
Structural and Thermal Analysis of Softwood Lignins from a Pressurized Hot Water Extraction Biorefinery Process and Modified Derivatives

 ,
,
Abstract
:
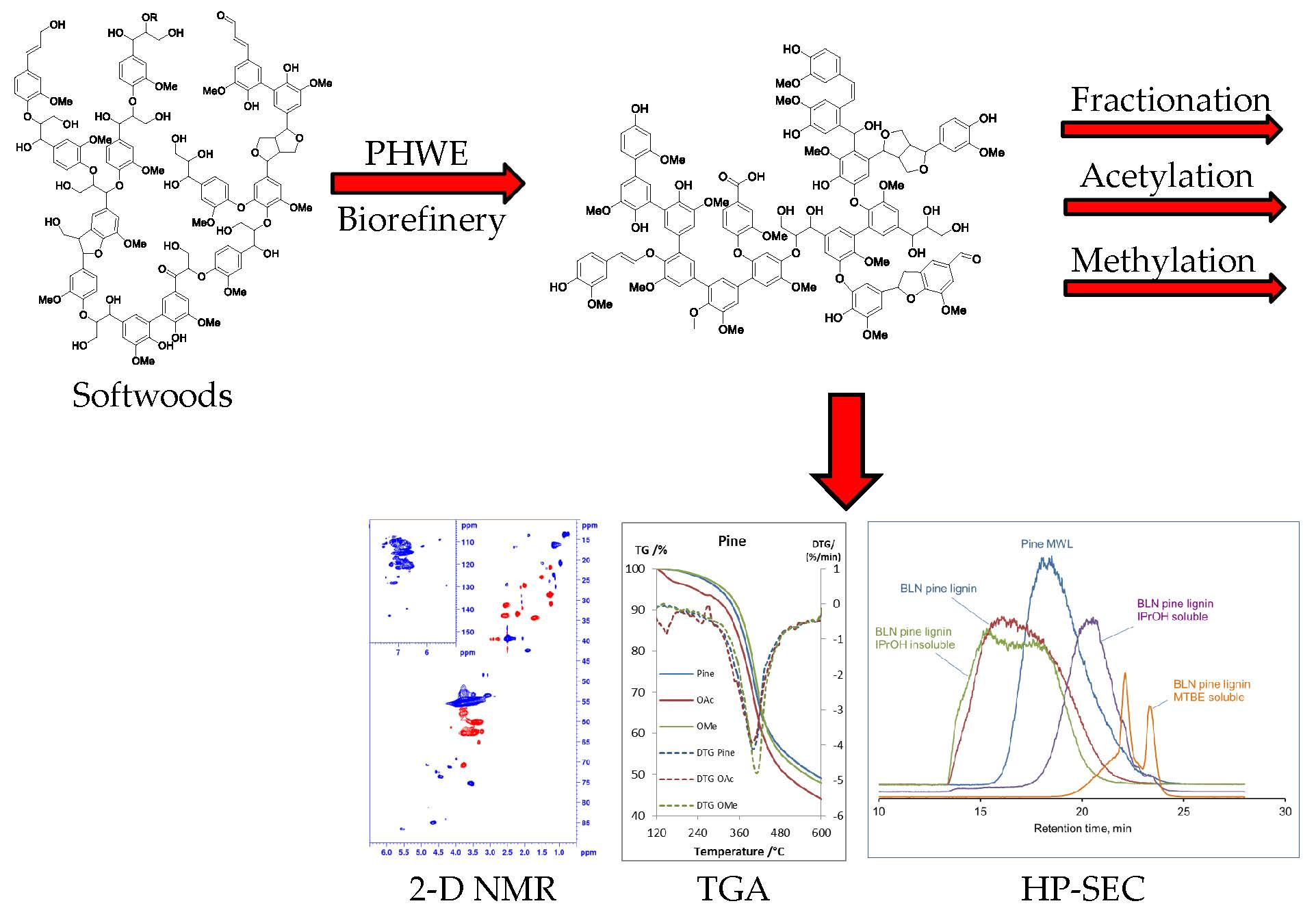
1. Introduction
2. Results and Discussion
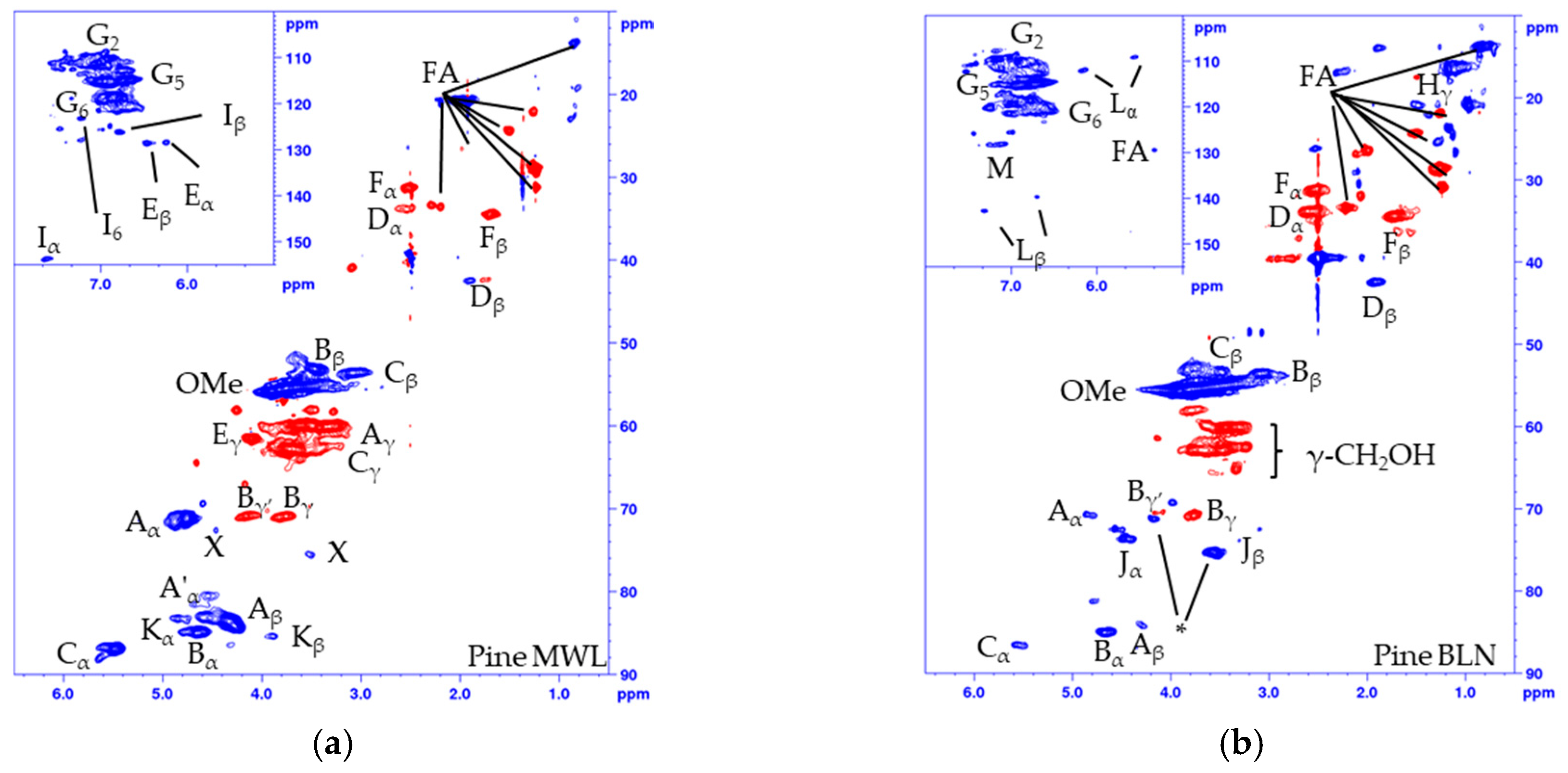
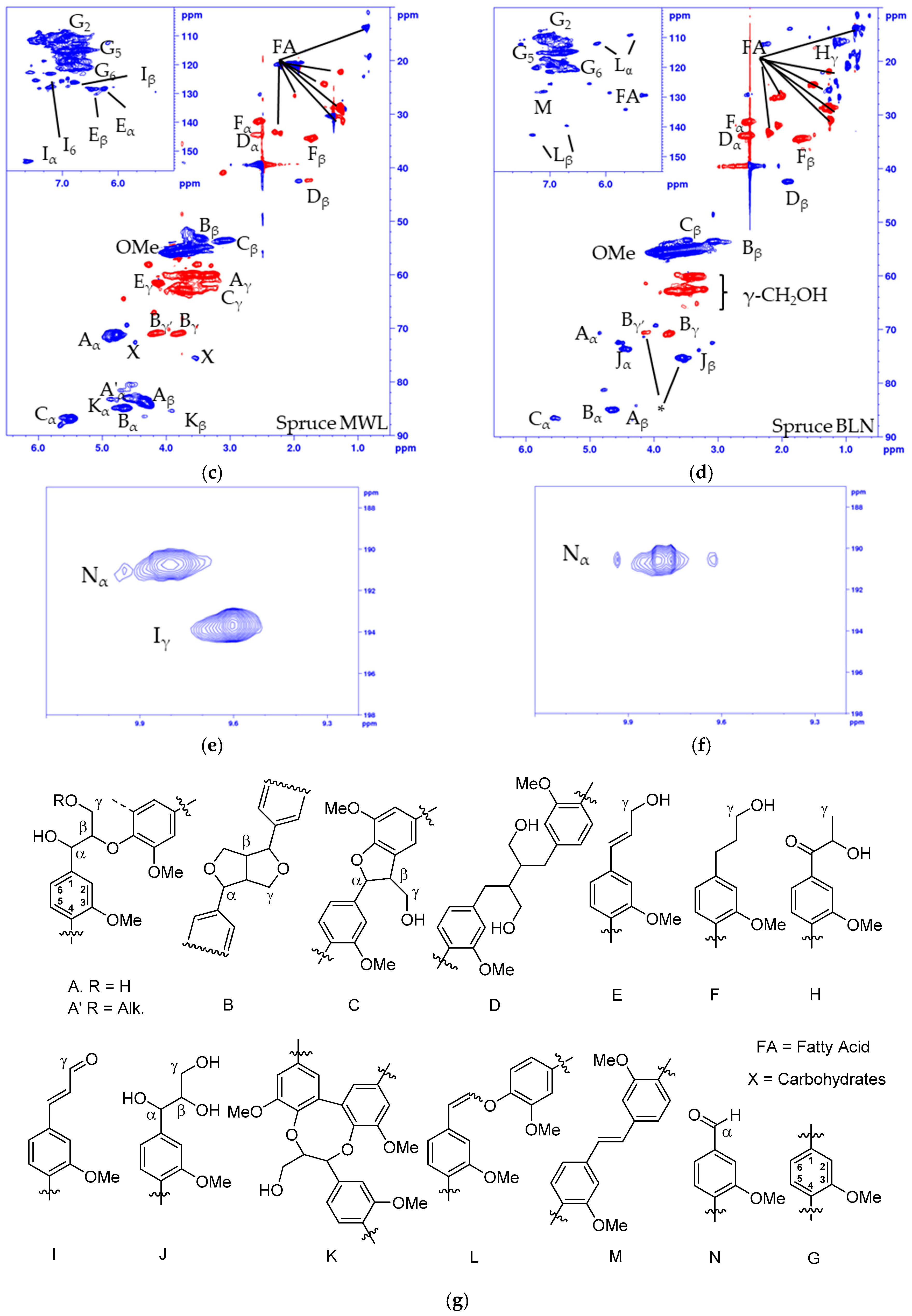
2.1. Analysis of the Unmodified Lignin
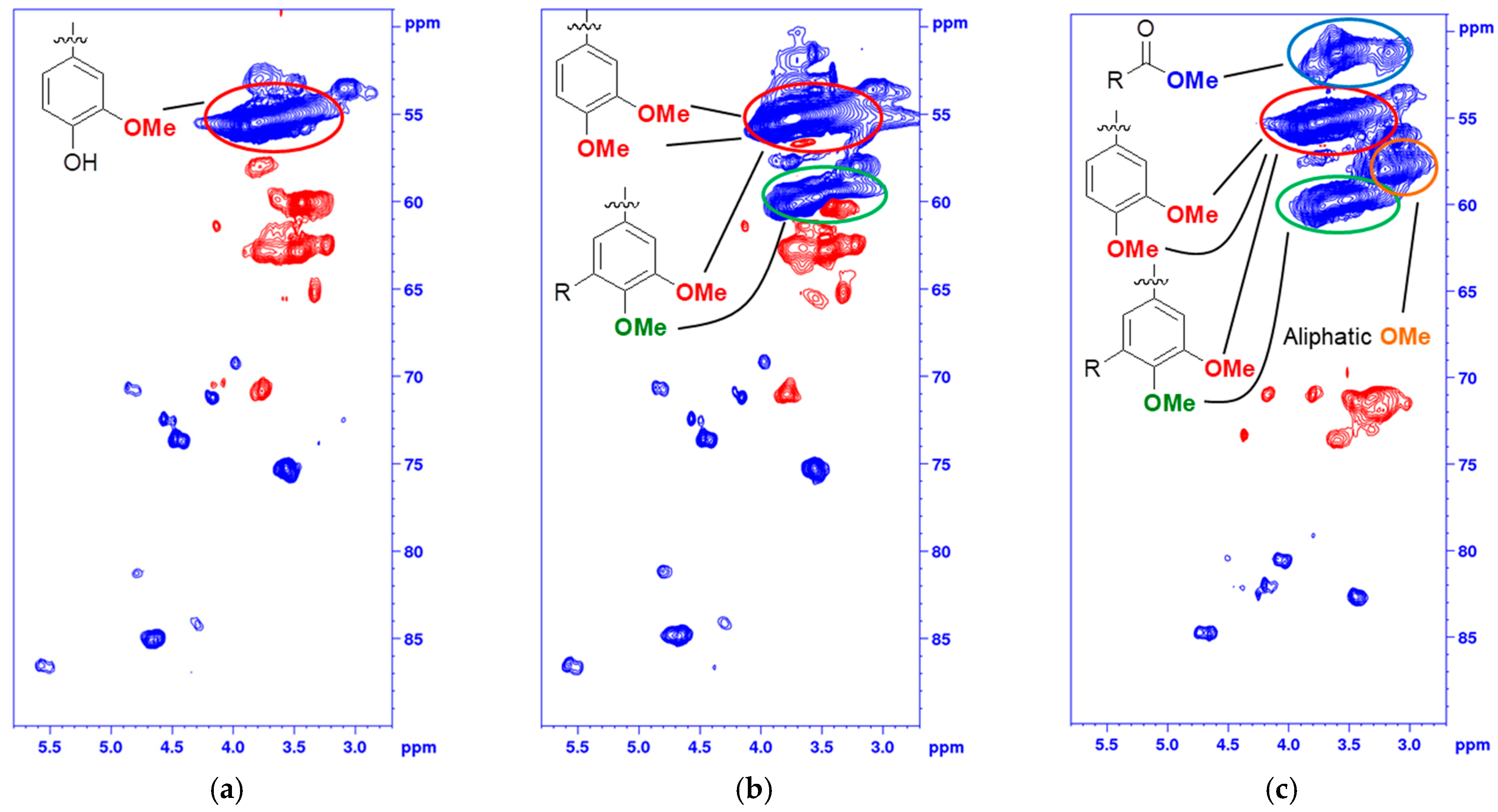
2.2. Acetylated and Methylated Lignin
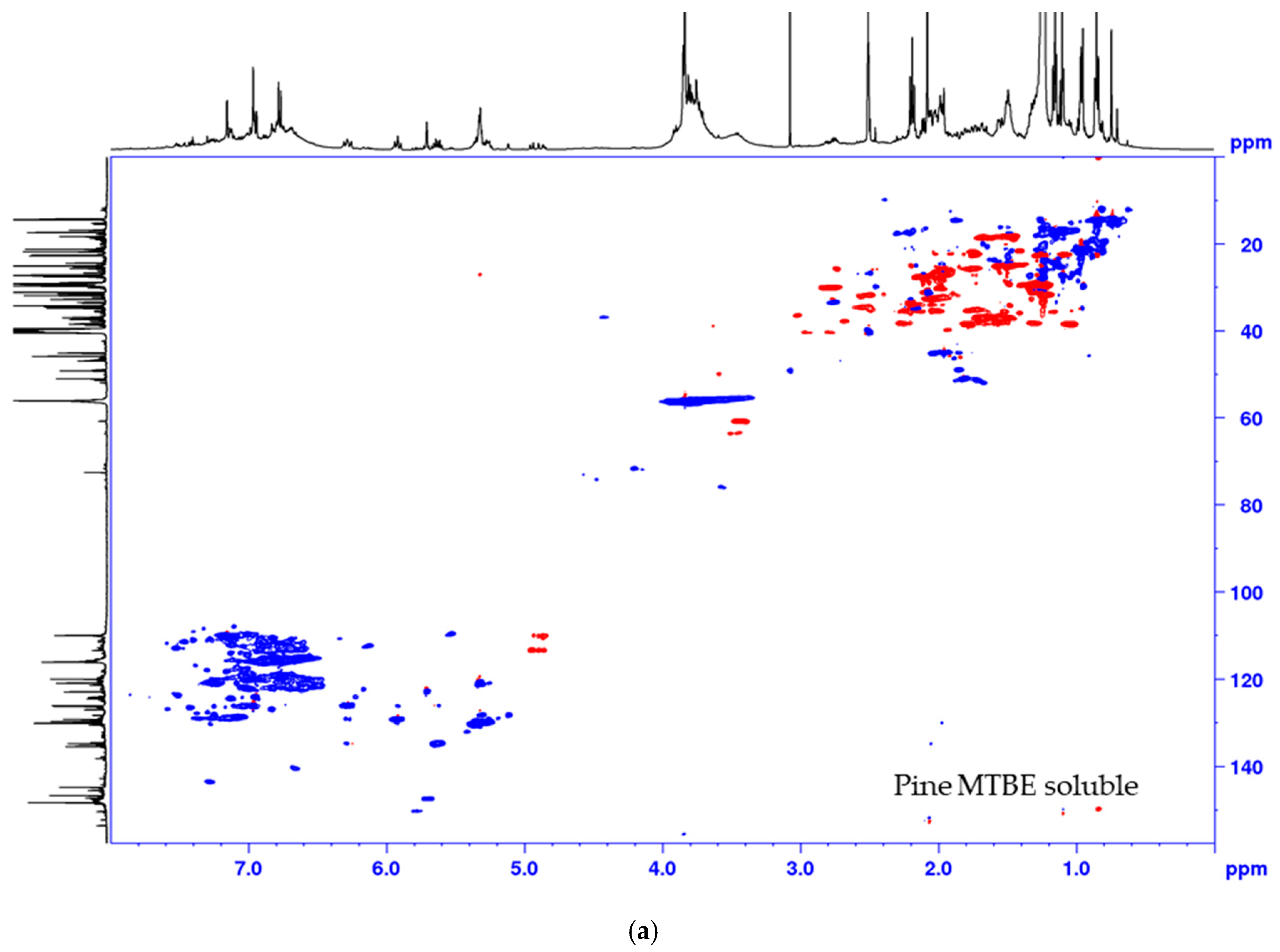
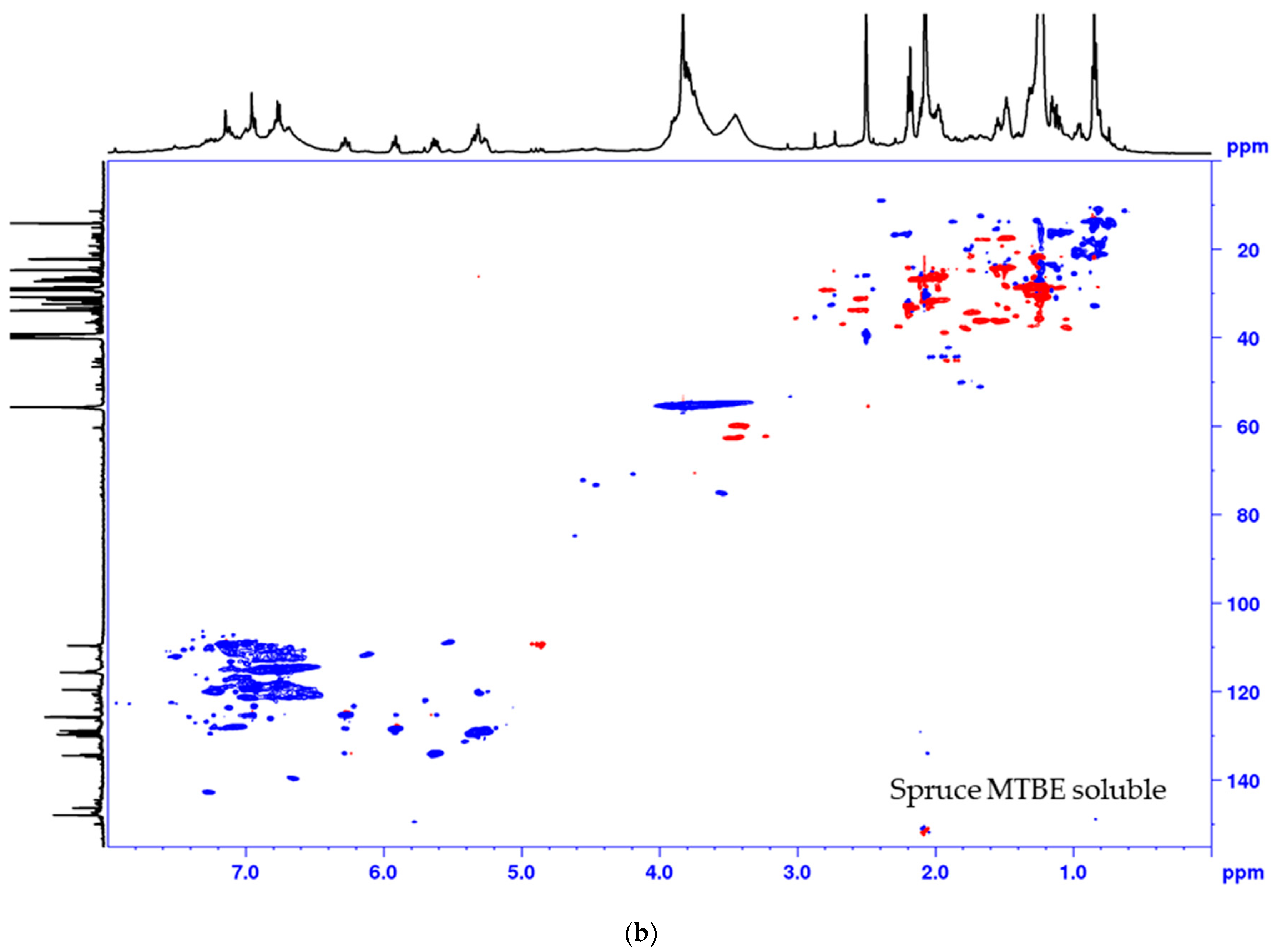
2.3. MTBE-Soluble and i-PrOH Fractions
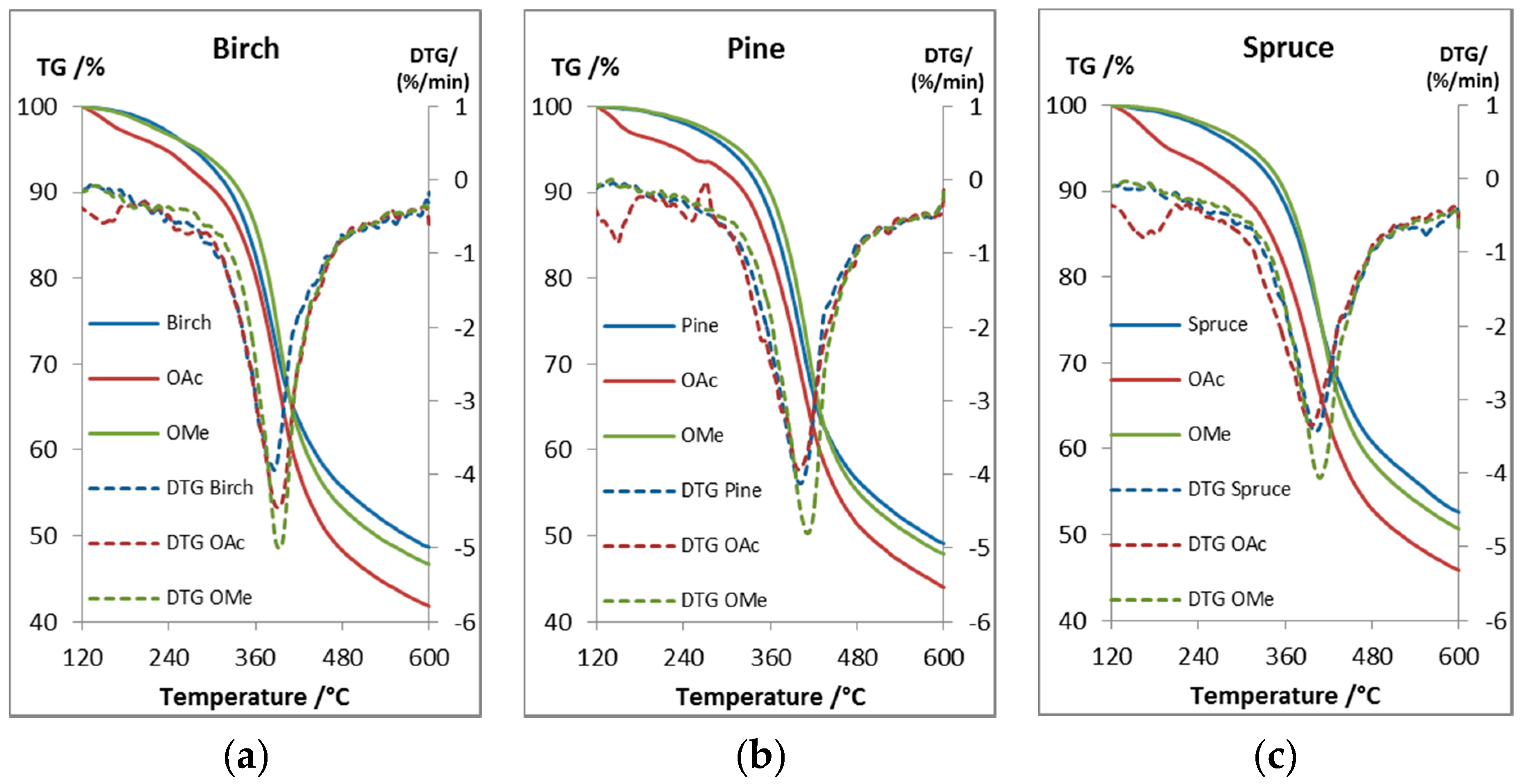
2.4. Thermal Properties
3. Materials and Methods
3.1. Materials
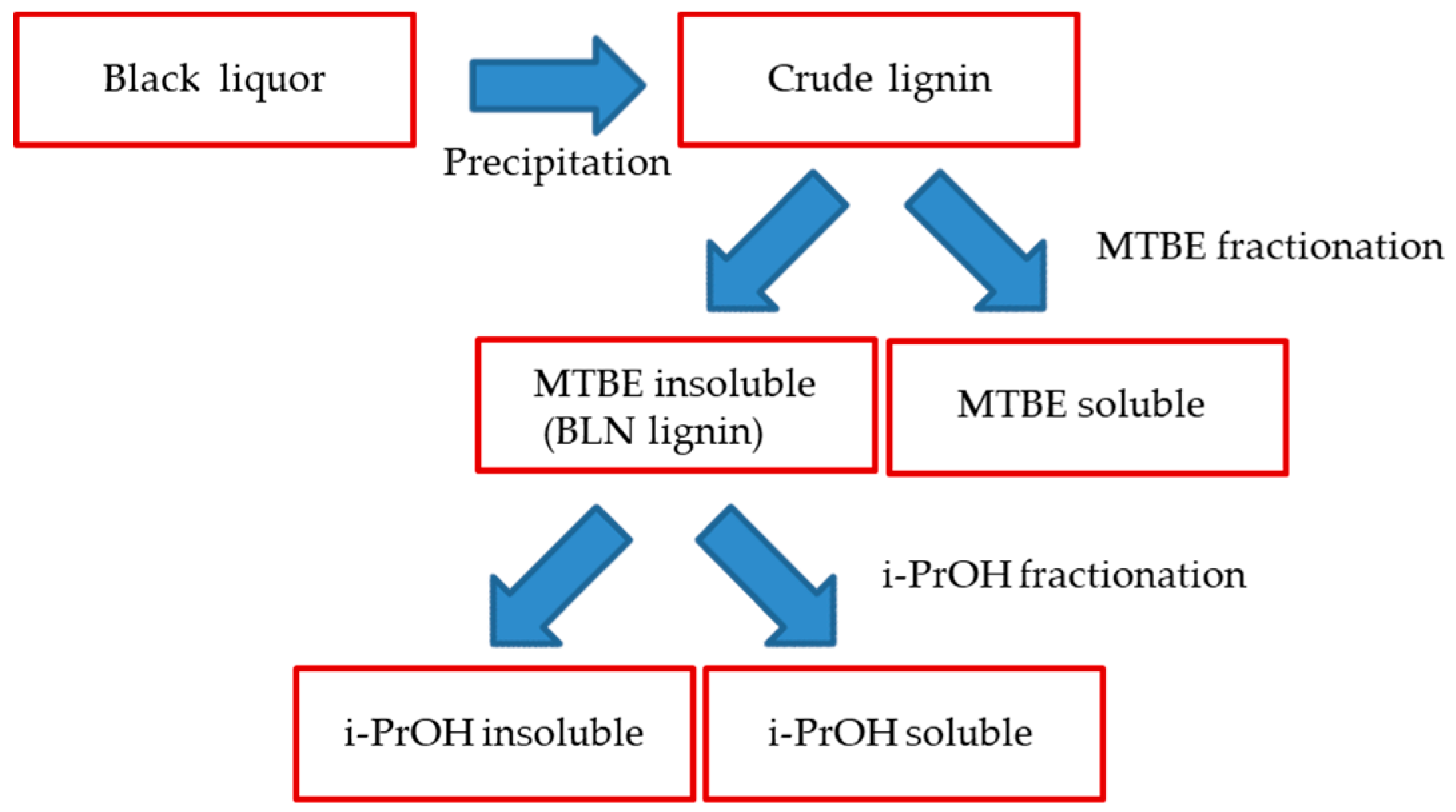
3.2. BLN Process
3.2.1. Precipitation, Purification and Fractionation
3.2.2. iPrOH Fractionation
3.3. Elemental Analysis
3.4. Molar Mass Distribution
3.5. NMR Spectroscopy
3.6. Thermal Analysis
3.7. Acetylation
3.8. Methylation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin Biosynthesis and Structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlin, A.; Balakshin, M. Chapter 18—Industrial Lignins: Analysis, Properties, and Applications. In Bioenergy Research: Advances and Applications; Gupta, V.K., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 315–336. ISBN 978-0-444-59561-4. [Google Scholar]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis. Angew. Chem. Int. Ed. 2016, 55, 8164–8215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherubini, F. The biorefinery concept: Using biomass instead of oil for producing energy and chemicals. Energy. Convers. Manag. 2010, 51, 1412–1421. [Google Scholar] [CrossRef]
- Tomani, P. The lignoboost process. Cellul. Chem. Technol. 2010, 44, 53–58. [Google Scholar]
