
Photocatalytic Degradation of Pharmaceuticals Carbamazepine, Diclofenac, and Sulfamethoxazole by Semiconductor and Carbon Materials: A Review



Abstract
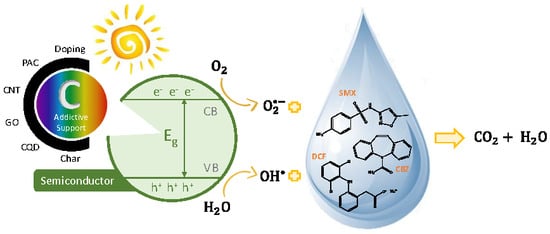
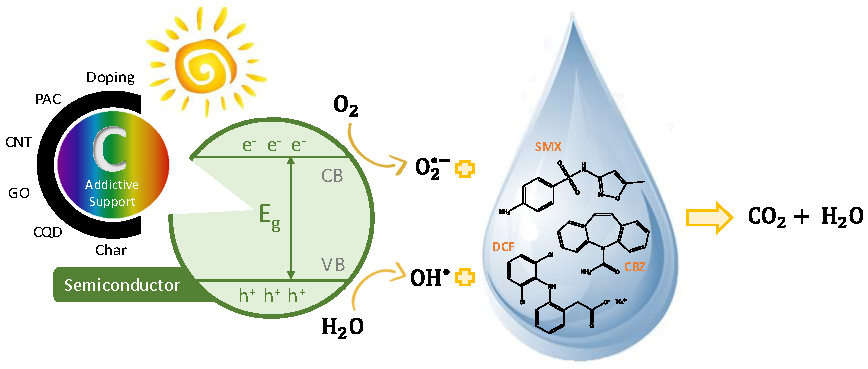
:
1. Introduction
2. Pharmaceutical Compounds as Pollutants of Emergent Concern
2.1. CBZ, DCF, and SMX Detections in Wastewater and Environmental Water
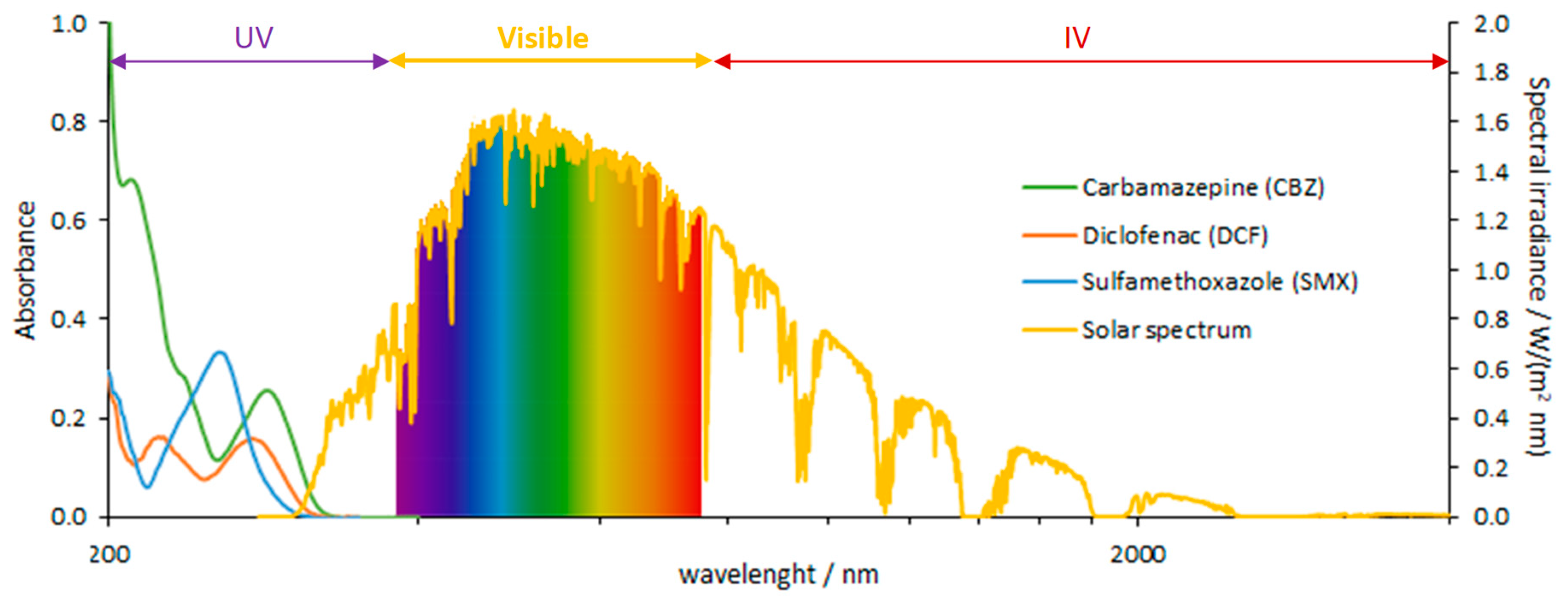
2.2. Photodegradation
3. Photocatalysis
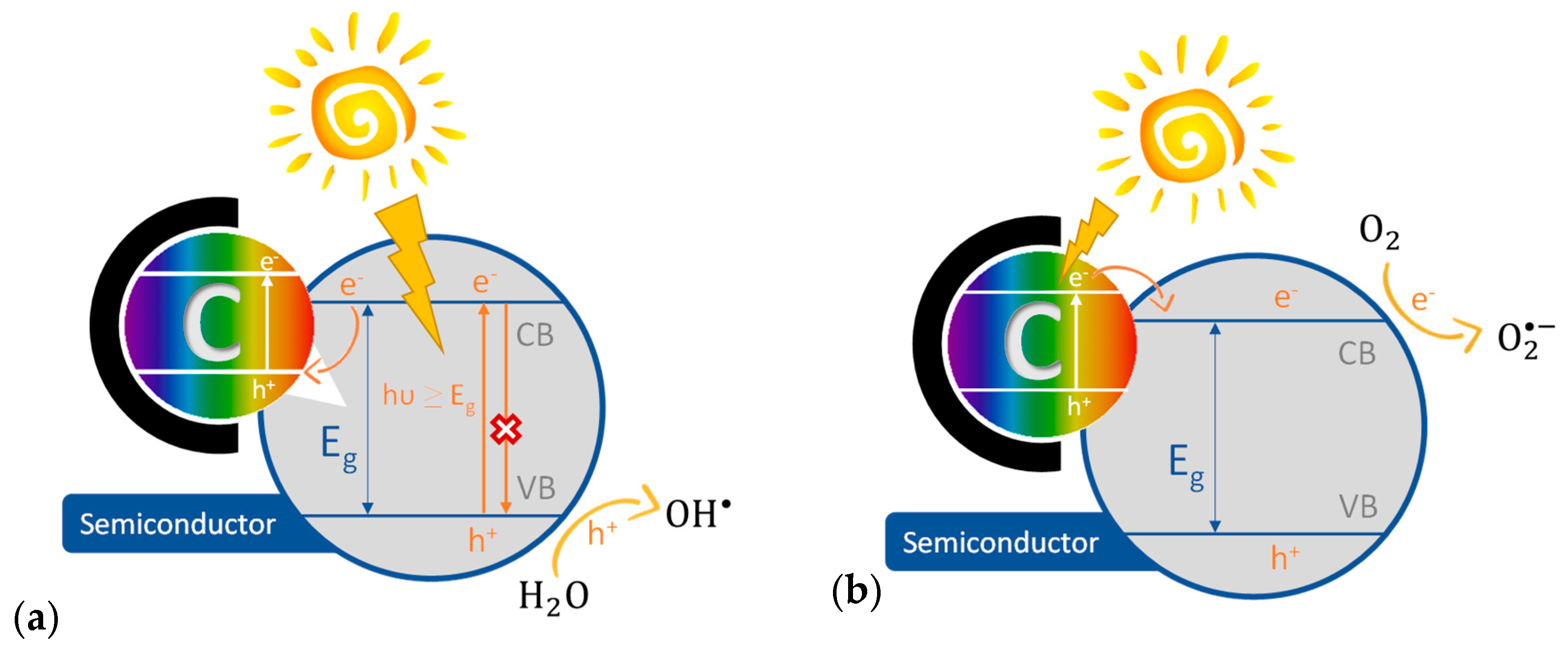
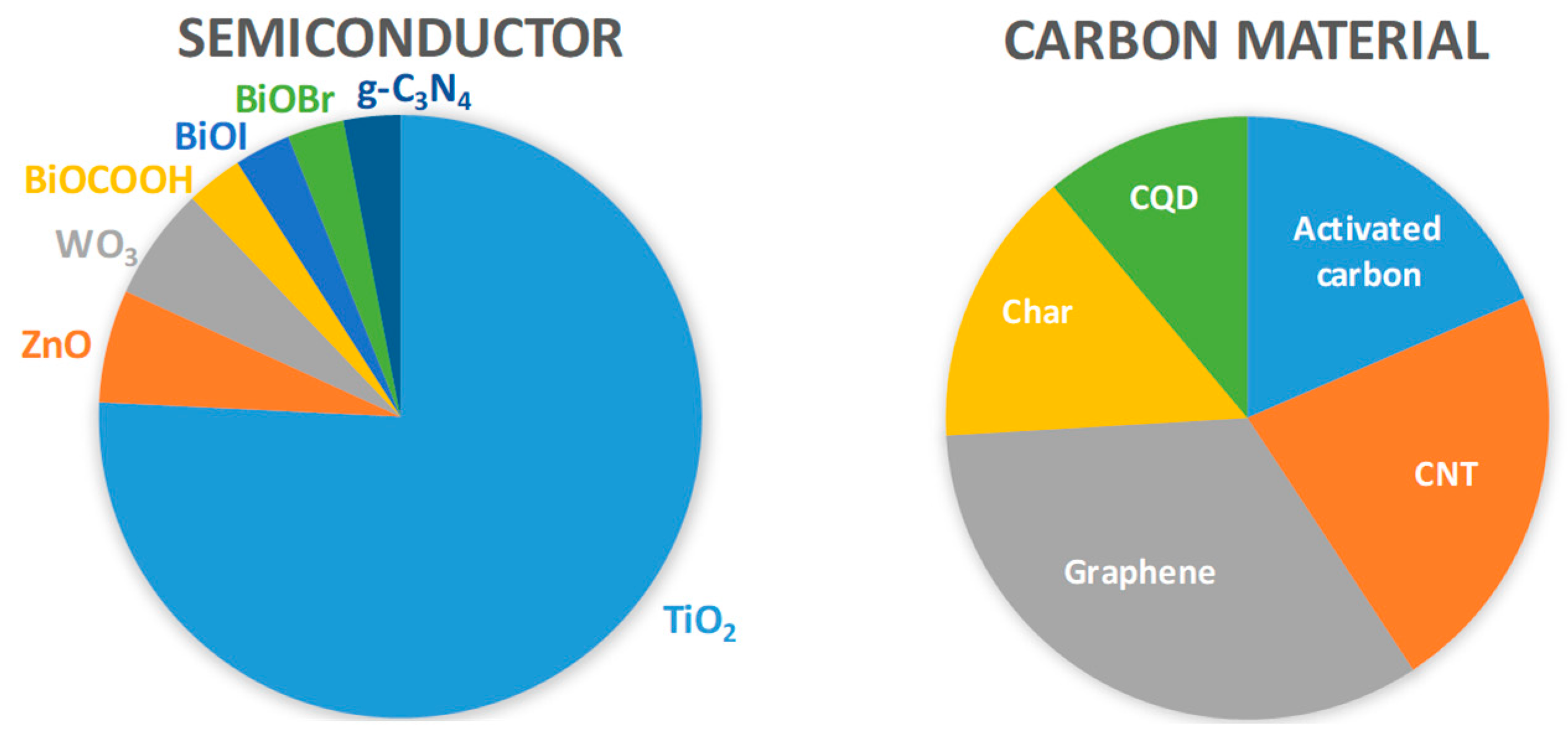
3.1. Semiconductor/C
3.2. Semiconductor/Activated Carbon
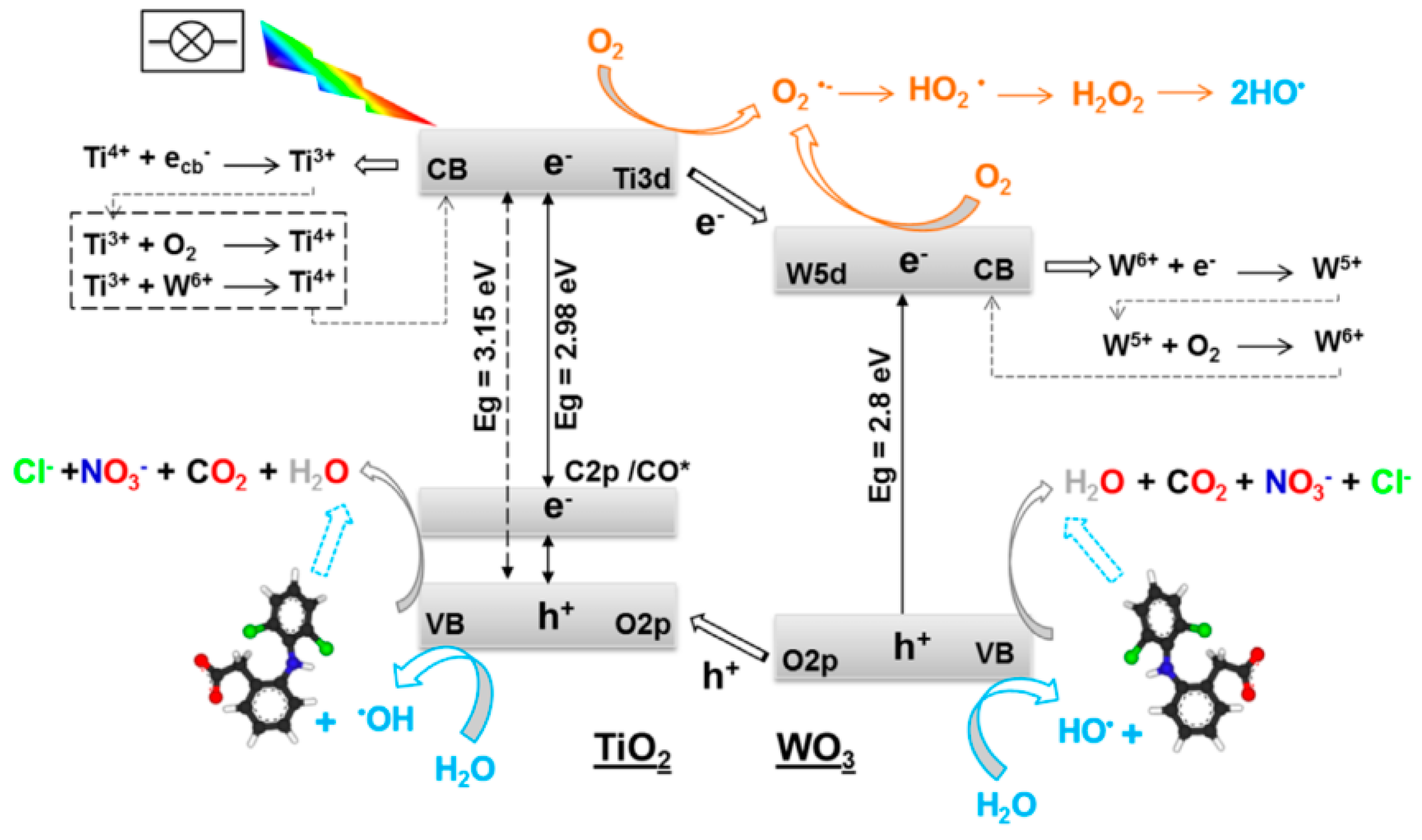
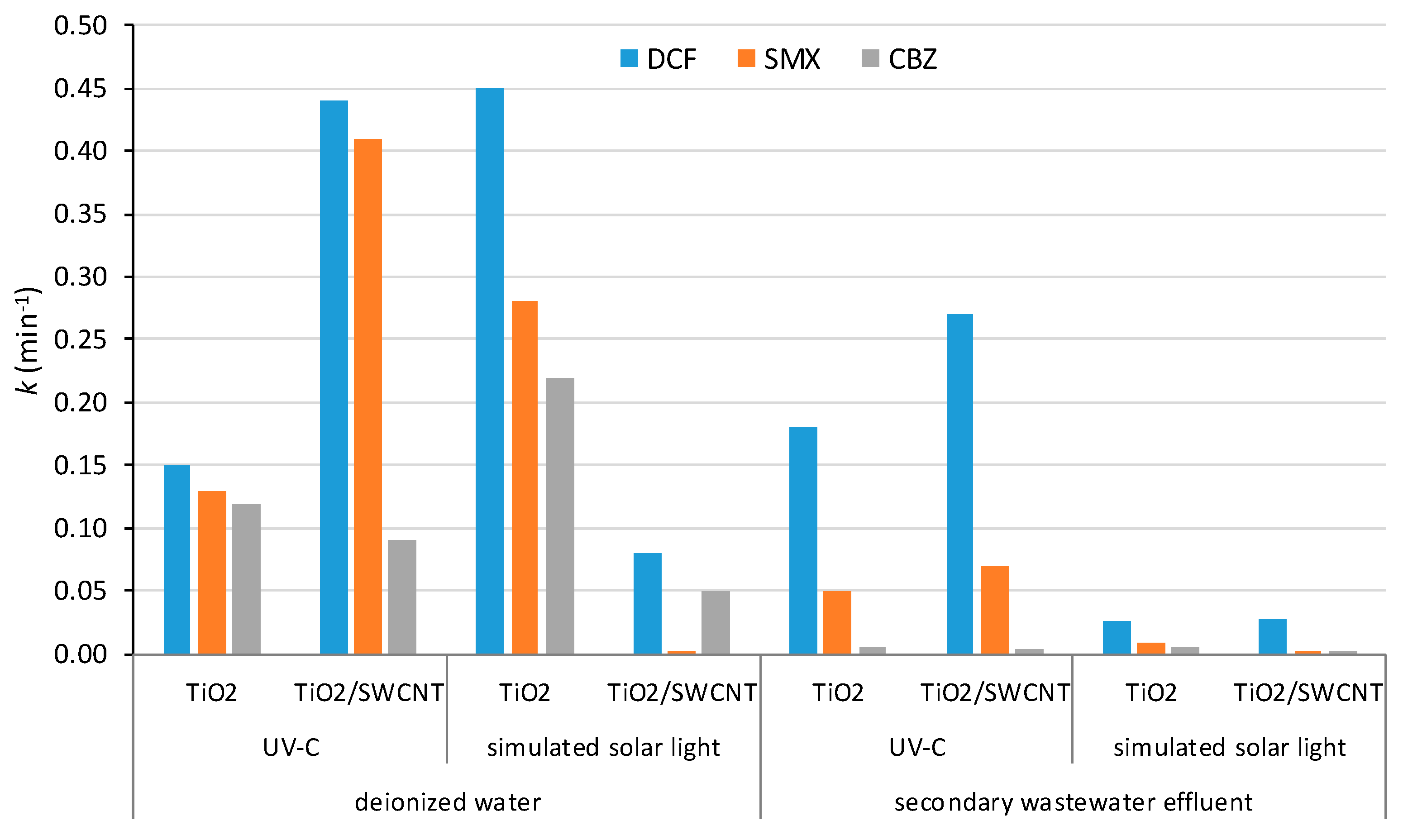
3.3. Semiconductor/CNT
3.4. Semiconductor/(r)GO
3.5. Semiconductor/Carbon Quantum Dots (CQDs)
3.6. Semiconductor/Char
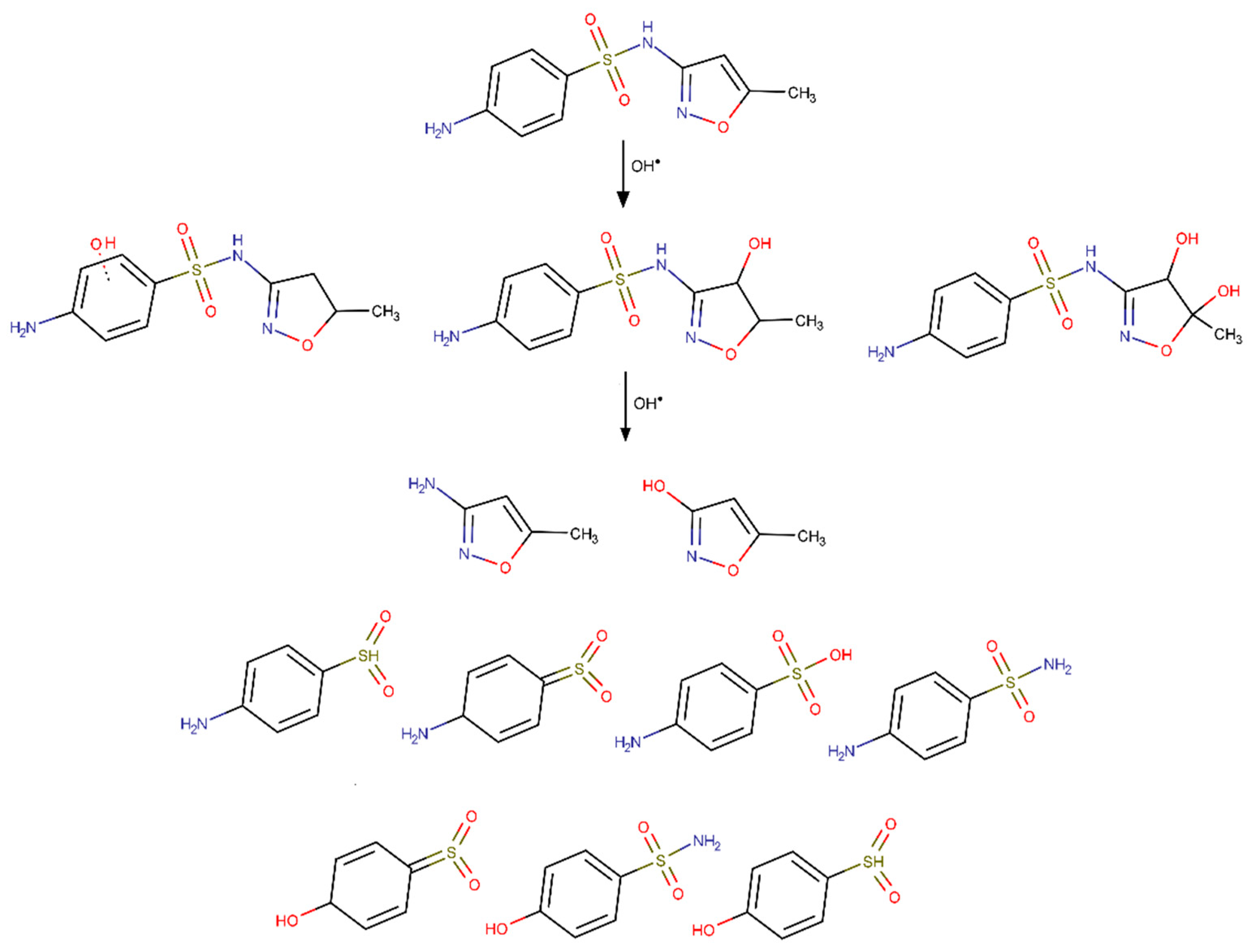
3.7. Overview of the Degradation Pathways and Intermediates/Products
4. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Jägerskog, A.; Clausen, T.J.; Holmgren, T.; Lexén, K. Energy and Water: The Vital Link for a Sustainable Future (http://www.worldwaterweek.org/); Stockholm International Water Institute, SIWI: Stockholm, Swedish, 2014. [Google Scholar]
- Poyatos, J.M.; Munio, M.M.; Almecija, M.C.; Torres, J.C.; Hontoria, E.; Osorio, F. Advanced oxidation processes for wastewater treatment: State of the art. Water Air Soil Pollut. 2010, 205, 187–204. [Google Scholar] [CrossRef]
- Ribeiro, A.R.; Nunes, O.C.; Pereira, M.F.R.; Silva, A.M.T. An overview on the advanced oxidation processes applied for the treatment of water pollutants defined in the recently launched Directive 2013/39/EU. Environ. Int. 2015, 75, 33–51. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Database. Carbamazepine, CID=2554. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Carbamazepine (accessed on 29 August 2019).
- Ternes, T.A.; Herrmann, N.; Bonerz, M.; Knacker, T.; Siegrist, H.; Joss, A. A rapid method to measure the solid-water distribution coefficient (K-d) for pharmaceuticals and musk fragrances in sewage sludge. Water Res. 2004, 38, 4075–4084. [Google Scholar] [CrossRef] [PubMed]
- Joss, A.; Zabczynski, S.; Göbel, A.; Hoffmann, B.; Löffler, D.; McArdell, C.S.; Ternes, T.A.; Thomsen, A.; Siegrist, H. Biological degradation of pharmaceuticals in municipal wastewater treatment: Proposing a classification scheme. Water Res. 2006, 40, 1686–1696. [Google Scholar] [CrossRef]
- Huber, M.M.; Canonica, S.; Park, G.-Y.; von Gunten, U. Oxidation of pharmaceuticals during ozonation and advanced oxidation processes. Environ. Sci. Technol. 2003, 37, 1016–1024. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Database. Diclofenac, CID=3033. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Diclofenac (accessed on 29 August 2019).
- National Center for Biotechnology Information. PubChem Database. Sulfamethoxazole CID=5329. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Sulfamethoxazole (accessed on 29 August 2019).
