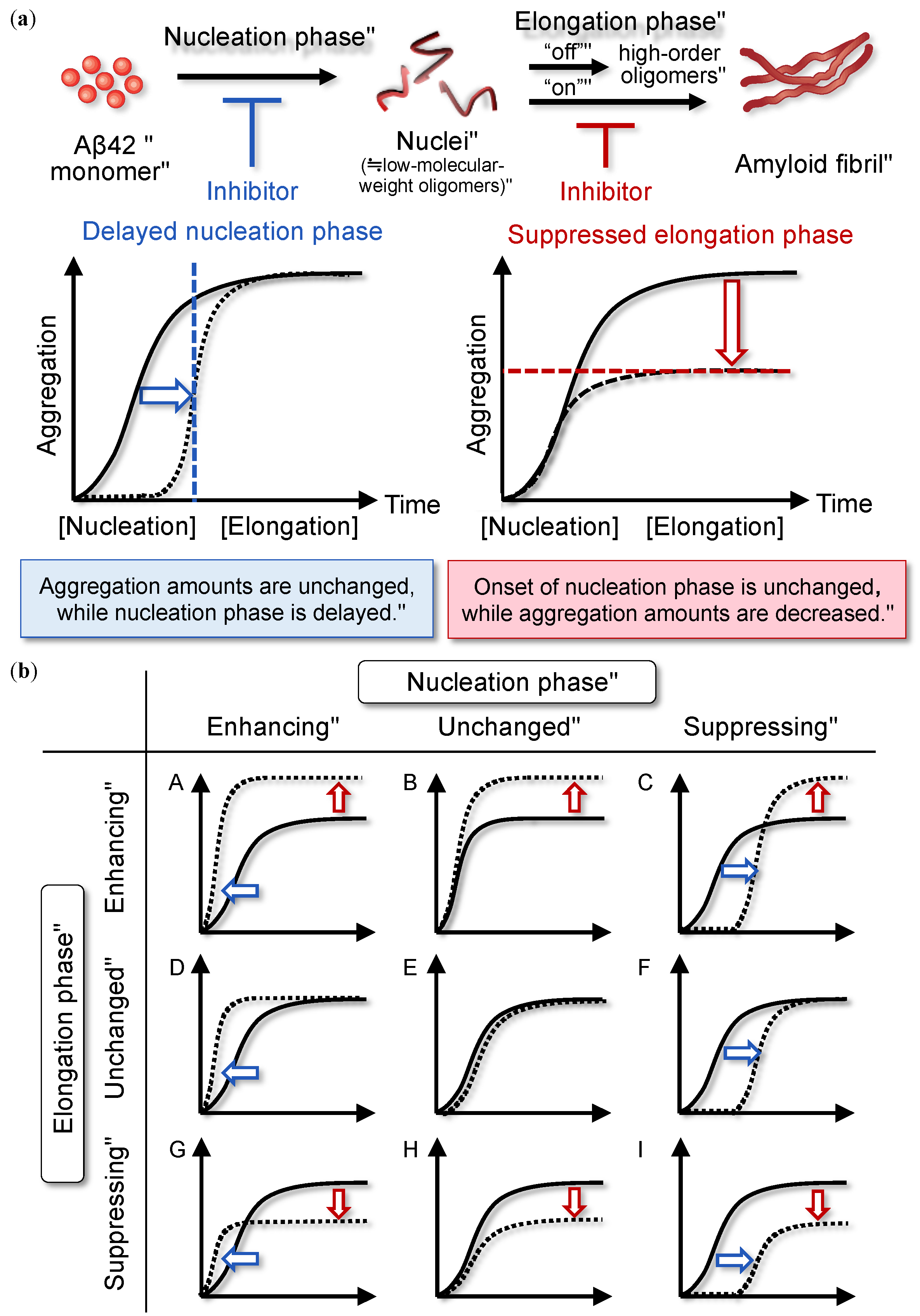
2. Nucleation-Dependent Polymerization Mechanism of Aβ42
2.1. Overview
2.2. Classification of Aggregation Inhibitors
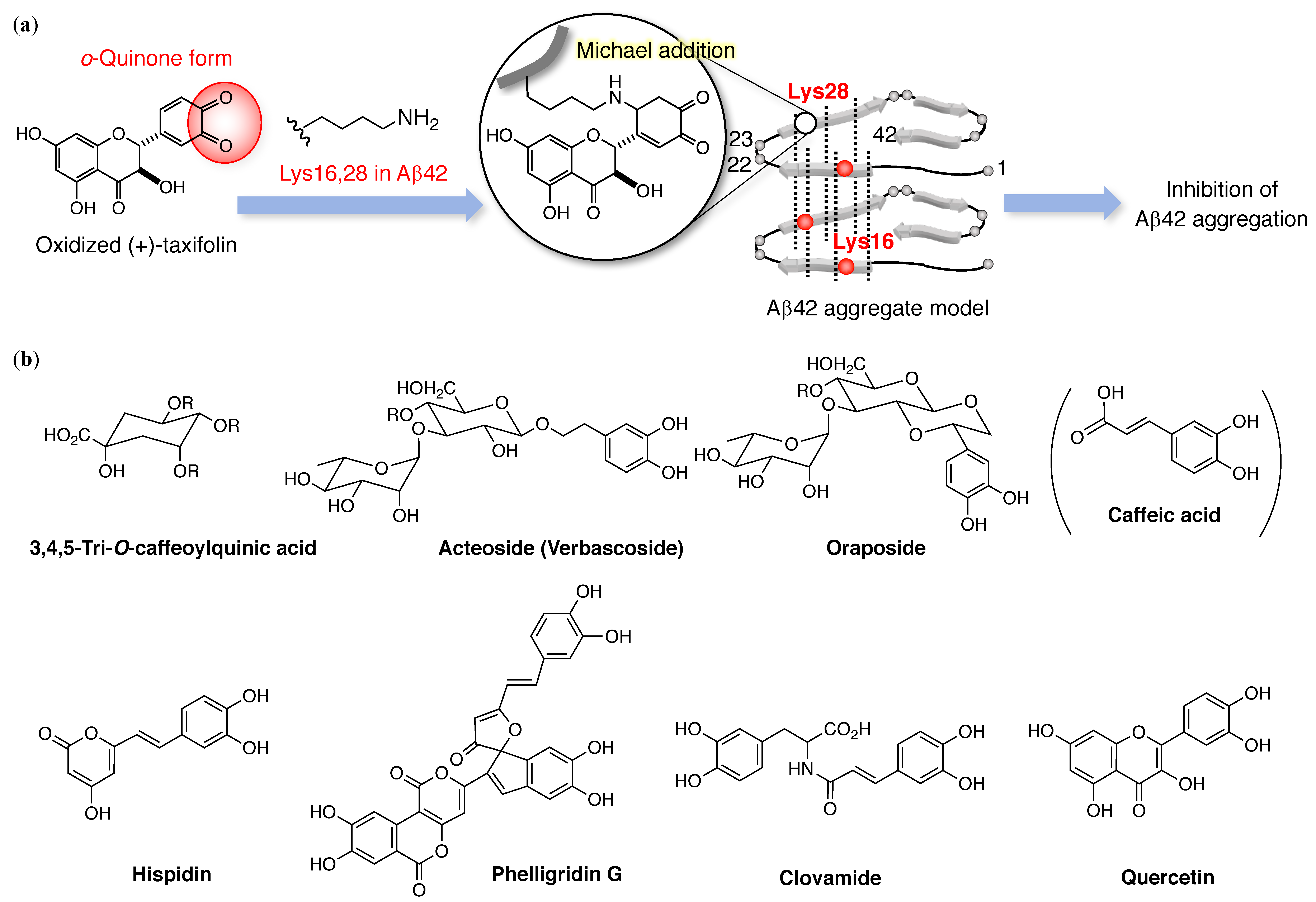
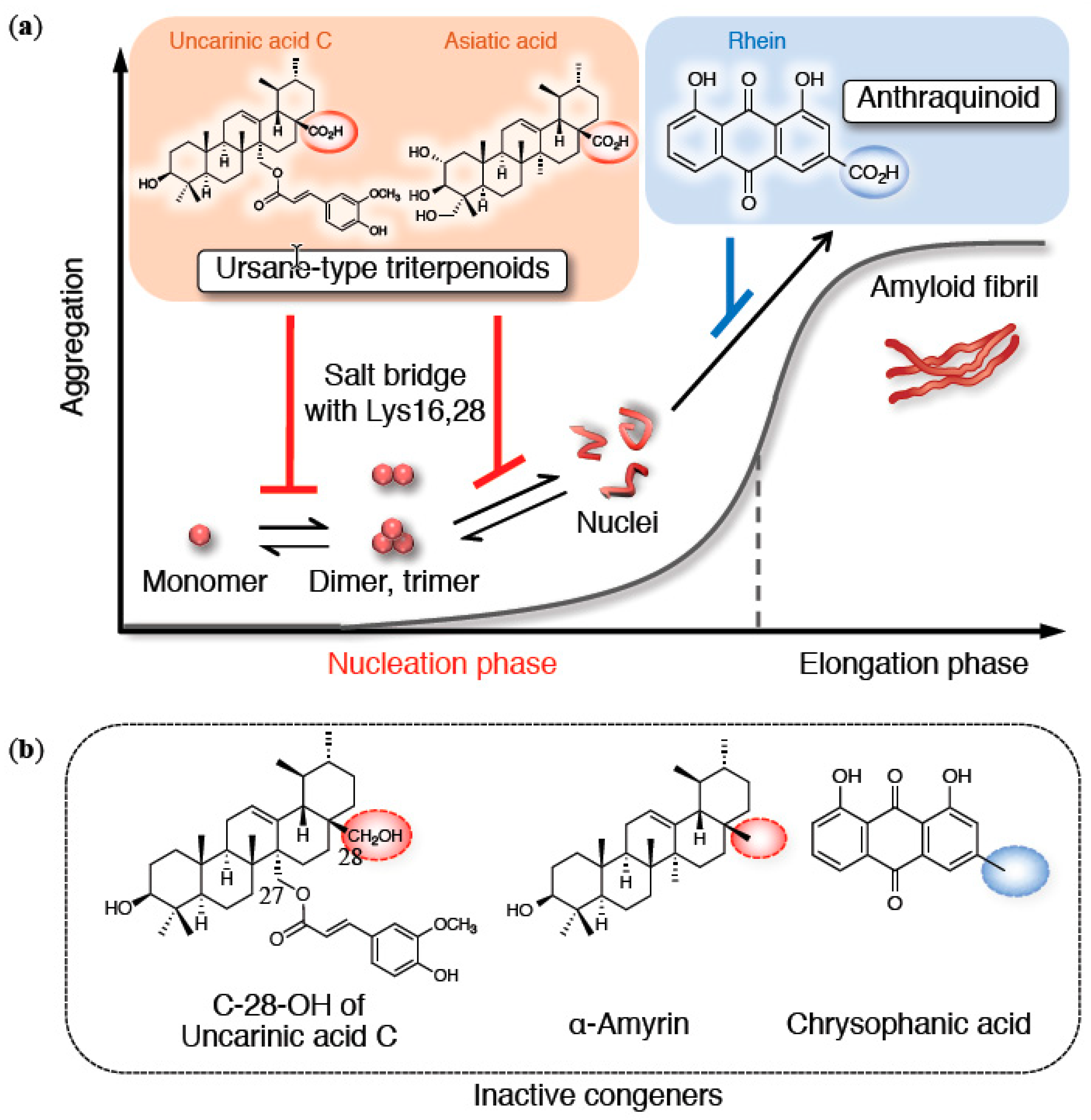
3. Structural Features of Anti-Aβ42 Aggregative Compounds
3.1. Catechol-Type Flavonoid: Catechol Moiety
3.2. Non-Catechol-Type Flavonoid: Planarity
3.3. Triterpenoid Carboxy Acid: Carboxy Group
4. In Vivo Metabolism of Anti-Aβ42 Aggregative Compounds
5. Conclusions and Perspective
Funding
Conflicts of Interest
References
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Roychaudhuri, R.; Yang, M.; Hoshi, M.M.; Teplow, D.B. Amyloid β-protein assembly and Alzheimer disease. J. Biol. Chem. 2009, 284, 4749–4753. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.D.; Wong, S.S.; Lieber, C.M.; Lansbury, P.T. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997, 4, 119–125. [Google Scholar] [CrossRef]
- Walsh, D.M.; Lomakin, A.; Benedek, G.B.; Condron, M.M.; Teplow, D.B. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 1997, 272, 22364–22372. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, M.; Sato, M.; Matsumoto, S.; Noguchi, A.; Yasutake, K.; Yoshida, N.; Sato, K. Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. USA 2003, 100, 6370–6375. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J.; Spencer, J.P. Flavonoids, cognition, and dementia: Actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radic. Biol. Med. 2012, 52, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hamaguchi, T.; Naiki, H.; Yamada, M. Anti-amyloidogenic effects of antioxidants: Implications for the prevention and therapeutics of Alzheimer’s disease. Biochim. Biophys. Acta 2006, 1762, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Yamin, G.; Ono, K.; Inayathullah, M.; Teplow, D.B. Amyloid β-protein assembly as a therapeutic target of Alzheimer’s disease. Curr. Pharm. Design 2008, 14, 3231–3246. [Google Scholar] [CrossRef]
- Williams, P.; Sorribas, A.; Howes, M.J. Natural products as a source of Alzheimer’s drug leads. Nat. Prod. Rep. 2011, 28, 48–77. [Google Scholar] [CrossRef] [PubMed]
- Ushikubo, H.; Watanabe, S.; Tanimoto, Y.; Abe, K.; Hiza, A.; Ogawa, T.; Asakawa, T.; Kan, T.; Akaishi, T. 3,3’,4’,5,5’-Pentahydroxyflavone is a potent inhibitor of amyloid β fibril formation. Neurosci. Lett. 2012. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Orhan, I.E.; Daglia, M.; Manayi, A.; Gortzi, O.; Nabavi, S.M. Neuroprotective effects of chrysin: From chemistry to medicine. Neurochem. Int. 2015, 90, 224–231. [Google Scholar] [CrossRef]