- Pu, Y.; Hu, F.; Huang, F.; Ragauskas, A.J. Lignin Structural Alterations in Thermochemical Pretreatments with Limited Delignification. Bioenergy Res. 2015, 8, 992–1003. [Google Scholar] [CrossRef]
- De Menezes, F.F.; Rencoret, J.; Nakanishi, S.C.; Nascimento, V.M.; Silva, V.F.N.; Gutiérrez, A.; Del Río, J.C.; De Moraes Rocha, G.J. Alkaline Pretreatment Severity Leads to Different Lignin Applications in Sugar Cane Biorefineries. ACS Sustain. Chem. Eng. 2017, 5, 5702–5712. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Dai, Z.; Cao, Q.; Shi, X.; Wang, X.; Li, H.; Han, Y.; Li, Y.; Zhou, J. Lignin/Polyacrylonitrile Carbon Fibers: The Effect of Fractionation and Purification on Properties of Derived Carbon Fibers. ACS Sustain. Chem. Eng. 2018, 6, 8554–8562. [Google Scholar] [CrossRef]
- Tejado, A.; Peña, C.; Labidi, J.; Echeverria, J.M.; Mondragon, I. Physico-chemical characterization of lignins from different sources for use in phenol-formaldehyde resin synthesis. Bioresour. Technol. 2007, 98, 1655–1663. [Google Scholar] [CrossRef]
- Kärkäs, M.D.; Matsuura, B.S.; Monos, T.M.; Magallanes, G.; Stephenson, C.R.J. Transition-metal catalyzed valorization of lignin: The key to a sustainable carbon-neutral future. Org. Biomol. Chem. 2016, 14, 1853–1914. [Google Scholar] [CrossRef]
- Deuss, P.J.; Barta, K. From models to lignin: Transition metal catalysis for selective bond cleavage reactions. Coord. Chem. Rev. 2016, 306, 510–532. [Google Scholar] [CrossRef]
- Sen, S.; Patil, S.; Argyropoulos, D.S. Thermal properties of lignin in copolymers, blends, and composites: A review. Green Chem. 2015, 17, 4862–4887. [Google Scholar] [CrossRef]
- Doherty, W.O.S.; Mousavioun, P.; Fellows, C.M. Value-adding to cellulosic ethanol: Lignin polymers. Ind. Crops Prod. 2011, 33, 259–276. [Google Scholar] [CrossRef] [Green Version]