- Göbel, A.; Thomsen, A.; McArdell, C.S.; Joss, A.; Giger, W. Occurrence and sorption behavior of sulfonamides, macrolides, and trimethoprim in activated sludge treatment. Environ. Sci. Technol. 2005, 39, 3981–3989. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Geissen, S.U.; Gal, C. Carbamazepine and diclofenac: Removal in wastewater treatment plants and occurrence in water bodies. Chemosphere 2008, 73, 1151–1161. [Google Scholar] [CrossRef]
- Zhou, W.; Moore, D.E. Photochemical decomposition of sulfamethoxazole. Int. J. Pharm. 1994, 110, 55–63. [Google Scholar] [CrossRef]
- Martini, J.; Orge, C.A.; Faria, J.L.; Pereira, M.F.R.; Soares, O.S.G.P. Sulfamethoxazole degradation by combination of advanced oxidation processes. J. Environ. Chem. Eng. 2018, 6, 4054–4060. [Google Scholar] [CrossRef]
- Stumm-Zollinger, E.; Fair, G.M. Biodegradation of steroid hormones. J. Water Pollut. Cont. Fed. 1965, 37, 1506–1510. [Google Scholar]
- Hignite, C.; Azarnoff, D.L. Drugs and drug metabolites as environmental contaminants: Chlorophenoxyisobutyrate and salicylic acid in sewage water effluent. Life Sci. 1977, 20, 337–341. [Google Scholar] [CrossRef]
- Fent, K.; Weston, A.A.; Caminada, D. Ecotoxicology of human pharmaceuticals. Aquat. Toxicol. 2006, 76, 122–159. [Google Scholar] [CrossRef] [PubMed]
- Heberer, T. Tracking persistent pharmaceutical residues from municipal sewage to drinking water. J. Hydrol. 2002, 266, 175–189. [Google Scholar] [CrossRef]
- Ternes, T. Pharmaceuticals and metabolites as contaminants of the aquatic environment. In Pharmaceuticals and Care Products in the Environment; American Chemical Society: Washington, DC, USA, 2001; Volume 791, pp. 39–54. [Google Scholar]
- Ternes, T.A. Occurrence of drugs in German sewage treatment plants and rivers. Water Res. 1998, 32, 3245–3260. [Google Scholar] [CrossRef]
- Stumpf, M.; Ternes, T.A.; Wilken, R.-D.; Silvana Vianna, R.; Baumann, W. Polar drug residues in sewage and natural waters in the state of Rio de Janeiro, Brazil. Sci. Total Environ. 1999, 225, 135–141. [Google Scholar] [CrossRef]
- Halling-Sørensen, B.; Nors Nielsen, S.; Lanzky, P.F.; Ingerslev, F.; Holten Lützhøft, H.C.; Jørgensen, S.E. Occurrence, fate and effects of pharmaceutical substances in the environment-a review. Chemosphere 1998, 36, 357–393. [Google Scholar] [CrossRef]
- Buser, H.-R.; Poiger, T.; Müller, M.D. Occurrence and fate of the pharmaceutical drug diclofenac in surface waters: Rapid photodegradation in a lake. Environ. Sci. Technol. 1998, 32, 3449–3456. [Google Scholar] [CrossRef]
- Andreozzi, R.; Raffaele, M.; Nicklas, P. Pharmaceuticals in STP effluents and their solar photodegradation in aquatic environment. Chemosphere 2003, 50, 1319–1330. [Google Scholar] [CrossRef]
- Heberer, T.; Reddersen, K.; Mechlinski, A. From municipal sewage to drinking water: Fate and removal of pharmaceutical residues in the aquatic environment in urban areas. Water Sci. Technol. 2002, 46, 81–88. [Google Scholar] [CrossRef]
- Hirsch, R.; Ternes, T.; Haberer, K.; Kratz, K.-L. Occurrence of antibiotics in the aquatic environment. Sci. Total Environ. 1999, 225, 109–118. [Google Scholar] [CrossRef]
- Sacher, F.; Lange, F.T.; Brauch, H.-J.; Blankenhorn, I. Pharmaceuticals in groundwaters: Analytical methods and results of a monitoring program in Baden-Württemberg, Germany. J. Chromatogr. A 2001, 938, 199–210. [Google Scholar] [CrossRef]
- Stumpf, M.; Terries, T.A.; Haberer, K.P.S.; Baumann, W. Nachweis von arzneimittelrückänden in kläranlagen und flicssgewässern. Vom Wasser 1996, 86, 291–303. [Google Scholar]
- Watts, C.D.; Craythorne, M.; Fielding, M.; Steel, C.P. Identification of non-volatile organics in water using field desorption mass spectrometry and high performance liquid chromatography. In Analysis of Organic Micropollutants in Water; Ageletti, G., Bjørseth, A., Eds.; Springer: Dordrech, The Netherlands, 1984; pp. 120–131. [Google Scholar]
- Loos, R.; Gawlik, B.M.; Locoro, G.; Rimaviciute, E.; Contini, S.; Bidoglio, G. EU-wide survey of polar organic persistent pollutants in European river waters. Environ. Pollut. 2009, 157, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.; Locoro, G.; Comero, S.; Contini, S.; Schwesig, D.; Werres, F.; Balsaa, P.; Gans, O.; Weiss, S.; Blaha, L.; et al. Pan-European survey on the occurrence of selected polar organic persistent pollutants in ground water. Water Res. 2010, 44, 4115–4126. [Google Scholar] [CrossRef] [PubMed]
- Clara, M.; Strenn, B.; Kreuzinger, N. Carbamazepine as a possible anthropogenic marker in the aquatic environment: Investigations on the behaviour of Carbamazepine in wastewater treatment and during groundwater infiltration. Water Res. 2004, 38, 947–954. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.Y.; Liu, X.S.; Ding, J. Ranking and prioritizing pharmaceuticals in the aquatic environment of China. Sci. Total Environ. 2019, 658, 333–342. [Google Scholar] [CrossRef]
- Guo, J.; Sinclair, C.J.; Selby, K.; Boxall, A.B.A. Toxicological and ecotoxicological risk-based prioritization of pharmaceuticals in the natural environment. Environ. Toxicol. Chem. 2016, 35, 1550–1559. [Google Scholar] [CrossRef] [Green Version]
- Besse, J.-P.; Kausch-Barreto, C.; Garric, J. Exposure assessment of pharmaceuticals and their metabolites in the aquatic environment: application to the french situation and preliminary prioritization AU–Besse, Jean-Philippe. Hum. Ecol. Risk Assess. Int. J. 2008, 14, 665–695. [Google Scholar] [CrossRef]
- de Voogt, P.; Janex-Habibi, M.L.; Sacher, F.; Puijker, L.; Mons, M. Development of a common priority list of pharmaceuticals relevant for the water cycle. Water Sci. Technol. 2009, 59, 39–46. [Google Scholar] [CrossRef]
- Standard Tables for Reference Solar Spectral Irradiances: Direct Normal and Hemispherical on 37° Tilted Surface. ASTM G173-03: 2012. Available online: https://www.nrel.gov/grid/solar-resource/spectra-am1.5.html (accessed on 11 October 2019).