- Habtemariam, S. Rutin as a natural therapy for Alzheimer’s disease: Insights into its mechanisms of action. Curr. Med. Chem. 2016, 23, 860–873. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Sureda, A.; Manayi, A.; Nabavi, S.M. Neuroprotective effects of fisetin in Alzheimer’s and Parkinson’s diseases: From chemistry to medicine. Curr. Top Med. Chem. 2016, 16, 1910–1915. [Google Scholar] [CrossRef]
- Braidy, N.; Jugder, B.E.; Poljak, A.; Jayasena, T.; Mansour, H.; Nabavi, S.M.; Sachdev, P.; Grant, R. Resveratrol as a potential therapeutic candidate for the treatment and management of Alzheimer’s disease. Curr. Top Med. Chem. 2016, 16, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Aggarwal, A. Therapeutic potentials of herbal drugs for Alzheimer’s disease-An overview. Indian J. Exp. Biol. 2017, 55, 63–73. [Google Scholar] [PubMed]
- Nabavi, S.F.; Khan, H.; D’Onofrio, G.; Samec, D.; Shirooie, S.; Dehpour, A.R.; Arguelles, S.; Habtemariam, S.; Sobarzo-Sanchez, E. Apigenin as neuroprotective agent: Of mice and men. Pharmacol. Res. 2018, 128, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Yamaguchi, I.; Omata, S.; Gejyo, F.; Naiki, H. Interaction between Aβ(1-42) and A β(1-40) in Alzheimer’s β-amyloid fibril formation in vitro. Biochemistry 1999, 38, 15514–15521. [Google Scholar] [CrossRef] [PubMed]
- Serio, T.R.; Cashikar, A.G.; Kowal, A.S.; Sawicki, G.J.; Moslehi, J.J.; Serpell, L.; Arnsdorf, M.F.; Lindquist, S.L. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 2000, 289, 1317–1321. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003, 26, 207–214. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef]
- Kotler, S.A.; Walsh, P.; Brender, J.R.; Ramamoorthy, A. Differences between amyloid-β aggregation in solution and on the membrane: Insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6692–6700. [Google Scholar] [CrossRef]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef]
- Bieschke, J.; Herbst, M.; Wiglenda, T.; Friedrich, R.P.; Boeddrich, A.; Schiele, F.; Kleckers, D.; Lopez del Amo, J.M.; Gruning, B.A.; Wang, Q.; et al. Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat. Chem. Biol. 2011, 8, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Culyba, E.K.; Powers, E.T.; Kelly, J.W. Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 2011, 7, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Bitan, G.; Fradinger, E.A.; Spring, S.M.; Teplow, D.B. Neurotoxic protein oligomers--what you see is not always what you get. Amyloid 2005, 12, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Hepler, R.W.; Grimm, K.M.; Nahas, D.D.; Breese, R.; Dodson, E.C.; Acton, P.; Keller, P.M.; Yeager, M.; Wang, H.; Shughrue, P.; et al. Solution state characterization of amyloid β-derived diffusible ligands. Biochemistry 2006, 45, 15157–15167. [Google Scholar] [CrossRef] [PubMed]
- Teplow, D.B. Preparation of amyloid β-protein for structural and functional studies. Methods Enzymol. 2006, 413, 20–33. [Google Scholar]
- Murakami, K.; Yoshioka, T.; Horii, S.; Hanaki, M.; Midorikawa, S.; Taniwaki, S.; Gunji, H.; Akagi, K.I.; Kawase, T.; Hirose, K.; et al. Role of the carboxy groups of triterpenoids in their inhibition of the nucleation of amyloid β42 required for forming toxic oligomers. Chem. Commun. 2018, 54, 6272–6275. [Google Scholar] [CrossRef]
- Shigemitsu, Y.; Iwaya, N.; Goda, N.; Matsuzaki, M.; Tenno, T.; Narita, A.; Hoshi, M.; Hiroaki, H. Nuclear magnetic resonance evidence for the dimer formation of β amyloid peptide 1-42 in 1,1,1,3,3,3-hexafluoro-2-propanol. Anal. Biochem. 2016, 498, 59–67. [Google Scholar] [CrossRef]
- Sato, M.; Murakami, K.; Uno, M.; Nakagawa, Y.; Katayama, S.; Akagi, K.; Masuda, Y.; Takegoshi, K.; Irie, K. Site-specific inhibitory mechanism for amyloid β42 aggregation by catechol-type flavonoids targeting the Lys residues. J. Biol. Chem. 2013, 288, 23212–23224. [Google Scholar] [CrossRef]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 2003, 87, 172–181. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Zhu, M.; Rajamani, S.; Kaylor, J.; Han, S.; Zhou, F.; Fink, A.L. The flavonoid baicalein inhibits fibrillation of α-synuclein and disaggregates existing fibrils. J. Biol. Chem. 2004, 279, 26846–26857. [Google Scholar] [CrossRef] [PubMed]
- Palhano, F.L.; Lee, J.; Grimster, N.P.; Kelly, J.W. Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J. Am. Chem. Soc. 2013, 135, 7503–7510. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Braymer, J.J.; Nanga, R.P.; Ramamoorthy, A.; Lim, M.H. Design of small molecules that target metal-Aβ species and regulate metal-induced Aβ aggregation and neurotoxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 21990–21995. [Google Scholar] [CrossRef] [PubMed]
- DeToma, A.S.; Salamekh, S.; Ramamoorthy, A.; Lim, M.H. Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem. Soc. Rev. 2012, 41, 608–621. [Google Scholar] [CrossRef] [PubMed]
- DeToma, A.S.; Krishnamoorthy, J.; Nam, Y.; Lee, H.J.; Brender, J.R.; Kochi, A.; Lee, D.; Onnis, V.; Congiu, C.; Manfredini, S.; et al. Synthetic flavonoids, aminoisoflavones: Interaction and reactivity with metal-free and metal-associated amyloid-β species. Chem. Sci. 2014, 5, 4851–4862. [Google Scholar] [CrossRef] [PubMed]
- Korshavn, K.J.; Jang, M.; Kwak, Y.J.; Kochi, A.; Vertuani, S.; Bhunia, A.; Manfredini, S.; Ramamoorthy, A.; Lim, M.H. Reactivity of metal-free and metal-associated amyloid-β with glycosylated polyphenols and their esterified derivatives. Sci. Rep. 2015, 5, 17842. [Google Scholar] [CrossRef] [PubMed]
- Lenhart, J.A.; Ling, X.; Gandhi, R.; Guo, T.L.; Gerk, P.M.; Brunzell, D.H.; Zhang, S. “Clicked” bivalent ligands containing curcumin and cholesterol as multifunctional Aβ oligomerization inhibitors: Design, synthesis, and biological characterization. J. Med. Chem. 2010, 53, 6198–6209. [Google Scholar] [CrossRef] [PubMed]
- Ginex, T.; Trius, M.; Luque, F.J. Computational study of the aza-michael addition of the flavonoid (+)-taxifolin in the inhibition of β-amyloid fibril aggregation. Chemistry 2018, 24, 5813–5824. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Yamamoto, Y.; Maki, T.; Hattori, Y.; Ito, H.; Mizuno, K.; Harada-Shiba, M.; Kalaria, R.N.; Fukushima, M.; Takahashi, R.; et al. Taxifolin inhibits amyloid-β oligomer formation and fully restores vascular integrity and memory in cerebral amyloid angiopathy. Acta Neuropathol. Commun. 2017, 5, 26. [Google Scholar] [CrossRef]
- Inoue, T.; Saito, S.; Tanaka, M.; Yamakage, H.; Kusakabe, T.; Shimatsu, A.; Ihara, M.; Satoh-Asahara, N. Pleiotropic neuroprotective effects of taxifolin in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 10031–10038. [Google Scholar] [CrossRef] [Green Version]
- Miyamae, Y.; Kurisu, M.; Murakami, K.; Han, J.; Isoda, H.; Irie, K.; Shigemori, H. Protective effects of caffeoylquinic acids on the aggregation and neurotoxicity of the 42-residue amyloid β-protein. Bioorg. Med. Chem. 2012, 20, 5844–5849. [Google Scholar] [CrossRef] [PubMed]
- Kurisu, M.; Miyamae, Y.; Murakami, K.; Han, J.; Isoda, H.; Irie, K.; Shigemori, H. Inhibition of amyloid β aggregation by acteoside, a phenylethanoid glycoside. Biosci. Biotech. Biochem. 2013, 77, 1329–1332. [Google Scholar] [CrossRef] [PubMed]
- Kidachi, E.; Kurisu, M.; Miyamae, Y.; Hanaki, M.; Murakami, K.; Irie, K.; Shigemori, H. Structure-activity relationship of phenylethanoid glycosides on the inhibition of amyloid β aggregation. HeteroCycles 2016, 92, 1976–1982. [Google Scholar]
- Aihara, Y.; Kawaguchi, A.; Hanaki, M.; Murakami, K.; Irie, K.; Shigemori, H. Inhibitory activity of hispidin derivatives isolated from Inonotus obliquus on amyloid β aggregation. HeteroCycles 2017, 94, 1280–1287. [Google Scholar]
- Tsunoda, T.; Takase, M.; Shigemori, H. Structure-activity relationship of clovamide and its related compounds for the inhibition of amyloid β aggregation. Bioorg. Med. Chem. 2018, 26, 3202–3209. [Google Scholar] [CrossRef] [PubMed]
- Ben Hmidene, A.; Hanaki, M.; Murakami, K.; Irie, K.; Isoda, H.; Shigemori, H. Inhibitory activities of antioxidant flavonoids from Tamarix gallica on amyloid aggregation related to Alzheimer’s and type 2 diabetes diseases. Biol. Pharm. Bull. 2017, 40, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Fukuchi, M.; Yatagawa, T.; Tada, M.; Takeda, K.; Irie, K.; Akagi, K.; Monobe, Y.; Imazawa, T.; Takegoshi, K. Solid-state NMR analysis of interaction sites of curcumin and 42-residue amyloid β-protein fibrils. Bioorg. Med. Chem. 2011, 19, 5967–5974. [Google Scholar] [CrossRef]