- Kun, D.; Pukánszky, B. Polymer/lignin blends: Interactions, properties, applications. Eur. Polym. J. 2017, 93, 618–641. [Google Scholar] [CrossRef]
- Upton, B.M.; Kasko, A.M. Strategies for the conversion of lignin to high-value polymeric materials: Review and perspective. Chem. Rev. 2016, 116, 2275–2306. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Suhr, J.; Seo, H.; Sun, H.; Kim, S.; Park, I.; Kim, S.; Lee, Y.; Kim, K.; Nam, J. All Biomass and UV Protective Composite Composed of Compatibilized Lignin and Poly (Lactic-acid). Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Koivu, K.A.Y.; Sadeghifar, H.; Nousiainen, P.A.; Argyropoulos, D.S.; Sipilä, J. Effect of Fatty Acid Esterification on the Thermal Properties of Softwood Kraft Lignin. ACS Sustain. Chem. Eng. 2016, 4, 5238–5247. [Google Scholar] [CrossRef]
- Dehne, L.; Vila Babarro, C.; Saake, B.; Schwarz, K.U. Influence of lignin source and esterification on properties of lignin-polyethylene blends. Ind. Crops Prod. 2016, 86, 320–328. [Google Scholar] [CrossRef]
- Duval, A.; Lawoko, M. A review on lignin-based polymeric, micro- and nano-structured materials. React. Funct. Polym. 2014, 85, 78–96. [Google Scholar] [CrossRef]
- Luo, S.; Cao, J.; Mcdonald, A.G. Esterification of industrial lignin and its effect on the resulting poly (3-hydroxybutyrate-co-3-hydroxyvalerate) or polypropylene blends. Ind. Crops Prod. 2017, 97, 281–291. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Yang, M.; Huang, Z.; Hu, H.; Huang, A.; Feng, Z. Acylation of Lignin with Different Acylating Agents by Mechanical Activation-Assisted Solid Phase Synthesis: Preparation and Properties. Polymers 2018, 10, 907. [Google Scholar] [CrossRef]
