
Late-Stage Copper-Catalyzed Radiofluorination of an Arylboronic Ester Derivative of Atorvastatin

 ,
,
Abstract
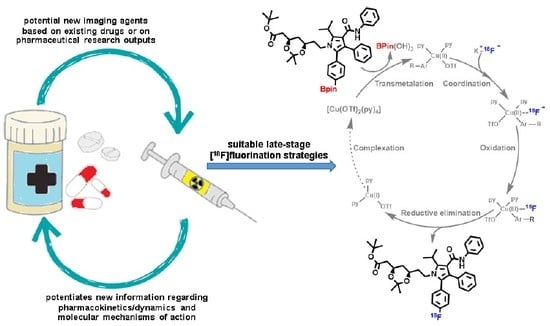
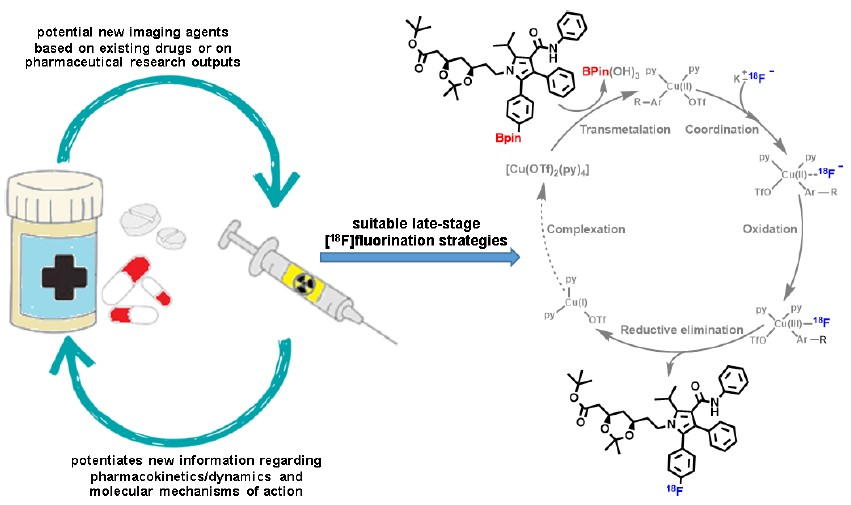
:
{kind=link}
{kind=link}
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
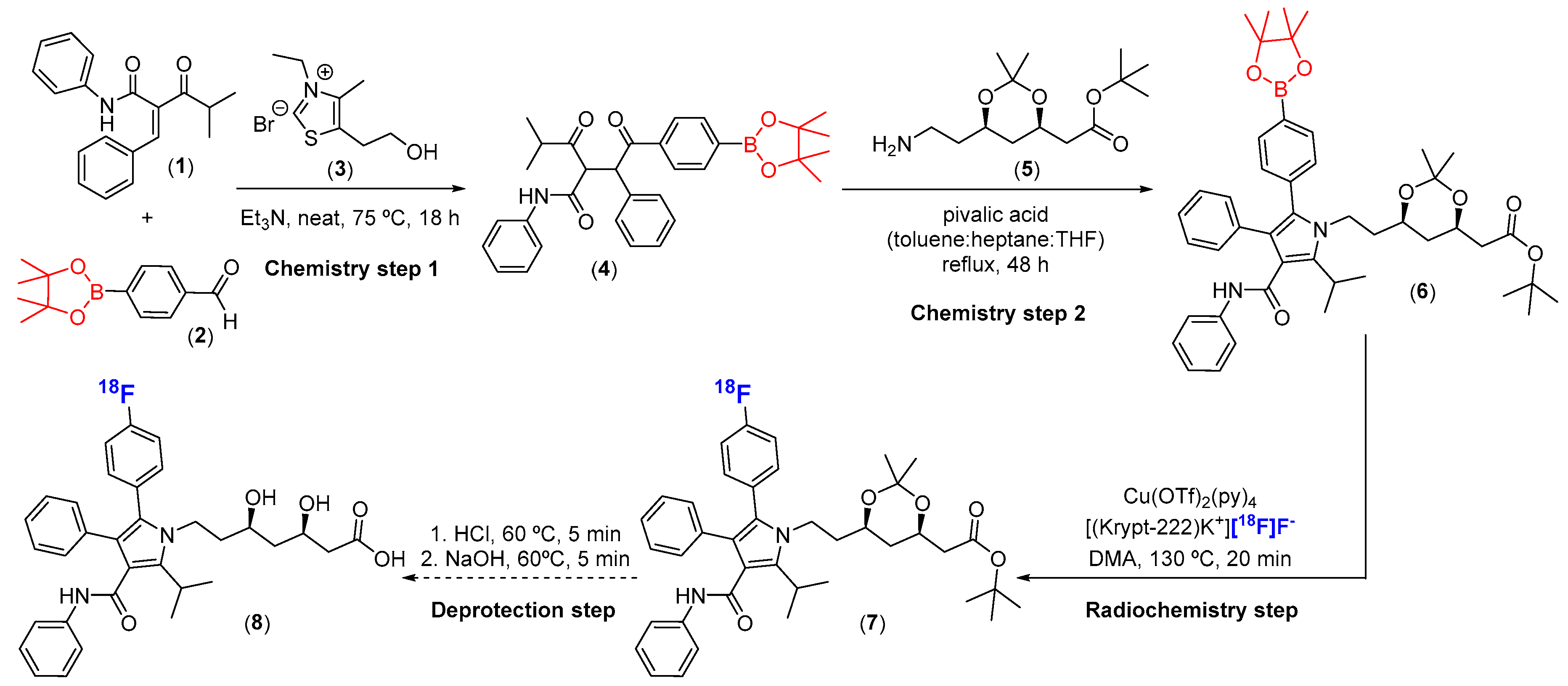
4.1. General Procedure for the Synthesis of the Arylboronic Pinacol Ester (Bpin) Labeling Precursor (6)
4.2. Characterization Data
4.3. General Procedure for the Cu-mediated Radiosynthesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Elsinga, P.H.; van Waarde, A.; Paans, A.M.J.; Dierckx, R.A.J.O. