Normal Mode Analysis as a Routine Part of a Structural Investigation


Abstract
:1. Introduction
2. Normal Mode Analysis
2.1. The Elastic Network Model
2.2. Concerns and Limitations
2.3. Web-Based Tools Using ENMs
3. Applications
3.1. Studies on Overall Flexible Motions
3.2. Correlations with Ligand Binding and Catalytic Activation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANM | Anisotropic Network Model |
| BPTI | Bovine Pancreatic Trypsin Inhibitor |
| EM | Electron Microscopy |
| ENM | Elastic Network Model |
| GNM | Gaussian Network Model |
| MD | Molecular Dynamics |
| MMTK | Molecular Modeling Toolkit |
| NMA | Normal Mode Analysis |
| NMR | Nuclear Magnetic Resonance |
| PCA | Principal Components Analysis |
| PDB | Protein Data Bank |
| RMSD | Root-Mean Square Deviation |
| RTB | Rotating-Translating Blocks |
| SAXS | Small Angle X-ray Scattering |
References
- Case, D.A. Normal mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 1994, 4, 285–290. [Google Scholar] [CrossRef]
- Brooks, B.R.; Janežič, D.; Karplus, M. Harmonic analysis of large systems. I. Methodology. J. Comput. Chem. 1995, 16, 1522–1542. [Google Scholar] [CrossRef]
- Ma, J. Usefulness and Limitations of Normal Mode Analysis in Modeling Dynamics of Biomolecular Complexes. Structure 2005, 13, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skjaerven, L.; Hollup, S.M.; Reuter, N. Normal mode analysis for proteins. J. Mol. Struct. THEOCHEM 2009, 898, 42–48. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.R.; Bakan, A.; Shrivastava, I.H. Normal Mode Analysis of Biomolecular Structures: Functional Mechanisms of Membrane Proteins. Chem. Rev. 2010, 110, 1463–1497. [Google Scholar] [CrossRef] [PubMed]
- Dykeman, E.C.; Sankey, O.F. Normal mode analysis and applications in biological physics. J. Phys. Condens. Matter 2010, 22, 423202. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Sanejouand, Y.H. On the relationship between low-frequency normal modes and the large-scale conformational changes of proteins. Arch. Biochem. Biophys. 2015, 567, 59–65. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Chacón, P. New generation of elastic network models. Curr. Opin. Struct. Biol. 2016, 37, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.P.; Reuter, N. Conservation of intrinsic dynamics in proteins—What have computational models taught us? Curr. Opin. Struct. Biol. 2018, 50, 75–81. [Google Scholar] [CrossRef]
- Cui, Q.; Bahar, I. (Eds.) Normal Mode Analysis. Theory and Applications to Biological and Chemical Systems; Chapman & Hall/CRC: Boca Raton, FL, USA, 2006. [Google Scholar]
- Suhre, K.; Sanejouand, Y.H. On the potential of normal-mode analysis for solving difficult molecular-replacement problems. Acta Cryst. 2004, D60, 796–799. [Google Scholar] [CrossRef]
- Delarue, M. Dealing with structural variability in molecular replacement and crystallographic refinement through normal-mode analysis. Acta Cryst. 2008, D64, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Diamond, R. On the Use of Normal Modes in Thermal Parameter Refinement: Theory and Application to the Bovine Pancreatic Trypsin Inhibitor. Acta Cryst. 1990, A46, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Kidera, A.; Gō, N. Refinement of protein dynamic structure: Normal mode refinement. Proc. Natl. Acad. Sci. USA 1990, 87, 3718–3772. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Melton, J.S.; Sorensen, D.C.; Phillips, G.N., Jr. Dynamics of Proteins in Crystals: Comparison of Experiment with Simple Models. Biophys. J. 2002, 83, 723–732. [Google Scholar] [CrossRef] [Green Version]
- Delarue, M.; Dumas, P. On the use of low-frequency normal modes to enforce collective movements in refining macromolecular structural models. Proc. Natl. Acad. Sci. USA 2004, 101, 6957–6962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, G.F.; Brunger, A.T.; Levitt, M. Combining Efficient Conformational Sampling with a Deformable Elastic Network Model Facilitates Structure Refinement at Low Resolution. Structure 2007, 15, 1630–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, B.K.; Chen, X.; Lu, M.; Vyas, N.K.; Quiocho, F.A.; Wang, Q.; Ma, J. Normal mode refinement of anisotropic thermal parameters for a supramolecular complex at 3.42-Å crystallographic resolution. Proc. Natl. Acad. Sci. USA 2007, 104, 7869–7874. [Google Scholar] [CrossRef]
- Tama, F.; Miyashita, O.; Brooks, C.L., III. Flexible Multi-scale Fitting of Atomic Structures into Low-resolution Electron Density Maps with Elastic Network Normal Mode Analysis. J. Mol. Biol. 2004, 337, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Navaza, J.; Sanejouand, Y.H. NORMA: A tool for flexible fitting of high-resolution protein structures into low-resolution electron-microscopy-derived density maps. Acta Cryst. 2006, D62, 1098–1100. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W. Accurate Flexible Fitting of High-Resolution Protein Structures into Cryo-Electron Microscopy Maps Using Coarse-Grained Pseudo-Energy Minimization. Biophys. J. 2011, 100, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Tekpinar, M. Accurate Flexible Fitting of High-Resolution Protein Structures to Small-Angle X-Ray Scattering Data Using a Coarse-Grained Model with Implicit Hydration Shell. Biophys. J. 2011, 101, 2981–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panjkovich, A.; Svergun, D.I. Deciphering conformational transitions of proteins by small angle X-ray scattering and normal mode analysis. Phys. Chem. Chem. Phys. 2016, 18, 5707. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, H. Classical Mechanics, 2nd ed.; Addison-Wesley: Reading, MA, USA, 1980; pp. 243–274. [Google Scholar]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar] [PubMed]
- González, M.A. Force fields and molecular dynamics simulations. Collect. SFN 2011, 12, 169–200. [Google Scholar] [CrossRef]
- Knapp, E.W.; Fischer, S.F.; Parak, F. Protein Dynamics from Mössbauer Spectra. The Temperature Dependence. J. Phys. Chem. 1982, 86, 5042–5047. [Google Scholar] [CrossRef]
- Zaccai, G. How Soft Is a Protein? A Protein Dynamics Force Constant Measured by Neutron Scattering. Science 2000, 288, 1604–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, J.H.; Novikov, V.N.; Gregory, R.B.; Curtis, J.E.; Chowdhuri, Z.; Sokolov, A.P. Onsets of Anharmonicity in Protein Dynamics. Phys. Rev. Lett. 2005, 95, 038101. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B., Jr.; Decius, J.C.; Cross, P.C. Molecular Vibrations: The Theory of Infrared and Raman Vibrational Spectra; McGraw-Hill: New York, NY, USA, 1955. [Google Scholar]
- Shimanouchi, T. Stable conformations of polymer chains and model compound molecules. Discuss. Faraday Soc. 1970, 49, 60–69. [Google Scholar] [CrossRef]
- Itoh, K.; Shimanouchi, T. Vibrational frequencies and modes of α-helix. Biopolymers 1970, 9, 383–399. [Google Scholar] [CrossRef]
- Levy, R.M.; Karplus, M. Vibrational Approach to the Dynamics of an α-helix. Biopolymers 1979, 18, 2465–2495. [Google Scholar] [CrossRef]
- Noguti, T.; Gō, N. Collective variable description of small-amplitude conformational fluctuations in a globular protein. Nature 1982, 296, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.; Karplus, M. Harmonic dynamics of proteins: Normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA 1983, 80, 6571–6575. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.; Sander, C.; Stern, P.S. The normal modes of a protein: Native bovine pancreatic trypsin inhibitor. Int. J. Quant. Chem. 1983, 24, 181–199. [Google Scholar] [CrossRef]
- Harrison, R.W. Vibrational Calculation of the Normal Modes of a Large Macromolecule: Methods and some Initial Results. Biopolymers 1984, 23, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.; Sander, C.; Stern, P.S. Protein Normal-mode Dynamics: Trypsin Inhibitor, Crambin, Ribonuclease and Lysozyme. J. Mol. Biol. 1985, 181, 423–447. [Google Scholar] [CrossRef]
- Brooks, B.; Karplus, M. Normal modes for specific motions of macromolecules: Application to the hinge-bending mode of lysozyme. Proc. Natl. Acad. Sci. USA 1985, 82, 4995–4999. [Google Scholar] [CrossRef] [PubMed]
- Seno, Y.; Gō, N. Deoxymyoglobin Studied by the Conformational Normal Mode Analysis I. Dynamics of Globin and the Heme–Globin Interaction. J. Mol. Biol. 1990, 216, 95–109. [Google Scholar] [CrossRef]
- Seno, Y.; Gō, N. Deoxymyoglobin Studied by the Conformational Normal Mode Analysis II. The Conformational Change upon Oxygenation. J. Mol. Biol. 1990, 216, 111–126. [Google Scholar] [CrossRef]
- Zheng, W. Toward decrypting the allosteric mechanism of the ryanodine receptor based on coarse-grained structural and dynamic modeling. Proteins 2015, 83, 2307–2318. [Google Scholar] [CrossRef]
- Lee, B.H.; Jo, S.; Choi, M.k.; Kim, M.H.; Choi, J.B.; Kim, M.K. Normal mode analysis of Zika virus. Comput. Biol. Chem. 2018, 72, 53–61. [Google Scholar] [CrossRef]