- Gordobil, O.; Egüés, I.; Labidi, J. Modification of Eucalyptus and Spruce organosolv lignins with fatty acids to use as filler in PLA. React. Funct. Polym. 2016, 104, 45–52. [Google Scholar] [CrossRef]
- Gordobil, O.; Egüés, I.; Llano-ponte, R.; Labidi, J. Physicochemical properties of PLA lignin blends. Polym. Degrad. Stab. 2014, 108, 330–338. [Google Scholar] [CrossRef]
- Mu, C.; Xue, L.; Zhu, J.; Jiang, M.; Zhou, Z. Mechanical and Thermal Properties of Toughened Poly(l-lactic) Acid and Lignin Blends. BioResources 2014, 9, 5557–5566. [Google Scholar] [CrossRef]
- Kubo, S.; Kadla, J.F. Poly (Ethylene Oxide)/Organosolv Lignin Blends: Relationship between Thermal Properties, Chemical Structure, and Blend Behavior. Macromolecules 2004, 37, 6904–6911. [Google Scholar] [CrossRef]
- Cicala, G.; Saccullo, G.; Blanco, I.; Samal, S.; Battiato, S.; Dattilo, S.; Saake, B. Polylactide/lignin blends. J. Therm. Anal. Calorim. 2017, 130, 515–524. [Google Scholar] [CrossRef]
- Von Schoultz, S. Method for Extracting Biomass. WO 2014/009604 A1, 16 January 2014. [Google Scholar]
- Von Schoultz, S. Method for Extracting Lignin. WO 2015/104460 A1, 16 July 2015. [Google Scholar]
- Lagerquist, L.; Pranovich, A.; Smeds, A.; von Schoultz, S.; Vähäsalo, L.; Rahkila, J.; Kilpeläinen, I.; Tamminen, T.; Willför, S.; Eklund, P. Structural characterization of birch lignin isolated from a pressurized hot water extraction and mild alkali pulped biorefinery process. Ind. Crops Prod. 2018, 111, 306–316. [Google Scholar] [CrossRef]
- Cui, C.; Sadeghifar, H.; Sen, S.; Argyropoulos, D.S. Toward thermoplastic lignin polymers; Part II: Thermal & polymer characteristics of kraft lignin & derivatives. BioResources 2013, 8, 864–886. [Google Scholar] [CrossRef]
- Gierer, J. Chemical Aspects of Kraft Pulping. Wood Sci. Technol. 1980, 14, 241–266. [Google Scholar] [CrossRef]