- Doll, T.E.; Frimmel, F.H. Fate of pharmaceuticals––photodegradation by simulated solar UV-light. Chemosphere 2003, 52, 1757–1769. [Google Scholar] [CrossRef]
- Andreozzi, R.; Marotta, R.; Pinto, G.; Pollio, A. Carbamazepine in water: Persistence in the environment, ozonation treatment and preliminary assessment on algal toxicity. Water Res. 2002, 36, 2869–2877. [Google Scholar] [CrossRef]
- Yamamoto, H.; Nakamura, Y.; Moriguchi, S.; Nakamura, Y.; Honda, Y.; Tamura, I.; Hirata, Y.; Hayashi, A.; Sekizawa, J. Persistence and partitioning of eight selected pharmaceuticals in the aquatic environment: Laboratory photolysis, biodegradation, and sorption experiments. Water Res. 2009, 43, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.W.; Young, C.J.; Brain, R.A.; Johnson, D.J.; Hanson, M.A.; Wilson, C.J.; Richards, S.M.; Solomon, K.R.; Mabury, S.A. Aquatic persistence of eight pharmaceuticals in a microcosm study. Environ. Toxicol. Chem. 2004, 23, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Chiron, S.; Minero, C.; Vione, D. Photodegradation processes of the Antiepileptic drug carbamazepine, relevant to estuarine waters. Environ. Sci. Technol. 2006, 40, 5977–5983. [Google Scholar] [CrossRef]
- Pereira, V.J.; Linden, K.G.; Weinberg, H.S. Evaluation of UV irradiation for photolytic and oxidative degradation of pharmaceutical compounds in water. Water Res. 2007, 41, 4413–4423. [Google Scholar] [CrossRef]
- Matamoros, V.; Duhec, A.; Albaigés, J.; Bayona, J.M. Photodegradation of Carbamazepine, Ibuprofen, Ketoprofen and 17α-Ethinylestradiol in Fresh and Seawater. Water Air Soil Pollut. 2008, 196, 161. [Google Scholar] [CrossRef]
- Poiger, T.; Buser, H.-R.; Müller, M.D. Photodegradation of the pharmaceutical drug diclofenac in a lake: Pathway, field measurements, and mathematical modeling. Environ. Toxicol. Chem. 2001, 20, 256–263. [Google Scholar] [CrossRef]
- Boreen, A.L.; Arnold, W.A.; McNeill, K. Photodegradation of pharmaceuticals in the aquatic environment: A review. Aquat. Sci. 2003, 65, 320–341. [Google Scholar] [CrossRef]
- Tixier, C.; Singer, H.P.; Oellers, S.; Müller, S.R. Occurrence and Fate of Carbamazepine, Clofibric Acid, Diclofenac, Ibuprofen, Ketoprofen, and Naproxen in Surface Waters. Environ. Sci. Technol. 2003, 37, 1061–1068. [Google Scholar] [CrossRef]
- Poirier-Larabie, S.; Segura, P.A.; Gagnon, C. Degradation of the pharmaceuticals diclofenac and sulfamethoxazole and their transformation products under controlled environmental conditions. Sci. Total Environ. 2016, 557–558, 257–267. [Google Scholar] [CrossRef]
- Moore, D.E.; Zhou, W. Photodegradation of sulfamethoxazole: A chemical system capable of monitoring seasonal changes in UVB intensity. Photochem. Photobiol. 1994, 59, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Trovo, A.G.; Nogueira, R.F.P.; Aguera, A.; Sirtori, C.; Fernandez-Alba, A.R. Photodegradation of sulfamethoxazole in various aqueous media: Persistence, toxicity and photoproducts assessment. Chemosphere 2009, 77, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Zhang, L.; Li, Y.; Zhao, J.; Lv, S.; Xiao, K. Effects of environmental factors on sulfamethoxazole photodegradation under simulated sunlight irradiation: Kinetics and mechanism. J. Environ. Sci. 2013, 25, 1098–1106. [Google Scholar] [CrossRef]
- Ryan, C.C.; Tan, D.T.; Arnold, W.A. Direct and indirect photolysis of sulfamethoxazole and trimethoprim in wastewater treatment plant effluent. Water Res. 2011, 45, 1280–1286. [Google Scholar] [CrossRef] [PubMed]
- Gmurek, M.; Horn, H.; Majewsky, M. Phototransformation of sulfamethoxazole under simulated sunlight: Transformation products and their antibacterial activity toward Vibrio fischeri. Sci. Total Environ. 2015, 538, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 370. [Google Scholar] [CrossRef] [PubMed]
- Ollis, D.F.; Al-Ekabi, H. Photocatalytic Purification and Treatment of Water and Air; Elsevier: Amsterdam, The Netherlands, 1993. [Google Scholar]
- Pelizzetti, E.; Serpone, N. Photocatalysis: Fundamental and applications; Wiley: New York, USA, 1989. [Google Scholar]
- Legrini, O.; Oliveros, E.; Braun, A.M. Photochemical processes for water treatment. Chem. Rev. 1993, 93, 671–698. [Google Scholar] [CrossRef]
- Chong, M.N.; Jin, B.; Chow, C.W.K.; Saint, C. Recent developments in photocatalytic water treatment technology: A review. Water Res. 2010, 44, 2997–3027. [Google Scholar] [CrossRef]
- Kumaravel, V.; Mathew, S.; Bartlett, J.; Pillai, S.C. Photocatalytic hydrogen production using metal doped TiO2: A review of recent advances. Appl.Catal. B-Environ. 2019, 244, 1021–1064. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Tong, H.; Ouyang, S.X.; Bi, Y.P.; Umezawa, N.; Oshikiri, M.; Ye, J.H. Nano-photocatalytic materials: Possibilities and challenges. Adv. Mater. 2012, 24, 229–251. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Irie, H.; Fujishima, A. TiO2 photocatalysis: A historical overview and future prospects. Jpn. J. Appl. Phys. Part. 1-Regul. Pap. Brief. Commun. Rev. Pap. 2005, 44, 8269–8285. [Google Scholar] [CrossRef]
- Irie, H.; Watanabe, Y.; Hashimoto, K. Carbon-doped anatase TiO2 powders as a visible-light sensitive photocatalyst. Chem. Lett. 2003, 32, 772–773. [Google Scholar] [CrossRef]
- Pan, J.H.; Dou, H.; Xiong, Z.; Xu, C.; Ma, J.; Zhao, X.S. Porous photocatalysts for advanced water purifications. J. Mater. Chem. 2010, 20, 4512–4528. [Google Scholar] [CrossRef]
- Wang, X.C.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.J.; Tan, L.L.; Ng, Y.H.; Yong, S.T.; Chai, S.P. Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: Are we a step closer to achieving sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.C.; Antonietti, M. Polymeric graphitic carbon nitride as a heterogeneous organocatalyst: From Photochemistry to multipurpose catalysis to sustainable chemistry. Angew. Chem.-Int. Ed. 2012, 51, 68–89. [Google Scholar] [CrossRef]
- Cao, S.W.; Low, J.X.; Yu, J.G.; Jaroniec, M. polymeric photocatalysts based on graphitic carbon nitride. Adv. Mater. 2015, 27, 2150–2176. [Google Scholar] [CrossRef]
- Sapurina, I.Y.; Shishov, M.A. Oxidative polymerization of aniline: Molecular synthesis of polyaniline and the formation of supramolecular structures. In New Polymers for Special Applications; Gomes, A.D.S., Ed.; IntechOpen: London, UK, 12 September 2012; pp. 252–312. Available online: https://www.intechopen.com/books/new-polymers-for-special-applications/oxidative-polymerization-of-aniline-molecular-synthesis-of-polyaniline-and-the-formation-of-supramol (accessed on 2 October 2019).