- Richard, T.; Papastamoulis, Y.; Waffo-Teguo, P.; Monti, J.P. 3D NMR structure of a complex between the amyloid β peptide (1–40) and the polyphenol ε-viniferin glucoside: Implications in Alzheimer’s disease. Biochim. Biophys. Acta 2013, 1830, 5068–5074. [Google Scholar] [CrossRef]
- Hanaki, M.; Murakami, K.; Akagi, K.; Irie, K. Structural insights into mechanisms for inhibiting amyloid β42 aggregation by non-catechol-type flavonoids. Bioorg. Med. Chem. 2016, 24, 304–313. [Google Scholar] [CrossRef]
- Schanda, P.; Brutscher, B. Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J. Am. Chem. Soc. 2005, 127, 8014–8015. [Google Scholar] [CrossRef]
- Morimoto, A.; Irie, K.; Murakami, K.; Ohigashi, H.; Shindo, M.; Nagao, M.; Shimizu, T.; Shirasawa, T. Aggregation and neurotoxicity of mutant amyloid β (Aβ) peptides with proline replacement: Importance of turn formation at positions 22 and 23. Biochem. Biophys. Res. Commun. 2002, 295, 306–311. [Google Scholar] [CrossRef]
- Morimoto, A.; Irie, K.; Murakami, K.; Masuda, Y.; Ohigashi, H.; Nagao, M.; Fukuda, H.; Shimizu, T.; Shirasawa, T. Analysis of the secondary structure of β-amyloid (Aβ42) fibrils by systematic proline replacement. J. Biol. Chem. 2004, 279, 52781–52788. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Uemura, S.; Ohashi, R.; Nakanishi, A.; Takegoshi, K.; Shimizu, T.; Shirasawa, T.; Irie, K. Identification of physiological and toxic conformations in Aβ42 aggregates. ChemBioChem 2009, 10, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Luhrs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Dobeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Irie, K.; Ohigashi, H.; Hara, H.; Nagao, M.; Shimizu, T.; Shirasawa, T. Formation and stabilization model of the 42-mer Aβ radical: Implications for the long-lasting oxidative stress in Alzheimer’s disease. J. Am. Chem. Soc. 2005, 127, 15168–15174. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Qu, J.; Zhang, W.; Bai, M.; Zhou, Q.; Zhang, Z.; Li, Z.; Miao, J. Morin reverses neuropathological and cognitive impairments in APPswe/PS1dE9 mice by targeting multiple pathogenic mechanisms. Neuropharmacology 2016, 108, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ano, Y.; Dohata, A.; Taniguchi, Y.; Hoshi, A.; Uchida, K.; Takashima, A.; Nakayama, H. Iso-α-acids, bitter components of beer, prevent inflammation and cognitive decline induced in a mouse model of Alzheimer’s disease. J. Biol. Chem. 2017, 292, 3720–3728. [Google Scholar] [CrossRef]