- Lundquist, K.; Von Unge, S. Stability of arylglycerols during alkaline cooking. Holzforschung 2004, 58, 330–333. [Google Scholar] [CrossRef]
- Sun, S.; Yang, H.; Sun, R.; Tu, M. The strong association of condensed phenolic moieties in isolated lignins with their inhibition of enzymatic hydrolysis. Green Chem. 2016, 18, 4276–4286. [Google Scholar] [CrossRef]
- Ralph, S.A.; Ralph, J.; Landucci, L.L. NMR Database of Lignin and Cell Wall Model Compounds. Available online: https://www.glbrc.org/databases_and_software/nmrdatabase/ (accessed on 6 August 2018).
- Domínguez-Robles, J.; Tamminen, T.; Liitiä, T.; Peresin, M.S.; Rodríguez, A.; Jääskeläinen, A.S. Aqueous acetone fractionation of kraft, organosolv and soda lignins. Int. J. Biol. Macromol. 2018, 106, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Jääskeläinen, A.S.; Liitiä, T.; Mikkelson, A.; Tamminen, T. Aqueous organic solvent fractionation as means to improve lignin homogeneity and purity. Ind. Crops Prod. 2017, 103, 51–58. [Google Scholar] [CrossRef]
- Brebu, M.; Vasile, C. Thermal degradation of lignin—A review. Cellul. Chem. Technol. 2010, 44, 353–363. [Google Scholar]
- Gordobil, O.; Delucis, R.; Egüés, I.; Labidi, J. Kraft lignin as filler in PLA to improve ductility and thermal properties. Ind. Crops Prod. 2015, 72, 46–53. [Google Scholar] [CrossRef]
- Björkman, A. Studies on Finely Divided Wood. Part I. Extraction of Lignin with Neutral Solvents. Sven. Papperstidn. 1956, 59, 477–485. [Google Scholar]
- Granata, A.; Argyropoulos, D.S. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a Reagent for the Accurate Determination of the Uncondensed and Condensed Phenolic Moieties in Lignins. J. Agric. Food Chem. 1995, 43, 1538–1544. [Google Scholar] [CrossRef]