- Tama, F.; Brooks, C.L., III. Diversity and Identity of Mechanical Properites of Icosahedral Viral Capsids Studied with Elastic Network Normal Mode Analysis. J. Mol. Biol. 2005, 345, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Van Vlijmen, H.W.T.; Karplus, M. Normal Mode Calculations of Icosahedral Viruses with Full Dihedral Flexibility by Use of Molecular Symmetry. J. Mol. Biol. 2005, 350, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Cui, Q. Analysis of Functional Motions in Brownian Molecular Machines with an Efficient Block Normal Mode Approach: Myosin-II and Ca2+-ATPase. Biophys. J. 2004, 86, 743–763. [Google Scholar] [CrossRef]
- Zheng, W.; Brooks, B. Identification of Dynamical Correlations within the Myosin Motor Domain by the Normal Mode Analysis of an Elastic Network Model. J. Mol. Biol. 2005, 346, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Thirumalai, D. Coupling between Normal Modes Drives Protein Conformational Dynamics: Illustrations Using Allosteric Transitions in Myosin II. Biophys. J. 2009, 96, 2128–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Q.; Li, G.; Ma, J.; Karplus, M. A Normal Mode Analysis of Structural Plasticity in the Biomolecular Motor F1-ATPase. J. Mol. Biol. 2004, 340, 345–372. [Google Scholar] [CrossRef]
- Ma, J.; Karplus, M. The allosteric mechanism of the chaperonin GroEL: A dynamic analysis. Proc. Natl. Acad. Sci. USA 1998, 95, 8502–8507. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Sigler, P.B.; Xu, Z.; Karplus, M. A Dynamic Model for the Allosteric Mechanism of GroEL. J. Mol. Biol. 2000, 302, 303–313. [Google Scholar] [CrossRef]
- Keskin, O.; Bahar, I.; Flatow, D.; Covell, D.G.; Jernigan, R.L. Molecular Mechanisms of Chaperonin GroEL–GroES Function. Biochemistry 2002, 41, 491–501. [Google Scholar] [CrossRef]
- Tama, F.; Valle, M.; Frank, J.; Brooks, C.L., III. Dynamic reorganization of the functionally active ribosome explored by normal mode analysis and cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 9319–9323. [Google Scholar] [CrossRef] [Green Version]
- Yan, A.; Wang, Y.; Klochkowski, A.; Jernigan, R.L. Effects of Protein Subunits Removal on the Computed Motions of Partial 30S Structures of the Ribosome. J. Chem. Theory Comput. 2008, 4, 1757–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurkcuoglu, O.; Kurkcuoglu, Z.; Doruker, P.; Jernigan, R.L. Collective dynamics of the ribosomal tunnel revealed by elastic network modeling. Proteins 2009, 75, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.k.; Jo, S.; Lee, B.H.; Kim, M.H.; Choi, J.B.; Kim, K.; Kim, M.K. Dynamic characteristics of a flagellar motor protein analyzed using an elastic network model. J. Mol. Graph. Model. 2017, 78, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Lezon, T.R.; Sali, A.; Bahar, I. Global Motions of the Nuclear Pore Complex: Insights from Elastic Network Models. PLoS Comput. Biol. 2009, 5, e1000496. [Google Scholar] [CrossRef] [PubMed]
- Tirion, M.M. Large Amplitude Elastic Motions in Proteins from a Single-Parameter, Atomic Analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, S.; Jernigan, R.L. Estimation of Effective Interresidue Contact Energies from Protein Crystal Structures: Quasi-Chemical Approximation. Macromolecules 1985, 18, 534–552. [Google Scholar] [CrossRef]
- Zheng, W. A Unification of the Elastic Network Model and the Gaussian Network Model for Optimal Description of Protein Conformational Motions and Fluctuations. Biophys. J. 2008, 94, 3853–3857. [Google Scholar] [CrossRef] [Green Version]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997, 2, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Haliloglu, T.; Bahar, I.; Erman, B. Gaussian Dynamics of Folded Proteins. Phys. Rev. Lett. 1997, 79, 3090–3093. [Google Scholar] [CrossRef]
- Hinsen, K. The Molecular Modeling Toolkit: A New Approach to Molecular Simulations. J. Comp. Chem. 2000, 21, 79–85. [Google Scholar] [CrossRef]
- Bahar, I.; Rader, A.J. Coarse-grained normal mode analysis in structural biology. Curr. Opin. Struct. Biol. 2005, 15, 586–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chennubhotla, C.; Rader, A.J.; Yang, L.W.; Bahar, I. Elastic network models for understanding biomolecular machinery: From enzymes to supramolecular assemblies. Phys. Biol. 2005, 2, S173–S180. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.L.; Song, G. Generalized spring tensor models for protein fluctuation dynamics and conformation changes. BMC Struct. Biol. 2010, 10 (Suppl. 1), S3. [Google Scholar] [CrossRef] [PubMed]
- Durand, P.; Trinquier, G.; Sanejouand, Y.H. A New Approach for Determining Low-Frequency Normal Modes in Macromolecules. Biopolymers 1994, 34, 759–771. [Google Scholar] [CrossRef]
- Suhre, K.; Sanejouand, Y.H. ElNémo: A normal mode web server for protein movements analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004, 32, W610–W614. [Google Scholar] [CrossRef] [PubMed]
- Balog, E.; Becker, T.; Oettl, M.; Lechner, R.; Daniel, R.; Finney, J.; Smith, J.C. Direct Determination of Vibrational Density of States Change on Ligand Binding to a Protein. Phys. Rev. Lett. 2004, 93, 028103. [Google Scholar] [CrossRef] [PubMed]
- Cusack, S.; Smith, J.; Finney, J.; Tidor, B.; Karplus, M. Inelastic Neutron Scattering Analysis of Picosecond Internal Protein Dynamics. Comparison of Harmonic Theory with Experiment. J. Mol. Biol. 1988, 202, 903–908. [Google Scholar] [CrossRef]
- Réat, V.; Dunn, R.; Ferrand, M.; Finney, J.L.; Daniel, R.M.; Smith, J.C. Solvent dependence of dynamic transitions in protein solutions. Proc. Natl. Acad. Sci. USA 2000, 97, 9961–9966. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.; Kuczera, K.; Karplus, M. Dynamics of myoglobin: Comparison of simulation results with neutron scattering spectra. Proc. Natl. Acad. Sci. USA 1990, 87, 1601–1605. [Google Scholar] [CrossRef]
- Smith, J.C. Protein dynamics: Comparison of simulations with inelastic neutron scattering experiments. Q. Rev. Biophys. 1991, 24, 227–291. [Google Scholar] [CrossRef]
- McQuarrie, D.A. Statistical Mechanics; Harper & Row: New York, NY, USA, 1976. [Google Scholar]
- Rueda, M.; Chacón, P.; Orozco, M. Thorough Validation of Protein Normal Mode Analysis: A Comparative Study with Essential Dynamics. Structure 2007, 15, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda, M.; Ferrer-Costa, C.; Meyer, T.; Pérez, A.; Camps, J.; Hospital, A.; Gelpi, J.L.; Orozco, M. A consensus view of protein dynamics. Proc. Natl. Acad. Sci. USA 2007, 104, 796–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skjaerven, L.; Martinez, A.; Reuter, N. Principal component and normal mode analysis of proteins; a quantitative comparison using the GroEL subunit. Proteins 2011, 79, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Marques, O.; Sanejouand, Y.H. Hinge-Bending Motion in Citrate Synthase Arising From Normal Mode Calculations. Proteins 1995, 23, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Hinsen, K. Analysis of Domain Motions by Approximate Normal Mode Calculations. Proteins 1998, 33, 417–429. [Google Scholar] [CrossRef]
- Hinsen, K.; Thomas, A.; Field, M.J. Analysis of Domain Motions in Large Proteins. Proteins 1999, 34, 369–382. [Google Scholar] [CrossRef]
- Mouawad, L.; Perahia, D. Motions in Hemoglobin Studied by Normal Mode Analysis and Energy Minimization: Evidence for the Existence of Tertiary T-like Quaternary R-like Intermediate Structures. J. Mol. Biol. 1996, 258, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Trobi, D.; Bahar, I. Allosteric Changes in Protein Structure Computed by a Simple Mechanical Model: Hemoglobin T ↔ R2 Transition. J. Mol. Biol. 2003, 333, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Brooks, B.R.; Thirumalai, D. Allosteric Transitions in the Chaperonin GroEL are Captured by a Dominant Normal Mode that is Most Robust to Sequence Variations. Biophys. J. 2007, 93, 2289–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Rader, A.J.; Bahar, I.; Jernigan, R.L. Global ribosome motions revealed with elastic network model. J. Struct. Biol. 2004, 147, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.W.; Bahar, I. Coupling between Catalytic Site and Collective Dynamics: A Requirement for Mechanochemical Activity of Enzymes. Structure 2005, 13, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tama, F.; Sanejouand, Y.H. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001, 14, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tobi, D.; Bahar, I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc. Natl. Acad. Sci. USA 2005, 102, 18908–18913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Avraham, D. Vibrational normal-mode spectrum of globular proteins. Phys. Rev. B 1993, 47, 14559–14560. [Google Scholar] [CrossRef] [PubMed]
- Doruker, P.; Jernigan, R.L.; Bahar, I. Dynamics of Large Proteins through Hierarchical Levels of Coarse-Grained Structures. J. Comput. Chem. 2002, 23, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ma, J. The Role of Shape in Determining Molecular Motions. Biophys. J. 2005, 89, 2395–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Brooks, B.R.; Thirumalai, D. Low-frequency normal modes that describe allosteric transitions in biological nanomachines are robust to sequence variations. Proc. Natl. Acad. Sci. USA 2006, 103, 7664–7669. [Google Scholar] [CrossRef] [Green Version]