- Herculano, B.; Tamura, M.; Ohba, A.; Shimatani, M.; Kutsuna, N.; Hisatsune, T. β-Alanyl-l-histidine rescues cognitive deficits caused by feeding a high fat diet in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, 983–997. [Google Scholar] [CrossRef]
- Grunblatt, E.; Mandel, S.; Gassen, M.; Youdim, M.B. Potent neuroprotective and antioxidant activity of apomorphine in MPTP and 6-hydroxydopamine induced neurotoxicity. J. Neural Transm. Suppl. 1999, 55, 57–70. [Google Scholar]
- Hanaki, M.; Murakami, K.; Katayama, S.; Akagi, K.I.; Irie, K. Mechanistic analyses of the suppression of amyloid β42 aggregation by apomorphine. Bioorg. Med. Chem. 2018, 26, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Himeno, E.; Ohyagi, Y.; Ma, L.; Nakamura, N.; Miyoshi, K.; Sakae, N.; Motomura, K.; Soejima, N.; Yamasaki, R.; Hashimoto, T.; et al. Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation. Ann. Neurol. 2011, 69, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Ohyagi, Y.; Imamura, T.; Yanagihara, Y.T.; Iinuma, K.M.; Soejima, N.; Murai, H.; Yamasaki, R.; Kira, J.I. Apomorphine therapy for neuronal insulin resistance in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2017, 58, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Kanapathipillai, M.; Lentzen, G.; Park, C.B. Inhibition of β-amyloid peptide aggregation and neurotoxicity by α-d-mannosylglycerate, a natural extremolyte. Peptides 2008, 29, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, T.; Petrassi, H.M.; Oza, V.B.; Raman, P.; Kelly, J.W.; Sacchettini, J.C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 2000, 7, 312–321. [Google Scholar]
- Yoshioka, T.; Murakami, K.; Ido, K.; Hanaki, M.; Yamaguchi, K.; Midorikawa, S.; Taniwaki, S.; Gunji, H.; Irie, K. Semisynthesis and structure-activity studies of uncarinic acid C isolated from Uncaria rhynchophylla as a specific inhibitor of the nucleation phase in amyloid β42 aggregation. J. Nat. Prod. 2016, 79, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, S.L.; Dupuis, N.F.; Lazo, N.D.; Wyttenbach, T.; Condron, M.M.; Bitan, G.; Teplow, D.B.; Shea, J.E.; Ruotolo, B.T.; Robinson, C.V.; et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 2009, 1, 326–331. [Google Scholar] [CrossRef]
- Kloniecki, M.; Jablonowska, A.; Poznanski, J.; Langridge, J.; Hughes, C.; Campuzano, I.; Giles, K.; Dadlez, M. Ion mobility separation coupled with MS detects two structural states of Alzheimer’s disease Aβ1-40 peptide oligomers. J. Mol. Biol. 2011, 407, 110–124. [Google Scholar] [CrossRef]
- Young, L.M.; Saunders, J.C.; Mahood, R.A.; Revill, C.H.; Foster, R.J.; Tu, L.H.; Raleigh, D.P.; Radford, S.E.; Ashcroft, A.E. Screening and classifying small-molecule inhibitors of amyloid formation using ion mobility spectrometry-mass spectrometry. Nat. Chem. 2015, 7, 73–81. [Google Scholar] [CrossRef]
- Li, H.; Rahimi, F.; Murakami, K.; Maiti, P.; Sinha, S.; Bitan, G. Amyloids and protein aggregation–analytical methods. Encycl. Ana. Chem.: Appl., Theory Instrum. 2009, 635–666. [Google Scholar] [CrossRef]
- Rangachari, V.; Reed, D.K.; Moore, B.D.; Rosenberry, T.L. Secondary structure and interfacial aggregation of amyloid-β(1–40) on sodium dodecyl sulfate micelles. Biochemistry 2006, 45, 8639–8648. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, V.; Moore, B.D.; Reed, D.K.; Sonoda, L.K.; Bridges, A.W.; Conboy, E.; Hartigan, D.; Rosenberry, T.L. Amyloid-β(1–42) rapidly forms protofibrils and oligomers by distinct pathways in low concentrations of sodium dodecylsulfate. Biochemistry 2007, 46, 12451–12462. [Google Scholar] [CrossRef] [PubMed]
- Irie, Y.; Murakami, K.; Hanaki, M.; Hanaki, Y.; Suzuki, T.; Monobe, Y.; Takai, T.; Akagi, K.I.; Kawase, T.; Hirose, K.; et al. Synthetic models of quasi-stable amyloid β40 oligomers with significant neurotoxicity. ACS Chem. Neurosci. 2017, 8, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.W.; Tsai, C.W.; Mong, M.C.; Yin, M.C. Maslinic acid protected PC12 cells differentiated by nerve growth factor against β-amyloid-induced apoptosis. J. Agri. Food Chem. 2015, 63, 10243–10249. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, R.; Dierssen, M. Therapeutic approaches in the improvement of cognitive performance in Down syndrome: Past, present, and future. Prog. Brain Res. 2012, 197, 1–14. [Google Scholar] [PubMed]
- Salloway, S.; Sperling, R.; Keren, R.; Porsteinsson, A.P.; van Dyck, C.H.; Tariot, P.N.; Gilman, S.; Arnold, D.; Abushakra, S.; Hernandez, C.; et al. A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology 2011, 77, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef]
- Chow, S.-C.; Chiu, S.-T. A note on design and analysis of clinical trials. Drug Des.: Open Access 2013, 2, 102. [Google Scholar]
- Chin, D.; Huebbe, P.; Pallauf, K.; Rimbach, G. Neuroprotective properties of curcumin in Alzheimer’s disease--merits and limitations. Curr. Med. Chem. 2013, 20, 3955–3985. [Google Scholar] [CrossRef]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Bacteria, colonic fermentation, and gastrointestinal health. J. AOAC Int. 2012, 95, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type β-amyloid neuropathological mechanisms. Exp. Rev. Neurother. 