- Sadeghifar, H.; Cui, C.; Argyropoulos, D.S. Toward thermoplastic lignin polymers. Part 1. Selective masking of phenolic hydroxyl groups in kraft lignins via methylation and oxypropylation chemistries. Ind. Eng. Chem. Res. 2012, 51, 16713–16720. [Google Scholar] [CrossRef]
- Crestini, C.; Lange, H.; Sette, M.; Argyropoulos, D.S. on the Structure of Softwood Kraft Lignin. Green Chem. 2017, 19, 4104–4121. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | δC/δH (ppm) | Assignment (C-H Correlation) |
|---|---|---|
| Hγ | 21.0/1.48 | γ in G-hydroxyethyl ketone |
| Fα | 31.2/2.51 | α in G-propanol |
| Dα | 33.9/2.53 | α in secoisolariresinol |
| Fβ | 34.4/1.70 | β in G-propanol |
| Dβ | 42.5/1.89 | β in secoisolariresinol |
| Cβ | 53.1/3.47 | β in phenylcoumaran |
| Bβ | 53.5/3.06 | β in resinol (β-β) |
| OMe | 55.5/3.76 | Methoxy peaks |
| Aγ | 59.9/3.61 | γ in β-O-4 |
| Eγ | 61.5/4.10 | γ in cinnamyl alcohol |
| Cγ | 62.8/3.73 | γ in phenylcoumaran |
| Jγ | 62.8/3.47 | γ in arylglycerol |
| Bγ | 70.8/4.16 and 3.73 | γ in resinol |
| Aα | 71.06/4.76 | α in β-O-4 |
| Jα | 73.5/4.45 | α in arylglycerol |
| Jβ | 75.4/3.56 | β in arylglycerol |
| Kα | 83.2/4.84 | α in dibenzodioxocin |
| Aβ | 84.0/4.30 | β in β-O-4 |
| Bα | 84.9/4.64 | α in resinol (β-β) |
| Kβ | 85.4/3.89 | β in dibenzodioxocin |
| Cα | 87.0/5.47 | α in phenylcoumaran |
| Lα | 108.9/5.56 | aryl enol ether |
| G2 | 110.3/6.94 | 2 in the guaiacyl unit |
| Lβ | 112.1/6.17 | Aryl enol ether |
| G5 | 115.2/6.85 | 5 in the guaiacyl unit |
| G6 | 118.9/6.83 | 6 in the guaiacyl unit |
| I6 | 123.2/7.22 | 6 in cinnamyl aldehyde |
| Iβ | 126.3/6.77 | β in cinnamyl aldehyde |
| M | 128.3/7.12 | Stillbene |
| Eα | 128.4/6.24 | α in cinnamyl alcohol |
| Eβ | 128.5/6.46 | β in cinnamyl alcohol |