- Bell, A.T. The impact of nanoscience on heterogeneous catalysis. Science 2003, 299, 1688–1691. [Google Scholar] [CrossRef]
- Minella, M.; Fabbri, D.; Calza, P.; Minero, C. Selected hybrid photocatalytic materials for the removal of drugs from water. Curr. Opin. Green Sustain. Chem. 2017, 6, 11–17. [Google Scholar] [CrossRef]
- Shan, A.Y.; Ghazi, T.I.M.; Rashid, S.A. Immobilisation of titanium dioxide onto supporting materials in heterogeneous photocatalysis: A review. Appl. Catal. A-Gen. 2010, 389, 1–8. [Google Scholar] [CrossRef]
- Awfa, D.; Ateia, M.; Fujii, M.; Johnson, M.S.; Yoshimura, C. Photodegradation of pharmaceuticals and personal care products in water treatment using carbonaceous-TiO2 composites: A critical review of recent literature. Water Res. 2018, 142, 26–45. [Google Scholar] [CrossRef] [PubMed]
- Khalid, N.R.; Majid, A.; Tahir, M.B.; Niaz, N.A.; Khalid, S. Carbonaceous-TiO2 nanomaterials for photocatalytic degradation of pollutants: A review. Ceram. Int. 2017, 43, 14552–14571. [Google Scholar] [CrossRef]
- Lim, T.T.; Yap, P.S.; Srinivasan, M.; Fane, A.G. TiO2/AC Composites for synergistic adsorption-photocatalysis processes: Present challenges and further developments for water treatment and reclamation. Crit. Rev. Environ. Sci. Technol. 2011, 41, 1173–1230. [Google Scholar] [CrossRef]
- Meng, Z.D.; Choi, J.G.; Park, J.Y.; Zhu, L.; Oh, W.C. Review for fullerene materials enhanced TiO2 photocatalysis. J. Photocatl. Sci 2011, 2, 29–38. [Google Scholar]
- Woan, K.; Pyrgiotakis, G.; Sigmund, W. Photocatalytic carbon-nanotube–TiO2 composites. Adv. Mater. 2009, 21, 2233–2239. [Google Scholar] [CrossRef]
- Cao, Q.M.; Yu, Q.M.; Connell, D.W.; Yu, G. Titania/carbon nanotube composite (TiO2/CNT) and its application for removal of organic pollutants. Clean Technol. Environ. Policy 2013, 15, 871–880. [Google Scholar] [CrossRef]
- Lam, S.-M.; Sin, J.-C.; Abdullah, A.Z.; Mohamed, A.R. Photocatalytic TiO2/carbon nanotube nanocomposites for environmental applications: An overview and recent developments. Fuller. Nanotub. Carbon Nanostruct.S 2014, 22, 471–509. [Google Scholar] [CrossRef]
- Zhang, Y.; Utke, I.; Michler, J.; Ilari, G.; Rossell, M.D.; Erni, R. Growth and characterization of CNT–TiO2 heterostructures. Beilsteinj. Nanotechnol. 2014, 5, 946–955. [Google Scholar] [CrossRef]
- Morales-Torres, S.; Pastrana-Martínez, L.M.; Figueiredo, J.L.; Faria, J.L.; Silva, A.M.T. Design of graphene-based TiO2 photocatalysts—A review. Environ. Sci. Poll. Res. 2012, 19, 3676–3687. [Google Scholar] [CrossRef]
- Bhanvase, B.A.; Shende, T.P.; Sonawane, S.H. A review on graphene–TiO2 and doped graphene–TiO2 nanocomposite photocatalyst for water and wastewater treatment. Environ. Technol. Rev. 2017, 6, 1–14. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Wageh, S.; Al-Ghamdi, A.A.; Xie, J. Graphene in photocatalysis: A Review. Small 2016, 12, 6640–6696. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.-L.; Chai, S.-P.; Mohamed, A.R. Synthesis and applications of graphene-based TiO2 photocatalysts. ChemSusChem 2012, 5, 1868–1882. [Google Scholar] [CrossRef] [PubMed]
- Faraldos, M.; Bahamonde, A. Environmental applications of titania-graphene photocatalysts. Catal. Today 2017, 285, 13–28. [Google Scholar] [CrossRef]
- Leary, R.; Westwood, A. Carbonaceous nanomaterials for the enhancement of TiO2 photocatalysis. Carbon 2011, 49, 741–772. [Google Scholar] [CrossRef]
- Ania, C.O.; Velasco, L.F.; Valdés-Solís, T. Photochemical behavior of carbon adsorbents. In Novel Carbon Adsorbents; Tascón, J.M.D., Ed.; Elsevier: Oxford, UK, 2012; pp. 521–547. [Google Scholar]
- Wang, W.; Serp, P.; Kalck, P.; Faria, J.L. Visible light photodegradation of phenol on MWNT-TiO2 composite catalysts prepared by a modified sol–gel method. J. Mol. Catal. A Chem. 2005, 235, 194–199. [Google Scholar] [CrossRef]
- Chen, X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef]
- Valentin, C.D.; Pacchioni, G.; Selloni, A. Theory of carbon doping of titanium dioxide. Chem. Mater. 2005, 17, 6656–6665. [Google Scholar] [CrossRef]
- Sakthivel, S.; Kisch, H. Daylight photocatalysis by carbon-modified titanium dioxide. Angew. Chem. Int. Ed. 2003, 42, 4908–4911. [Google Scholar] [CrossRef]
- Surenjan, A.; Sambandam, B.; Pradeep, T.; Philip, L. Synthesis, characterization and performance of visible light active C-TiO2 for pharmaceutical photodegradation. J. Environ. Chem. Eng. 2017, 5, 757–767. [Google Scholar] [CrossRef]
- Sambandam, B.; Surenjan, A.; Philip, L.; Pradeep, T. Rapid synthesis of C-TiO2: Tuning the shape from spherical to rice grain morphology for visible light photocatalytic application. ACS Sustain. Chem. Eng. 2015, 3, 1321–1329. [Google Scholar] [CrossRef]
- Buda, W.; Czech, B. Preparation and characterization of C,N-codoped TiO2 photocatalyst for the degradation of diclofenac from wastewater. Water Sci. Technol. 2013, 68, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Garcia, A.; Guzman-Mar, J.L.; Hinojosa-Reyes, L.; Ruiz-Ruiz, E.; Hernandez-Ramirez, A. Effect of carbon doping on WO3/TiO2 coupled oxide and its photocatalytic activity on diclofenac degradation. Ceram. Int. 2016, 42, 9796–9803. [Google Scholar] [CrossRef]
- Nazarkovsky, M.A.; Bogatyrov, V.M.; Czech, B.; Galaburda, M.V.; Wojcik, G.; Kolomys, O.F.; Strelchuk, V.V.; Malysheva, M.L.; Oranska, O.I.; Gun’ko, V.M. Synthesis and properties of zinc oxide photocatalyst by high-temperature processing of resorcinol-formaldehyde/zinc acetate mixture. J. Photochem. Photobiol. A-Chem. 2017, 334, 36–46. [Google Scholar] [CrossRef]
- Schulze-Hennings, U.; Bruckner, I.; Gebhardt, W.; Groteklaes, M.; Bloss, S.P.; Wett, M.; Linnemann, V.; Montag, D.; Pinnekamp, J. Durability of a coating containing titanium dioxide for the photocatalytic degradation of diclofenac in water with UV-A irradiation. Water Environ. J. 2017, 31, 508–514. [Google Scholar] [CrossRef]
- Tanguay, J.F.; Suib, S.L.; Coughlin, R.W. Dichloromethane photodegradation using titanium catalysts. J. Catal. 1989, 117, 335–347. [Google Scholar] [CrossRef]
- Matos, J.; Laine, J.; Herrmann, J.M. Synergy effect in the photocatalytic degradation of phenol on a suspended mixture of titania and activated carbon. Appl. Catal. B-Environ. 1998, 18, 281–291. [Google Scholar] [CrossRef]
- Matos, J.; Laine, J.; Herrmann, J.M. Association of activated carbons of different origins with titania in the photocatalytic purification of water. Carbon 1999, 37, 1870–1872. [Google Scholar] [CrossRef]
- Matos, J.; Laine, J.; Herrmann, J.M. Effect of the type of activated carbons on the photocatalytic degradation of aqueous organic pollutants by UV-irradiated titania. J. Catal. 2001, 200, 10–20. [Google Scholar] [CrossRef]
- Ocampo-Pérez, R.; Sánchez-Polo, M.; Rivera-Utrilla, J.; Leyva-Ramos, R. Enhancement of the catalytic activity of TiO2 by using activated carbon in the photocatalytic degradation of cytarabine. Appl. Catal. B-Environ. 2011, 104, 177–184. [Google Scholar]
- Bandosz, T.J.; Ania, C.O. Origin and perspectives of the photochemical activity of nanoporous carbons. Adv. Sci. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Velasco, L.F.; Parra, J.B.; Ania, C.O. Role of activated carbon features on the photocatalytic degradation of phenol. Appl. Surf. Sci. 2010, 256, 5254–5258. [Google Scholar] [CrossRef] [Green Version]
- Velasco, L.F.; Maurino, V.; Laurenti, E.; Fonseca, I.M.; Lima, J.C.; Ania, C.O. Photoinduced reactions occurring on activated carbons. A combined photooxidation and ESR study. Appl. Catal. A-Gen.L 2013, 452, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Velasco, L.F.; Maurino, V.; Laurenti, E.; Ania, C. Light-induced generation of radicals on semiconductor-free carbon photocatalysts. Appl. Catal. A-Gen. 2013, 453, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Andrade, M.A.; Mestre, A.S.; Carmona, R.J.; Carvalho, A.P.; Ania, C.O. Effect of the irradiation wavelength on the performance of nanoporous carbon as an additive to TiO2. Appl. Catal. A-Gen. 2015, 507, 91–98. [Google Scholar] [CrossRef]
- Rioja, N.; Benguria, P.; Penas, F.J.; Zorita, S. Competitive removal of pharmaceuticals from environmental waters by adsorption and photocatalytic degradation. Environ. Sci. Pollut. Res. 2014, 21, 11168–11177. [Google Scholar] [CrossRef] [PubMed]
- Ziegmann, M.; Frimmel, F.H. Photocatalytic degradation of clofibric acid, carbamazepine and iomeprol using conglomerated TiO2 and activated carbon in aqueous suspension. Water Sci. Technol. 2010, 61, 273–281. [Google Scholar] [CrossRef]
- Alalm, M.G.; Tawfik, A.; Ookawara, S. Enhancement of photocatalytic activity of TiO2 by immobilization on activated carbon for degradation of pharmaceuticals. J. Environ. Chem. Eng. 2016, 4, 1929–1937. [Google Scholar] [CrossRef]
- Moreno-Valencia, E.I.; Paredes-Carrera, S.P.; Sanchez-Ochoa, J.C.; Flores-Valle, S.O.; Avendano-Gomez, J.R. Diclofenac degradation by heterogeneous photocatalysis with Fe3O4/TixOy/activated carbon fiber composite synthesized by ultrasound irradiation. Mater. Res. Express 2017, 4. [Google Scholar]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Yi, H.; Huang, D.L.; Qin, L.; Zeng, G.M.; Lai, C.; Cheng, M.; Ye, S.J.; Song, B.; Ren, X.Y.; Guo, X.Y. Selective prepared carbon nanomaterials for advanced photocatalytic application in environmental pollutant treatment and hydrogen production. Appl. Catal. B-Environ. 2018, 239, 408–424. [Google Scholar] [CrossRef]
- Luo, Y.; Heng, Y.; Dai, X.; Chen, W.; Li, J. Preparation and photocatalytic ability of highly defective carbon nanotubes. J. Solid State Chem. 2009, 182, 2521–2525. [Google Scholar] [CrossRef]
- Murgolo, S.; Petronella, F.; Ciannarella, R.; Comparelli, R.; Agostiano, A.; Curri, M.L.; Mascolo, G. UV and solar-based photocatalytic degradation of organic pollutants by nano-sized TiO2 grown on carbon nanotubes. Catal. Today 2015, 240, 114–124. [Google Scholar] [CrossRef]
- Zhu, W.Y.; Li, Z.; He, C.; Sun, F.Q.; Yan, Z. Enhanced photodegradation of sulfamethoxazole by a novel WO3-CNT composite under visible light irradiation. J. Alloy. Compd. 2018, 754, 153–162. [Google Scholar] [CrossRef]
- Martinez, C.; Canle, M.; Fernandez, M.I.; Santaballa, J.A.; Faria, J. Kinetics and mechanism of aqueous degradation of carbamazepine by heterogeneous photocatalysis using nanocrystalline TiO2, ZnO and multi-walled carbon nanotubes-anatase composites. Appl. Catal. B-Environ. L. 2011, 102, 563–571. [Google Scholar] [CrossRef]
- Czech, B.; Buda, W. Photocatalytic treatment of pharmaceutical wastewater using new multiwall-carbon nanotubes/TiO2/SiO2 nanocomposites. Environ. Res. 2015, 137, 176–184. [Google Scholar] [CrossRef]
- Martinez, C.; Canle, M.; Fernandez, M.I.; Santaballa, J.A.; Faria, J. Aqueous degradation of diclofenac by heterogeneous photocatalysis using nanostructured materials. Appl. Catal. B-Environ. 2011, 107, 110–118. [Google Scholar] [CrossRef]
- Czech, B.; Buda, W. Multicomponent nanocomposites for elimination of diclofenac in water based on an amorphous TiO2 active in various light sources. J. Photochem. Photobiol A-Chem. 2016, 330, 64–70. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Xiang, Q.J.; Yu, J.G.; Jaroniec, M. Graphene-based semiconductor photocatalysts. Chem. Soc. Rev. 2012, 41, 782–796. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, Y.H.; Xu, Y.J. Recent progress on graphene-based photocatalysts: Current status and future perspectives. Nanoscale 2012, 4, 5792–5813. [Google Scholar] [CrossRef] [PubMed]
- An, X.Q.; Yu, J.C. Graphene-based photocatalytic composites. RSC Adv. 2011, 1, 1426–1434. [Google Scholar] [CrossRef]
- Nawaz, M.; Miran, W.; Jang, J.; Lee, D.S. One-step hydrothermal synthesis of porous 3D reduced graphene oxide/TiO2 aerogel for carbamazepine photodegradation in aqueous solution. Appl. Catal. B-Environ. 2017, 203, 85–95. [Google Scholar] [CrossRef]
- Lin, L.; Wang, H.; Xu, P. Immobilized TiO2-reduced graphene oxide nanocomposites on optical fibers as high performance photocatalysts for degradation of pharmaceuticals. Chem. Eng. J. 2017, 310, 389–398. [Google Scholar] [CrossRef]
- Linley, S.; Liu, Y.Y.; Ptacek, C.J.; Blowes, D.W.; Gu, F.X. Recyclable Graphene Oxide-Supported Titanium Dioxide Photocatalysts with Tunable Properties. Acs Appl. Mater. Interfaces 2014, 6, 4658–4668. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.Y.; Feng, J.L.; Fan, M.H.; Pi, Y.Q.; Hu, L.M.; Han, X.; Liu, M.L.; Sun, J.Y.; Sun, J.H. Recent developments in heterogeneous photocatalytic water treatment using visible light-responsive photocatalysts: A review. Rsc Adv. 2015, 5, 14610–14630. [Google Scholar] [CrossRef]
- Karaolia, P.; Michael-Kordatou, I.; Hapeshi, E.; Drosou, C.; Bertakis, Y.; Christofilos, D.; Armatas, G.S.; Sygellou, L.; Schwartz, T.; Xekoukoulotakis, N.P.; et al. Removal of antibiotics, antibiotic-resistant bacteria and their associated genes by graphene-based TiO2 composite photocatalysts under solar radiation in urban wastewaters. Appl. Catal. B: Environ. 2018, 224, 810–824. [Google Scholar] [CrossRef]
- Yang, L.; Xu, L.; Bai, X.; Jin, P. Enhanced visible-light activation of persulfate by Ti3+ self-doped TiO2/graphene nanocomposite for the rapid and efficient degradation of micropollutants in water. J. Hazard. Mater. 2019, 365, 107–117. [Google Scholar] [CrossRef]
- Amalraj Appavoo, I.; Hu, J.; Huang, Y.; Li, S.F.Y.; Ong, S.L. Response surface modeling of Carbamazepine (CBZ) removal by Graphene-P25 nanocomposites/UVA process using central composite design. Water Res. 2014, 57, 270–279. [Google Scholar] [CrossRef]
- Cheng, X.; Deng, X.; Wang, P.; Liu, H. Coupling TiO2 nanotubes photoelectrode with Pd nano-particles and reduced graphene oxide for enhanced photocatalytic decomposition of diclofenac and mechanism insights. Sep. Purif. Technol. 2015, 154, 51–59. [Google Scholar] [CrossRef]
- Li, W.; Yu, R.; Li, M.; Guo, N.; Yu, H.; Yu, Y. Photocatalytical degradation of diclofenac by Ag-BiOI-rGO: Kinetics, mechanisms and pathways. Chemosphere 2019, 218, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhang, Q.; Shen, L.; Li, R.; Tan, C.; Chen, T.; Liu, H.; Liu, Y.; Cai, Z.; Liu, G.; et al. Insights into the synergetic mechanism of a combined vis-RGO/TiO2/peroxodisulfate system for the degradation of PPCPs: Kinetics, environmental factors and products. Chemosphere 2019, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ray, R.; Gu, Y.; Ploehn, H.J.; Gearheart, L.; Raker, K.; Scrivens, W.A. Electrophoretic analysis and purification of fluorescent single-walled carbon nanotube fragments. J. Am. Chem. Soc. 2004, 126, 12736–12737. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-P.; Zhou, B.; Lin, Y.; Wang, W.; Fernando, K.A.S.; Pathak, P.; Meziani, M.J.; Harruff, B.A.; Wang, X.; Wang, H.; et al. Quantum-sized carbon dots for bright and colorful photoluminescence. J. Am. Chem. Soc. 2006, 128, 7756–7757. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Shen, W.; Gao, Z.Q. Carbon quantum dots and their applications. Chem. Soc. Rev. 2015, 44, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Li, H.T.; Kang, Z.H.; Liu, Y.; Lee, S.T. Carbon nanodots: Synthesis, properties and applications. J. Mater. Chem. 2012, 22, 24230–24253. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, Q.X.; Su, Y.H.; Shen, L.Z.; Wang, F.L.; Liu, H.J.; Liu, Y.; Cai, Z.W.; Lv, W.Y.; Liu, G.G. Accelerated photocatalytic degradation of diclofenac by a novel CQDs/BiOCOOH hybrid material under visible-light irradiation: Dechloridation, detoxicity, and a new superoxide radical model study. Chem.Eng. J. 2018, 332, 737–748. [Google Scholar] [CrossRef]
- Wang, F.L.; Wu, Y.L.; Wang, Y.F.; Li, J.H.; Jin, X.Y.; Zhang, Q.X.; Li, R.B.; Yan, S.C.; Liu, H.J.; Feng, Y.P.; et al. Construction of novel Z-scheme nitrogen-doped carbon dots/{001} TiO2 nanosheet photocatalysts for broad-spectrum-driven diclofenac degradation: Mechanism insight, products and effects of natural water matrices. Chem. Eng. J. 2019, 356, 857–868. [Google Scholar] [CrossRef]
- Liu, W.; Li, Y.; Liu, F.; Jiang, W.; Zhang, D.; Liang, J. Visible-light-driven photocatalytic degradation of diclofenac by carbon quantum dots modified porous g-C3N4: Mechanisms, degradation pathway and DFT calculation. Water Res. 2019, 151, 8–19. [Google Scholar] [CrossRef]
- Song, Y.L.; Tian, J.Y.; Gao, S.S.; Shao, P.H.; Qi, J.Y.; Cui, F.Y. Photodegradation of sulfonamides by g-C3N4 under visible light irradiation: Effectiveness, mechanism and pathways. Appl. Catal. B-Environ. 2017, 210, 88–96. [Google Scholar] [CrossRef]
- Mestre, A.S.; Carvalho, A.P. Nanoporous carbons synthesis: An old story with exciting new chapters. In Porosity; Ghrib, T., Ed.; IntechOpen: London, UK, 26 April 2018; pp. 37–68. Available online: https://www.intechopen.com/books/porosity-process-technologies-and-applications/nanoporous-carbon-synthesis-an-old-story-with-exciting-new-chapters (accessed on 5 August 2019).