2018, 18, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fak, F.; Jucker, M.; Lasser, T.; et al. Reduction of Aβ amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Donovan, J.L. Pharmacokinetics and metabolism of dietary flavonoids in humans. Free Radic. Res. 2004, 38, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Krizkova, J.; Burdova, K.; Stiborova, M.; Kren, V.; Hodek, P. The effects of selected flavonoids on cytochromes P450 in rat liver and small intestine. Interdiscip. Toxicol. 2009, 2, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, T.; Hiza, A.; Nakayama, M.; Inai, M.; Oyama, D.; Koide, H.; Shimizu, K.; Wakimoto, T.; Harada, N.; Tsukada, H.; et al. PET imaging of nobiletin based on a practical total synthesis. Chem. Commun. 2011, 47, 2868–2870. [Google Scholar] [CrossRef] [PubMed]
- Cote, C.D.; Rasmussen, B.A.; Duca, F.A.; Zadeh-Tahmasebi, M.; Baur, J.A.; Daljeet, M.; Breen, D.M.; Filippi, B.M.; Lam, T.K. Resveratrol activates duodenal Sirt1 to reverse insulin resistance in rats through a neuronal network. Nat. Med. 2015, 21, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Duca, F.A.; Cote, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 2015, 21, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Mouse | Aβ Pathology | Memory Loss | Remarks | Ref. |
|---|---|---|---|---|---|
| (+)-Taxifolin | Tg-SwDI | SP, oligomer decreased | Improved (MWM) | Impaired CBF restored | [49,50] |
| Morin | APPswe/PS1dE9 | SP, Aβ production decreased | Improved (MWM) | Tau-P decreased | [67] |
| (R)-Apomorphine | 3xTg-AD | SP, intracellular Aβ decreased | Improved (MWM) | Tau-P decreased | [72,73] |
| Drug | Patient | Enrollment | Phase | Outcome | ClinicalTrials ID |
|---|---|---|---|---|---|
| EGCG | Early AD | 21 | II, III | Insufficient efficacy | NCT00951834 |
| scyllo-inositol (ELND005) | Moderate to severe AD | 350 | II | Insufficient efficacy | NCT01735630 |
| Curcumin C3 Complex | Mild to moderate AD | 33 | II | Insufficient efficacy | NCT00099710 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murakami, K.; Irie, K. Three Structural Features of Functional Food Components and Herbal Medicine with Amyloid β42 Anti-Aggregation Properties. Molecules 2019, 24, 2125. https://doi.org/10.3390/molecules24112125
Murakami K, Irie K. Three Structural Features of Functional Food Components and Herbal Medicine with Amyloid β42 Anti-Aggregation Properties. Molecules. 2019; 24(11):2125. https://doi.org/10.3390/molecules24112125
Chicago/Turabian StyleMurakami, Kazuma, and Kazuhiro Irie. 2019. "Three Structural Features of Functional Food Components and Herbal Medicine with Amyloid β42 Anti-Aggregation Properties" Molecules 24, no. 11: 2125. https://doi.org/10.3390/molecules24112125
APA StyleMurakami, K., & Irie, K. (2019). Three Structural Features of Functional Food Components and Herbal Medicine with Amyloid β42 Anti-Aggregation Properties. Molecules, 24(11), 2125. https://doi.org/10.3390/molecules24112125