- Lezon, T.R.; Banavar, J.R.; Lesk, A.M.; Maritan, A. What Determines the Spectrum of Protein Native State Structures? Proteins 2006, 63, 273–277. [Google Scholar] [CrossRef]
- Nicolay, S.; Sanejouand, Y.H. Functional Modes of Proteins Are among the Most Robust. Phys. Rev. Lett. 2006, 96, 078104. [Google Scholar] [CrossRef]
- Tama, F.; Brooks, C.L., III. Symmetry, Form, and Shape: Guiding Principles for Robustness in Macromolecular Machines. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 115–133. [Google Scholar] [CrossRef]
- Bertaccini, E.J.; Trudell, J.R.; Lindahl, E. Normal-Mode Analysis of the Glycine Alpha1 Receptor by Three Separate Methods. J. Chem. Inf. Model. 2007, 47, 1572–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Song, G.; Carriquiry, A.; Jernigan, R.L. Close Correspondence between the Motions from Principal Component Analysis of Multiple HIV-1 Protease Structures and Elastic Network Modes. Structure 2008, 16, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondrashov, D.A.; Van Wynsberghe, A.W.; Bannen, R.M.; Cui, Q.; Phillips, G.N., Jr. Protein Structural Variation in Computational Models and Crystallographic Data. Structure 2007, 15, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.A.; Borko, V.; Pavlović, J.; Kutejová, E.; Bauerová-Hlinková, V. Disease-associated mutations alter the dynamic motion of the N-terminal domain of the human cardiac ryanodine receptor. J. Biomol. Struct. Dyn. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Dutta, A.; Mao, W.; Liu, Y.; Chennubhotla, C.; Lezon, T.R.; Bahar, I. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 2014, 30, 2681–2683. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Field, M.J.; Perahia, D. Analysis of the Low-frequency Normal Modes of the R State of Aspartate Transcarbamylase and a Comparison with the T State Modes. J. Mol. Biol. 1996, 261, 490–506. [Google Scholar] [CrossRef] [PubMed]
- Sanejouand, Y.H. On the role of CD4 conformational change in the HIV-cell fusion process. Acad. Sci. Paris 1997, 320, 163–170. [Google Scholar]
- Van Vlijmen, H.W.T.; Karplus, M. Analysis of Calculated Normal Modes of a Set of Native and Partially Unfolded Proteins. J. Phys. Chem. B 1999, 103, 3009–3021. [Google Scholar] [CrossRef]
- Zheng, W.; Doniach, S. A comparative study of motor-protein motions by using a simple elastic-network model. Proc. Natl. Acad. Sci. USA 2003, 100, 13253–13258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Brooks, B. Normal-Modes-Based Prediction of Protein Conformational Changes Guided by Distance Constraints. Biophys. J. 2005, 88, 3109–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Brooks, B.R.; Hummer, G. Protein conformational transitions explored by mixed elastic network models. Proteins 2007, 69, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Azuara, C.; Koehl, P.; Delarue, M. NOMAD-Ref: Visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res. 2006, 34, W52–W56. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.W.; Rader, A.J.; Liu, X.; Jursa, C.J.; Chen, S.C.; Karimi, H.A.; Bahar, I. o GNM: Online computation of structural dynamics using the Gaussian Network Model. Nucleic Acids Res. 2006, 34, W24–W31. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.W.; Liu, X.; Jursa, C.J.; Holliman, M.; Rader, A.J.; Karimi, H.A.; Bahar, I. iGNM: A database of protein functional motions based on Gaussian Network Model. Bioinformatics 2005, 21, 2978–2987. [Google Scholar] [CrossRef]
- Li, H.; Chang, Y.Y.; Yang, L.W.; Bahar, I. IGNM 2.0: The Gaussian network model database for biomolecular structural dynamics. Nucleic Acids Res. 2016, 44, D415–D422. [Google Scholar] [CrossRef]
- Li, H.; Chang, Y.Y.; Lee, J.Y.; Bahar, I.; Yang, L.W. DynOmics: Dynamics of structural proteome and beyond. Nucleic Acids Res. 2017, 45, W374–W380. [Google Scholar]
- Eyal, E.; Yang, L.W.; Bahar, I. Anisotropic network model: Systematic evaluation and a new web interface. Bioinformatics 2006, 21, 2619–2627. [Google Scholar] [CrossRef]
- Eyal, E.; Lum, G.; Bahar, I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics 2015, 31, 1487–1489. [Google Scholar] [CrossRef]
- Emekli, U.; Schneidman-Duhovny, D.; Wolfson, H.J.; Nussinov, R.; Haliloglu, T. HingeProt: Automated prediction of hinges in protein structures. Proteins 2008, 70, 1219–1227. [Google Scholar] [CrossRef]