- Chen, N.; Huang, Y.; Hou, X.; Ai, Z.; Zhang, L. Photochemistry of hydrochar: Reactive oxygen species generation and sulfadimidine degradation. Environ. Sci. Technol. 2017, 51, 11278–11287. [Google Scholar] [CrossRef] [PubMed]
- Jäger, H.; Frohs, W.; Collin, G.; von Sturm, F.; Vohler, O.; Nutsch, G. Carbon, 1. General. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA, 2010; Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/14356007.a05_095.pub2 (accessed on 28 August 2019).
- Xie, X.; Li, S.; Zhang, H.; Wang, Z.; Huang, H. Promoting charge separation of biochar-based Zn-TiO2/pBC in the presence of ZnO for efficient sulfamethoxazole photodegradation under visible light irradiation. Sci. Total Environ. 2019, 659, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, Z.; Zhao, X.; Yang, X.; Liang, G.; Xie, X. Insight into enhanced carbamazepine photodegradation over biochar-based magnetic photocatalyst Fe3O4/BiOBr/BC under visible LED light irradiation. Chem. Eng. J. 2019, 360, 600–611. [Google Scholar] [CrossRef]
- Khraisheh, M.; Kim, J.; Campos, L.; Al-Muhtaseb, A.H.; Walker, G.M.; AlGhouti, M. Removal of carbamazepine from water by a novel TiO2-coconut shell powder/UV process: Composite preparation and photocatalytic activity. Environ. Eng. Sci. 2013, 30, 515–526. [Google Scholar] [CrossRef]
- Khraisheh, M.; Kim, J.; Campos, L.; Al-Muhtaseb, A.H.; Al-Hawari, A.; Al Ghouti, M.; Walker, G.M. Removal of pharmaceutical and personal care products (PPCPs) pollutants from water by novel TiO2-Coconut Shell Powder (TCNSP) composite. J. Ind. Eng. Chem. 2014, 20, 979–987. [Google Scholar] [CrossRef]
- Sanchez, V.C.; Pietruska, J.R.; Miselis, N.R.; Hurt, R.H.; Kane, A.B. Biopersistence and potential adverse health impacts of fibrous nanomaterials: What have we learned from asbestos? Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 511–529. [Google Scholar] [CrossRef]
- Jachak, A.C.; Creighton, M.; Qiu, Y.; Kane, A.B.; Hurt, R.H. Biological interactions and safety of graphene materials. MRS Bull. 2012, 37, 1307–1313. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
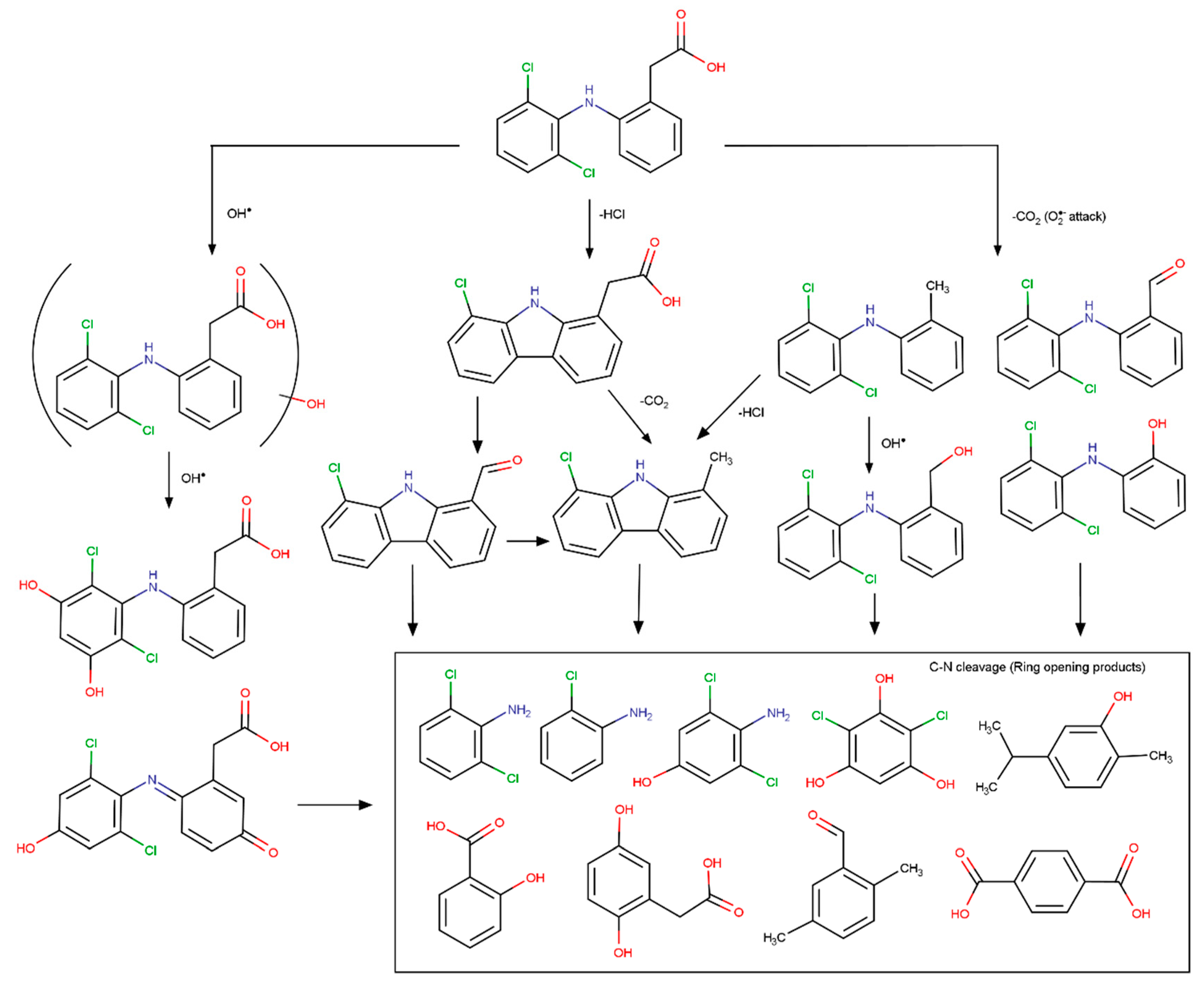{kind=link}
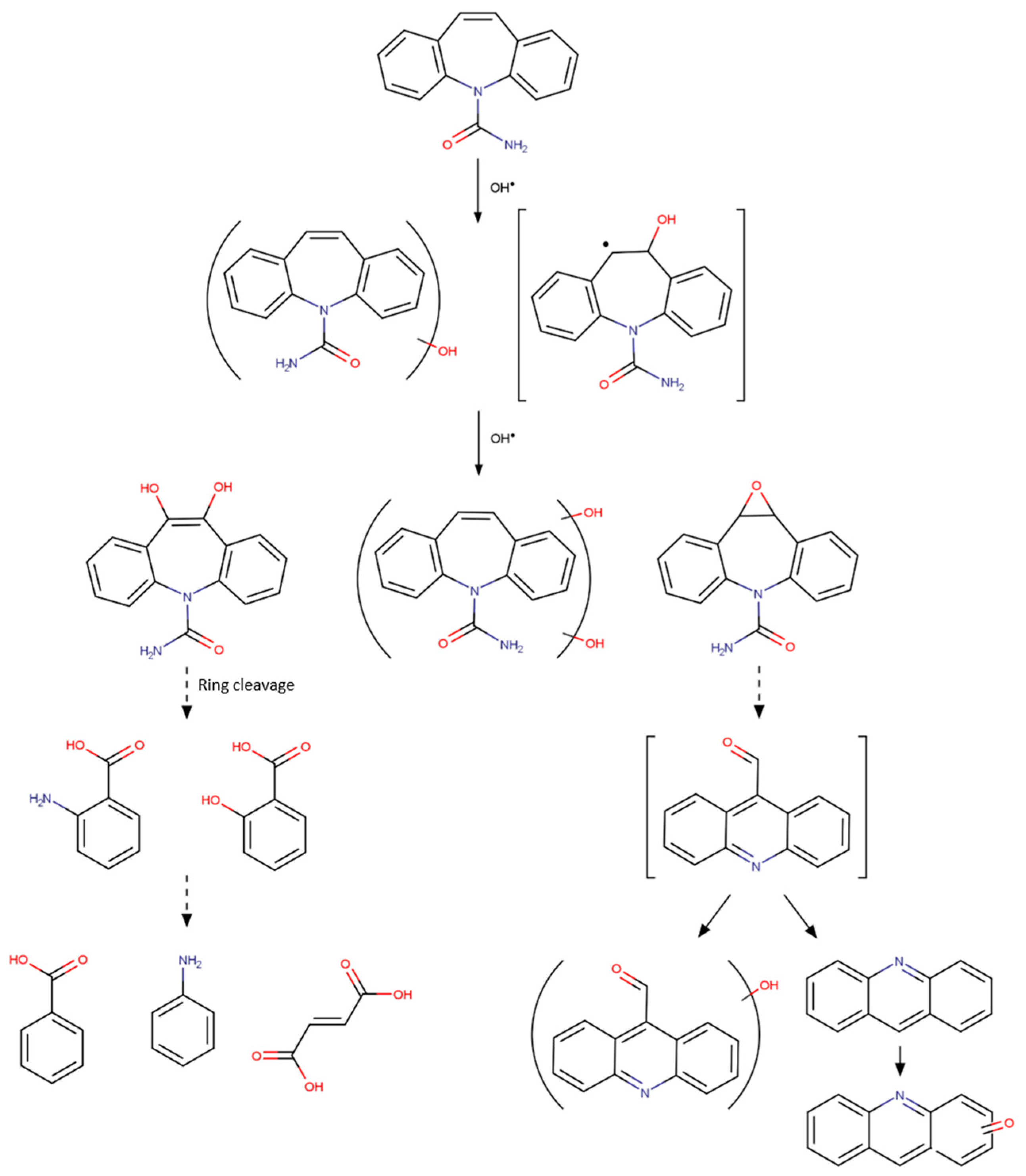{kind=link}
| Pharmaceutical Molecular Structure | Therapeutic Class | Properties |
|---|---|---|
Carbamazepine (CBZ) | Antiepileptic | MW = 236.3 g/mol pKa = 13.9 [4] Log Kow = 2.45 [4] Water solubility (25 °C) = 18 mg/dm3 [4] kH = 1.1 × 10−10 atm m3/mol [4] Kd,prim < 0.020 L/gss [5] Kd,sec = 0.0012 ± 0.0005 L/gss [5] kbiol ≤ 0.01 L/gss·d [6] KOH = 8.8 × 109 ± 1.2 × 109 1/M s [7] |
Diclofenac (DCF) | AnalgesicAnti-inflammatory | MW (acid) = 296.1 g/mol MW (sodium salt) = 318.1 g/mol pKa = 4.15 (acid) [8] Log Kow = 4.51 [8] Water solubility (25 °C) = 2.37 mg/dm3 [8] kH = 4.73 × 10−12 atm m3/mol [8] Kd,prim = 0.495 ± 0.032 L/gss [5] Kd,sec = 0.016 ± 0.003 L/gss [5] kbiol ≤ 0.1 L/gss·d [6] KOH = 7.5 × 109 ± 1.5 × 109 1/M s [7] |
Sulfamethoxazole (SMX) | Antibiotic | MW = 253.3 g/mol pKa1 = 1.6, pKa2 = 5.7 [9] Log Kow = 0.89 [9] Water solubility (37 °C) = 610 mg/dm3 [9] kH = 6.4 × 10−13 atm m3/mol [9] Kd = 0.256 ± 0.169 L/gss [10] kbiol ≤ 0.2 L/gss·d [6] KOH = 5.5 × 109 ± 7 × 109 1/M s [7] |
| PhC | Type of Water | Concentration (μg/L) | Publication Year | Ref. |
|---|---|---|---|---|
| CBZ | STP effluents | 3.7 (max 6.3) | 1998 | [19] |
| Rivers and streams | 0.82 (max 1.1) | |||
| Ground water | Up to 0.900 | 2001 | [26] | |
| STP influent (Frankfurt/Mains) | 2.2 | 2001 | [18] | |
| STP effluent (Frankfurt/Mains) | 2.0 | |||
| Ground water | Up to 1.1 | |||
| Drinking water | Up to 0.030 | |||
| STP influent (Berlin) | 1.78 (max 3.80) | 2002 | [17] | |
| STP effluent (Berlin) | 1.63 (max 5.00) | |||
| Surface water (Berlin) | 0.4–1.1 | |||
| Surface water (Berlin) | 0.025–1.075 | 2002 | [24] | |
| STP effluent (France) | 0.98–1.2 | 2003 | [23] | |
| STP effluent (Greece) | 1.03 | |||
| STP effluent (Italy) | 0.3–0.5 | |||
| STP effluent (Sweden) | 0.87 | |||
| DCF | Sedimentation tank effluent | ≤2 | 1996 | [27] (in [21]) |
| River Rhine | 0.015–0.304 | |||
| Different rivers | 0.038–0.489 | |||
| STP effluents | 1.6 (max 2.1) | 1998 | [19] | |
| Rivers and streams | 0.80 (max 1.2) | |||