| Lα’ | 139.8/6.71 | Aryl enol ether |
| Lβ’ | 143.0/7.31 | Aryl enol ether |
| Iα | 153.7/7.61 | α in cinnamyl aldehyde |
| Nα | 190.8/9.80 | Benzylic aldehyde |
| Iγ | 193.8/9.60 | γ in cinnamyl aldehyde |
| Lignin | Aliphatic (150.0–145.5 ppm) 1 | 5-subs (145.1–140.5 ppm) | G-units (140.5–136.8 ppm) | Ph-OH | OH Total | COOH (136.8–133.4 ppm) |
|---|---|---|---|---|---|---|
| Pine MWL | 4.7 | 0.8 | 1.2 | 2.0 | 6.7 | 0.3 |
| Pine BLN | 2.2 | 2.0 | 1.9 | 3.9 | 6.1 | 0.6 |
| Pine MTBE | 0.7 | 1.2 | 2.5 | 3.7 | 4.4 | 1.6 |
| Pine iPrOH insol | 1.9 | 1.8 | 1.5 | 3.3 | 5.2 | 0.4 |
| Pine iPrOH sol | 1.7 | 1.8 | 2.3 | 4.0 | 5.7 | 0.8 |
| Spruce MWL | 4.6 | 0.8 | 1.3 | 2.1 | 6.7 | 0.2 |
| Spruce BLN | 2.2 | 1.9 | 1.9 | 3.9 | 6.1 | 0.6 |
| Spruce MTBE | 1.0 | 1.3 | 2.3 | 3.6 | 4.6 | 1.4 |
| Spruce iPrOH insol | 2.1 | 1.9 | 1.6 | 3.5 | 5.6 | 0.5 |
| Spruce iPrOH sol | 1.6 | 1.7 | 2.1 | 3.8 | 5.4 | 0.8 |
| OMe Pine BLN | 2.0 | 0.8 | 2.8 | 0.7 | ||
| OMe Spruce BLN | 1.9 | 0.6 | 2.5 | 0.6 | ||
| OMe Birch BLN | 1.3 | 0.5 | 1.8 | 0.7 |
| Lignin | Mn (g/mol) | Mw (g/mol) | PDI | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pine MWL | 1500 | 2700 | 1.7 | |||||||||
| Pine BLN | 3200 | 6700 | 2.1 | |||||||||
| Pine MTBE | 380 | 480 | 1.3 | |||||||||
| Pine iPrOH insol | 4200 | 8000 | 1.9 | |||||||||
| Pine iPrOH sol | 860 | 1500 | 1.7 | |||||||||
| Spruce MWL | 2100 | 3000 | 1.5 | |||||||||
| Spruce BLN | 2100 | 5500 | 2.7 | |||||||||
| Spruce MTBE | 460 | 570 | 1.2 | |||||||||
| Spruce iPrOH insol | 3800 | 7000 | 1.9 | |||||||||
| Spruce iPrOH sol | 890 | 1200 | 1.3 | |||||||||
| C (%) | H (%) | N (%) | O (%) | |||||||||
| Pine MWL | 60.6 | 5.9 | 0.2 | 33.3 | ||||||||
| Pine BLN | 67.2 | 6.0 | 0.1 | 26.7 | ||||||||
| Spruce MWL | 59.5 | 5.7 | 0.1 | 34.7 | ||||||||