- Flores, S.; Echols, N.; Milburn, D.; Hespenheide, B.; Keating, K.; Lu, J.; Wells, S.; Yu, E.Z.; Thorpe, M. The Database of Macromolecular Motions: New features added at the decade mark. Nucleic Acids Res. 2006, 34, D296–D301. [Google Scholar] [CrossRef] [PubMed]
- Flores, S.C.; Gerstein, M.B. FlexOracle: Predicting flexible hinges by identification of stable domains. BMC Bioinform. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Lopéz-Blanco, J.R.; Garzón, J.I.; Chacón, P. iMod: Multipurpose normal mode analysis in internal coordinates. Bioinformatics 2011, 27, 2843–2850. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef] [PubMed]
- Garzón, J.I.; Kovacs, J.; Abagyan, R.; Chacón, P. DFprot: A webtool for predicting local chain deformability. Bioinformatics 2007, 23, 901–902. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.P.; Fugelbakk, E.; Hollup, S.M.; Skjærven, L.; Cragnolini, T.; Grindhaug, S.H.; Tekle, K.M.; Reuter, N. WEBnm v2.0: Web server and services for comparing protein flexibility. BMC Bioinform. 2014, 15, 427. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Brooks, B. Probing the Local Dynamics of Nucleotide-Binding Pocket Coupled to the Global Dynamics: Myosin versus Kinesin. Biophys. J. 2005, 89, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekpinar, M.; Zheng, W. Predicting order of conformational changes during protein conformational transitions using an interpolated elastic network model. Proteins 2010, 78, 2469–2481. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gerstein, M.; Krebs, W. A database of macromolecular motions. Nucleic Acids Res. 1998, 26, 4280–4290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, W.G.; Gerstein, M. The morph server: A standardized system for analyzing and visualizing macromolecular motions in a database framework. Nucleic Acids Res. 2000, 28, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.A.; Chacón, P.; Abagyan, R. Predictions of Protein Flexibility: First-Order Measures. Proteins 2004, 56, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Joti, Y.; Nakasako, M.; Kidera, A.; Go, N. Nonlinear temperature dependence of the crystal structure of lysozyme: Correlation between coordinate shifts and thermal factors. Acta Cryst. 2002, D58, 1421–1432. [Google Scholar] [CrossRef]
- Chen, X.; Lu, M.; Poon, B.K.; Wang, Q.; Ma, J. Structural improvement of unliganded simian immunodeficiency virus gp120 core by normal-mode-based X-ray crystallographic refinement. Acta Cryst. 2009, D65, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Takayama, Y.; Nakasako, M. A few low-frequency normal modes predominantly contribute to conformational responses of hen egg white lysozyme in the tetragonal crystal to variations of molecular packing controlled by environmental humidity. Biophys. Chem. 2011, 159, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Janowski, R.; Panjikar, S.; Eddine, A.N.; Kaufmann, S.H.E.; Weiss, M.S. Structural analysis reveals DNA binding properties of Rv2827c, a hypothetical protein from Mycobacterium tuberculosis. J. Struct. Funct. Genom. 2009, 10, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Huyton, T.; Pye, V.E.; Briggs, L.C.; Flynn, T.C.; Beuron, F.; Kondo, H.; Ma, J.; Zhang, X.; Freemont, P.S. The crystal structure of murine p97/VCP at 3.6 Å. J. Struct. Biol. 2003, 144, 337–348. [Google Scholar] [CrossRef]
- Burk, D.L.; Xiong, B.; Breitbach, C.; Berghuis, A.M. Structures of aminoglycoside acetyltransferase AAC(6′)-Ii in a novel crystal form: Structural and normal-mode analyses. Acta Cryst. 2005, D61, 1273–1279. [Google Scholar] [CrossRef]
- McCoy, J.G.; Bitto, E.; Bingman, C.A.; Wesenberg, G.E.; Bannen, R.M.; Kondrashov, D.A.; Phillips, G.N., Jr. Structure and Dynamics of UDP–Glucose Pyrophosphorylase from Arabidopsis thaliana with Bound UDP–Glucose and UTP. J. Mol. Biol. 2007, 366, 830–841. [Google Scholar] [CrossRef]
- Bitto, E.; Bingman, C.A.; Kondrashov, D.A.; McCoy, J.G.; Bannen, R.M.; Wesenberg, G.E.; Phillips, G.N., Jr. Structure and dynamics of γ-SNAP: Insight into flexibility of proteins from the SNAP family. Proteins 2008, 70, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Bitto, E.; Bingman, C.A.; Bittova, L.; Kondrashov, D.A.; Bannen, R.M.; Fox, B.G.; Markley, J.L.; Phillips, G.N., Jr. Structure of Human J-type Co-chaperone HscB Reveals a Tetracysteine Metal-binding Domain. J. Biol. Chem. 2008, 283, 30184–30192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, C.L.; Waterman, D.G.; Koonin, E.V.; Walters, A.D.; Chong, J.P.J.; Isupov, M.N.; Lebedev, A.A.; Bunka, D.H.J.; Stockley, P.G.; Oritiz-Lombardía, M.; et al. Conformational flexibility and molecular interactions of an archaeal homologue of the Schwachman-Bodian-Diamond syndrome protein. BMC Struct. Biol. 2009, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Fernández, D.; Boix, E.; Pallarès, I.; Avilés, F.X.; Vendrell, J. Analysis of a New Crystal Form of Procarboxypeptidase B: Further Insights into the Catalytic Mechanism. Biopolymers 2010, 93, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Kladova, A.V.; Bursakov, S.A.; Gavel, O.Y.; Calvete, J.J.; Shnyrov, V.L.; Moura, I.; Moura, J.J.G.; Romão, M.J.; Trincão, J. Crystal structure of the zinc-, cobalt-, and iron-containing adenylate kinase from Desulfovibrio gigas: A novel metal-containing adenylate kinase from Gram-negative bacteria. J. Biol. Inorg. Chem. 2011, 16, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lin, K.; Gao, L.; Chen, L.; Shi, X.; Wu, G. Crystal structure of the armadillo repeat domain of adenomatous polyposis coli which reveals its inherent flexibility. Biochem. Biophys. Res. Commun. 2011, 412, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Dian, C.; Bernaudat, F.; Langer, K.; Oliva, M.F.; Fornerod, M.; Schoehn, G.; Müller, C.W.; Petosa, C. Structure of a Truncation Mutant of the Nuclear Export Factor CRM1 Provides Insights into the Auto-Inhibitory Role of Its C-Terminal Helix. Structure 2013, 21, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Erskine, P.T.; Fokas, A.; Muriithi, C.; Rehman, H.; Yates, L.A.; Bowyer, A.; Findlow, I.S.; Hagan, R.; Werner, J.M.; Miles, A.J.; et al. X-ray, spectroscopic and normal-mode dynamics of calexcitin: structure–function studies of a neuronal calcium-signalling protein. Acta Cryst. 2015, D71, 615–631. [Google Scholar] [CrossRef]
- Yates, L.A.; Durrant, B.P.; Fleurdépine, S.; Harlos, K.; Norbury, C.J.; Gilbert, R.J.C. Structural plasticity of Cid1 provides a basis for its distributive RNA terminal uridylyl transferase activity. Nucleic Acids Res. 2015, 43, 2968–2979. [Google Scholar] [CrossRef]
- Are, V.N.; Ghosh, B.; Kumar, A.; Gadre, R.; Makde, R.D. Crystal structure and dynamics of Spt16N-domain of FACT complex from Cicer arietinum. Int. J. Biol. Macromol. 2016, 88, 36–43. [Google Scholar] [CrossRef]
- Waugh, B.; Sen, U.; Banerjee, R. Crystal structure of phosphoglucomutase from Leishmania major at 3.5 Å resolution. Biochimie 2016, 121, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Sathiyamoorthy, K.; Vijayalakshmi, J.; Tirupati, B.; Fan, L.; Saper, M.A. Structural analyses of the Haemophilus influenzae peptidoglycan synthase activator LpoA suggest multiple conformations in solution. J. Biol. Chem. 2017, 292, 17626–17642. [Google Scholar] [CrossRef] [PubMed]
- Stanek, K.A.; Patterson-West, J.; Randolph, P.S.; Mura, C. Crystal structure and RNA-binding properties of an Hfq homolog from the deep-branching Aquificae: conservation of the lateral RNA-binding mode. Acta Cryst. 2017, D73, 294–315. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Chakraborty, S.D.; Biswas, S. Not all pycnodysostosis-related mutants of human cathepsin K are inactive–crystal structure and biochemical studies of an active mutant I249T. FEBS J. 2018, 285, 4265–4280. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Roldan, J.E.; Freedman, R.B.; Römer, R.A.; Wells, S.A. Rapid simulation of protein motion: Merging flexibility, rigidity and normal mode analyses. Phys. Biol. 2012, 9, 016008. [Google Scholar] [CrossRef] [PubMed]
- Trakhanov, S.; Vyas, N.K.; Luecke, H.; Kristensen, D.M.; Ma, J.; Quiocho, F.A. Ligand-Free and -Bound Structures of the Binding Protein (LivJ) of the Escherichia coli ABC Leucine/Isoleucine/Valine Transport System: Trajectory and Dynamics of the Interdomain Rotation and Ligand Specificity. Biochemistry 2005, 44, 6597–6608. [Google Scholar] [CrossRef] [PubMed]
- Akif, M.; Suhre, K.; Verma, C.; Mande, S.C. Conformational flexibility of Mycobacterium tuberculosis thioredoxin reductase: Crystal structure and normal-mode analysis. Acta Cryst. 2005, D61, 1603–1611. [Google Scholar]
- Han, B.W.; Bingman, C.A.; Mahnke, D.K.; Bannen, R.M.; Bednarek, S.Y.; Sabina, R.L.; Phillips, G.N., Jr. Membrane Association, Mechanism of Action, and Structure of Arabidopsis Embryonic Factor 1 (FAC1). J. Biol. Chem. 2009, 281, 14939–14947. [Google Scholar] [CrossRef]
- Oruganti, S.; Zhang, Y.; Li, H.; Robinson, H.; Terns, M.P.; Terns, R.M.; Yang, W.; Li, H. Alternative Conformations of the Archaeal Nop56/58-Fibrillarin Complex Imply Flexibility in Box C/D RNPs. J. Mol. Biol. 2007, 371, 1141–1150. [Google Scholar] [CrossRef]
- Schmidberger, J.W.; Wilce, J.A.; Tsang, J.S.H.; Wilce, M.C.J. Crystal Structures of the Substrate Free-enzyme, and Reaction Intermediate of the HAD Superfamily Member, Haloacid Dehalogenase DehIVa from Burkholderia cepacia MBA4. J. Mol. Biol. 2007, 368, 706–717. [Google Scholar] [CrossRef]
- Kumar, P.; Joshi, D.C.; Akif, M.; Akhter, Y.; Hasnain, S.E.; Mande, S.C. Mapping Conformational Transitions in Cyclic AMP Receptor Protein: Crystal Structure and Normal-Mode Analysis of Mycobacterium tuberculosis apo-cAMP receptor protein. Biophys. J. 2010, 98, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Mehra-Chaudhary, R.; Mick, J.; Tanner, J.J.; Henzel, M.T.; Beamer, L.J. Crystal structure of a bacterial phosphoglucomutase, an enzyme involved in the virulence of multiple human pathogens. Proteins 2011, 79, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Koike, R.; Nitanai, Y.; Minakata, S.; Maéda, Y.; Ota, M. Actin capping protein and its inhibitor CARMIL: How intrinsically disordered regions function. Phys. Biol. 2011, 8, 035005. [Google Scholar] [CrossRef] [PubMed]
- Tarnawski, M.; Krzywda, S.; Bialek, W.; Jaskolski, M.; Szczepaniak, A. Structure of the RuBisCO chaperone RbcX from the thermophilic cyanobacterium Thermosynechococcus elongatus. Acta Cryst. 2011, F67, 851–857. [Google Scholar]
- Bloudoff, K.; Rodionov, D.; Schmeing, T.M. Crystal Structures of the First, Condensation Domain of CDA Synthetase Suggest Conformational Changes during the Synthetic Cycle of Nonribosomal Peptide Synthetases. J. Mol. Biol. 2013, 425, 3137–3150. [Google Scholar] [CrossRef] [PubMed]
- Takasuka, T.E.; Bianchetti, C.M.; Tobimatsu, Y.; Bergeman, L.F.; Ralph, J.; Fox, B.G. Structure-guided analysis of catalytic specificity of the abundantly secreted chitosanase SACTE_5457 from Streptomyces Sp. SirexAA-E. Proteins 2014, 82, 1245–1257. [Google Scholar] [CrossRef]
- Danyal, K.; Rasmussen, A.J.; Keable, S.M.; Inglet, B.S.; Shaw, S.; Zadvornyy, O.A.; Duval, S.; Dean, D.R.; Raugei, S.; Peters, J.W. Fe Protein-Independent Substrate Reduction by Nitrogenase MoFe Protein Variants. Biochemistry 2015, 54, 2456–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodra, N.; Young, D.; Rosado, L.A.; Pallo, A.; Wahni, K.; De Proft, F.; Huang, J.; Van Breusegem, F.; Messens, J. Arabidopsis thaliana dehydroascorbate reductase 2: Conformational flexibility during catalysis. Sci. Rep. 2017, 7, 42494. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Li, F.; Gao, F.; Wei, J.; Lin, Y.; Zhang, Y.; Lou, J.; Liu, G.; Dong, Y.; Liu, L.; et al. Structure of tRNA-Modifying Enzyme TiaS and Motions of Its Substrate Binding Zinc Ribbon. J. Mol. Biol. 2018, 430, 4183–4194. [Google Scholar] [CrossRef] [PubMed]
- Bruccoleri, R.E.; Karplus, M.; McCammon, J.A. The Hinge-Bending Mode of a Lysozyme–Inhibitor Complex. Biopolymers 1986, 25, 1767–1802. [Google Scholar] [CrossRef]



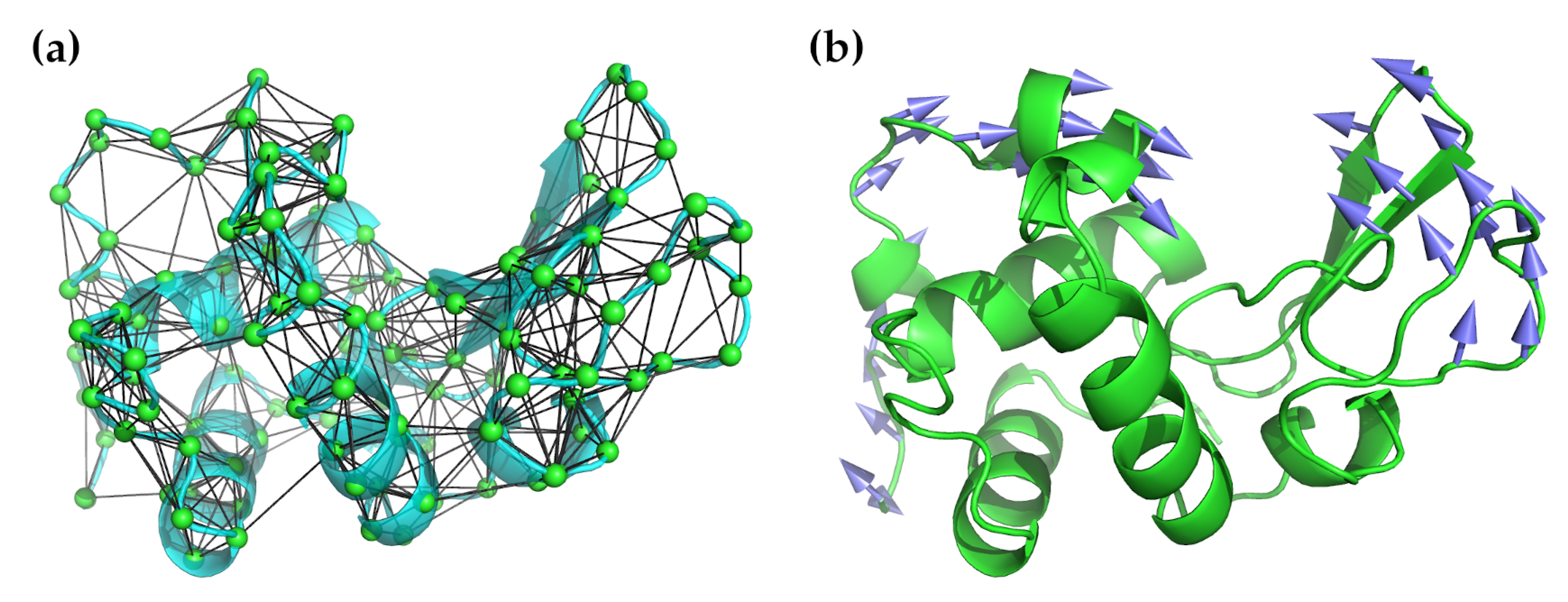{kind=link}
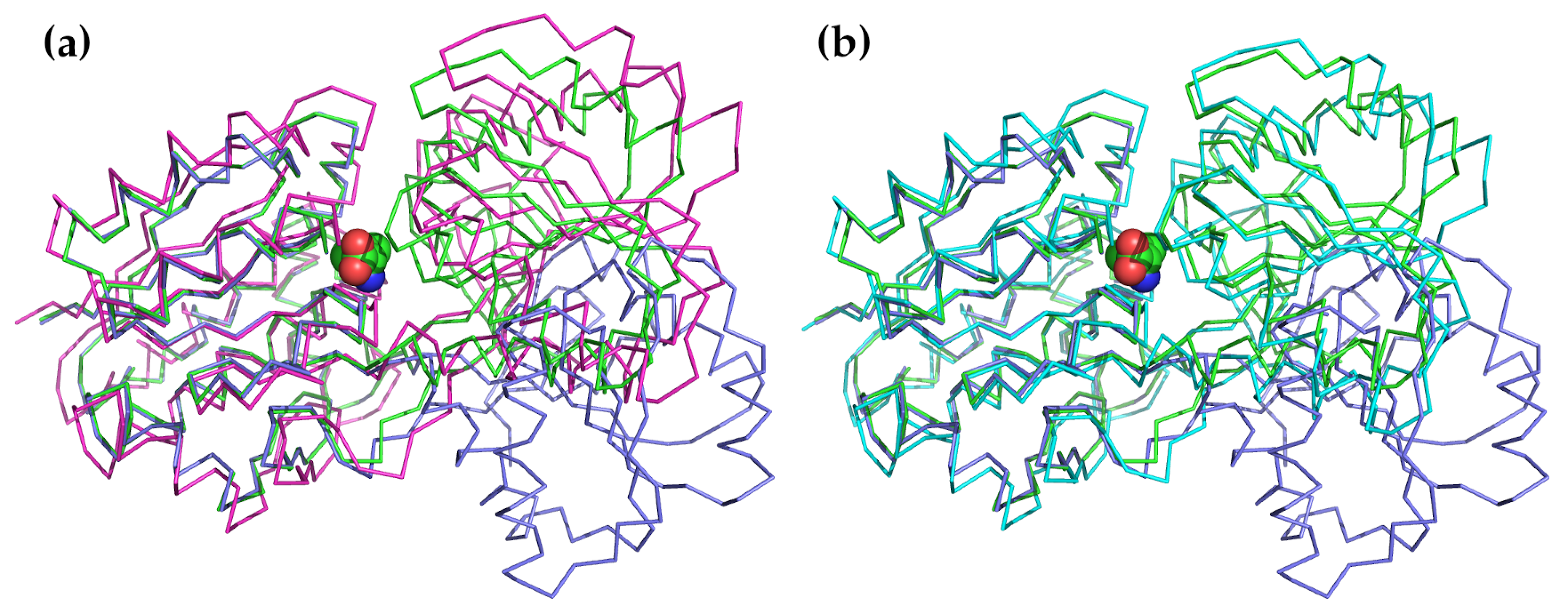{kind=link}
| Service | url | Reference |
|---|---|---|
| ElNémo | http://www.sciences.univ-nantes.fr/elnemo/ | [20,68] |
| AD-ENM | https://enm.lobos.nih.gov/index.html | [104,105,106] |
| NOMAD-Ref | http://lorentz.immstr.pasteur.fr/nomad-ref.php | [107] |
| oGNM | https://dyn.life.nthu.edu.tw/oGNM/oGNM.php | [108] |
| iGNM | http://gnm.csb.pitt.edu/index.php | [109,110] |
| DynOmics | http://gnm.csb.pitt.edu/index.php | [111] |
| ANM 2.1 | http://anm.csb.pitt.edu/ | [112,113] |
| HingeProt | http://www.prc.boun.edu.tr/appserv/prc/hingeprot/hingeprot.html | [114] |
| MolMovDB | http://molmovdb.org/ | [115,116] |
| iMODS | http://imods.chaconlab.org/ | [117,118] |
| WEBnm@ | http://apps.cbu.uib.no/webnma/home | [120] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, J.A.; Pavlović, J.; Bauerová-Hlinková, V. Normal Mode Analysis as a Routine Part of a Structural Investigation. Molecules 2019, 24, 3293. https://doi.org/10.3390/molecules24183293
Bauer JA, Pavlović J, Bauerová-Hlinková V. Normal Mode Analysis as a Routine Part of a Structural Investigation. Molecules. 2019; 24(18):3293. https://doi.org/10.3390/molecules24183293
Chicago/Turabian StyleBauer, Jacob A., Jelena Pavlović, and Vladena Bauerová-Hlinková. 2019. "Normal Mode Analysis as a Routine Part of a Structural Investigation" Molecules 24, no. 18: 3293. https://doi.org/10.3390/molecules24183293
APA StyleBauer, J. A., Pavlović, J., & Bauerová-Hlinková, V. (2019). Normal Mode Analysis as a Routine Part of a Structural Investigation. Molecules, 24(18), 3293. https://doi.org/10.3390/molecules24183293