| Influent Swiss STP | 0.47–1.92 | 1998 | [22] | |
| Effluent Swiss STP | 0.31–0.93 | |||
| Swiss lakes/rivers | 0.001–0.370 | |||
| Brazilian STP | 0.4 (max ≈ 1.4) | 1999 | [20] | |
| River water | 0.02–0.06 | |||
| STP influent (Frankfurt/Mains) | 1.9 | 2001 | [18] | |
| STP effluent (Frankfurt/Mains) | 0.58 | |||
| Ground water | 0.93 | |||
| Drinking water | Up to 0.006 | |||
| Ground water | Up to 0.590 | 2001 | [26] | |
| STP influent (Berlin) | 3.02 (max 7.10) | 2002 | [17] | |
| STP effluent (Berlin) | 2.51 (max 4.70) | |||
| Surface water | < 0.1–0.6 | |||
| Ground water (Berlin) | Up to 0.38 | |||
| Surface water (Berlin) | Up to 1.030 | 2002 | [24] | |
| STP effluent (France) | 0.25–0.41 | 2003 | [23] | |
| STP effluent (Greece) | 0.89 | |||
| STP effluent (Italy) | 0.47–5.45 | |||
| SMX | River water | ≈1 | 1983 | [28] (in [21]) |
| STP effluents | 0.9 (max 2.0) | 1999 | [25] | |
| Surface water | 0.14 (max 0.48) | |||
| Ground water | Up to 0.47 | |||
| Ground water | Up to 0.410 | 2001 | [26] | |
| STPs effluent (Berlin) | 0.9 | 2002 | [17] | |
| STP effluent (France) | 0.07–0.09 | 2003 | [23] | |
| STP effluent (Greece) | 0.09 | |||
| STP effluent (Italy) | 0.01–0.03 | |||
| STP effluent (Sweden) | 0.02 |
| PhC | Catalyst | Experimental Conditions | Concluding Remarks | Ref. |
|---|---|---|---|---|
| CBZ DCF | TiO2/C |
|
| [91] |
| CBZ | TiO2/C |
|
| [92] |
| DCF | TiO2/C TiO2/C,N |
|
| [93] |
| DCF | WO3/TiO2-C WO3/TiO2 TiO2 |
|
| [94] |
| DCF | ZnO/C ZnO TiO2 |
|
| [95] |
| DCF | TiO2/C |
|
| [96] |
| PhC | Catalyst | Experimental Conditions | Concluding Remarks | Ref. |
|---|---|---|---|---|
| SMX DCF CBZ | TiO2/PAC TiO2 |
|
| [107] |
| CBZ | TiO2/PAC TiO2 PAC |
|
| [108] |
| DCF | TiO2/AC TiO2 |
|
| [109] |
| DCF | Fe3O4/TixOy/ACfiber Fe3O4/TixOy TiO2 |
|
| [110] |
| PhC | Catalyst | Experimental Conditions | Concluding Remarks | Ref. |
|---|---|---|---|---|
| SMX DCF CBZ | TiO2/SWCNT TiO2 |
|
| [114] |
| SMX | WO3/MWCNT WO3 |
|
| [115] |
| CBZ | TiO2/MWCNTox TiO2 ZnO |
|
| [116] |
| CBZ | TiO2-SiO2/MWCNT TiO2 |
|
| [117] |
| DCF | TiO2/MWCNTox TiO2 |
|
| [118] |
| DCF | SiO2-TiO2/MWCNT SiO2-TiO2 TiO2 |
|
| [119] |
| PhC | Catalyst | Experimental Conditions | Concluding Remarks | Ref. |
|---|---|---|---|---|
| SMX CBZ | TiO2/rGO TiO2 |
|
| [125] |
| SMX | TiO2/rGO TiO2 |
|
| [128] |
| SMX | TiO2/rGO TiO2 |
|
| [129] |
| CBZ | TiO2/Fe3O4/rGO TiO2 |
|
| [126] |
| CBZ | TiO2/GO TiO2 |
|
| [130] |
| CBZ | TiO2/3D rGO TiO2 |
|
| [124] |
| DCF | TiO2 nanotubes/Pd-rGO |
|
| [131] |
| DCF | Ag-BiOI/rGO BiOI Ag-BiOI BiOI/rGO |
|
| [132] |
| DCF | TiO2/rGO |
|
| [133] |
| PhC | Catalyst | Experimental Conditions | Concluding Remarks | Ref. |
|---|---|---|---|---|
| DCF | BiOCOOH/CQD TiO2 |
|
| [138] |
| DCF | TiO2{001}/N-CQD TiO2{101}/N-CQD TiO2(P25)/N-CQD |
|
| [139] |
| DCF | g-C3N4/CQD g-C3N4 |
|
| [140] |
| PhC | Catalyst | Experimental Conditions | Concluding Remarks | Ref. |
|---|---|---|---|---|
| SMX | Zn-TiO2/biochar TiO2 TiO2/biochar |
|
| [145] |
| CBZ | Fe3O4/BiOBr/Biochar Fe3O4/BiOBr BiOBr |
|
| [146] |
| CBZ | TiO2/biochar TiO2 biochar |
|
| [147] |
| CBZ | TiO2/biochar TiO2 GAC biochar |
|
| [148] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mestre, A.S.; Carvalho, A.P. Photocatalytic Degradation of Pharmaceuticals Carbamazepine, Diclofenac, and Sulfamethoxazole by Semiconductor and Carbon Materials: A Review. Molecules 2019, 24, 3702. https://doi.org/10.3390/molecules24203702
Mestre AS, Carvalho AP. Photocatalytic Degradation of Pharmaceuticals Carbamazepine, Diclofenac, and Sulfamethoxazole by Semiconductor and Carbon Materials: A Review. Molecules. 2019; 24(20):3702. https://doi.org/10.3390/molecules24203702
Chicago/Turabian StyleMestre, Ana S., and Ana P. Carvalho. 2019. "Photocatalytic Degradation of Pharmaceuticals Carbamazepine, Diclofenac, and Sulfamethoxazole by Semiconductor and Carbon Materials: A Review" Molecules 24, no. 20: 3702. https://doi.org/10.3390/molecules24203702
APA StyleMestre, A. S., & Carvalho, A. P. (2019). Photocatalytic Degradation of Pharmaceuticals Carbamazepine, Diclofenac, and Sulfamethoxazole by Semiconductor and Carbon Materials: A Review. Molecules, 24(20), 3702. https://doi.org/10.3390/molecules24203702