| Spruce BLN | 66.5 | 5.8 | 0.0 | 27.6 | ||||||||
| Lignin | Tdst 95% (°C) | Tdmax (°C) | Char Residue at 600 °C (wt %) |
|---|---|---|---|
| Pine BLN | 302 | 401 | 49 |
| OAc Pine BLN | 233 | 398 | 44 |
| OMe Pine BLN | 317 | 411 | 48 |
| Spruce BLN | 296 | 401 | 53 |
| OAc Spruce BLN | 198 | 396 | 46 |
| OMe Spruce BLN | 312 | 408 | 51 |
| Birch BLN | 273 | 386 | 49 |
| OAc Birch BLN | 234 | 391 | 42 |
| OMe Birch BLN | 280 | 392 | 47 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagerquist, L.; Pranovich, A.; Sumerskii, I.; von Schoultz, S.; Vähäsalo, L.; Willför, S.; Eklund, P. Structural and Thermal Analysis of Softwood Lignins from a Pressurized Hot Water Extraction Biorefinery Process and Modified Derivatives. Molecules 2019, 24, 335. https://doi.org/10.3390/molecules24020335
Lagerquist L, Pranovich A, Sumerskii I, von Schoultz S, Vähäsalo L, Willför S, Eklund P. Structural and Thermal Analysis of Softwood Lignins from a Pressurized Hot Water Extraction Biorefinery Process and Modified Derivatives. Molecules. 2019; 24(2):335. https://doi.org/10.3390/molecules24020335
Chicago/Turabian StyleLagerquist, Lucas, Andrey Pranovich, Ivan Sumerskii, Sebastian von Schoultz, Lari Vähäsalo, Stefan Willför, and Patrik Eklund. 2019. "Structural and Thermal Analysis of Softwood Lignins from a Pressurized Hot Water Extraction Biorefinery Process and Modified Derivatives" Molecules 24, no. 2: 335. https://doi.org/10.3390/molecules24020335
APA StyleLagerquist, L., Pranovich, A., Sumerskii, I., von Schoultz, S., Vähäsalo, L., Willför, S., & Eklund, P. (2019). Structural and Thermal Analysis of Softwood Lignins from a Pressurized Hot Water Extraction Biorefinery Process and Modified Derivatives. Molecules, 24(2), 335. https://doi.org/10.3390/molecules24020335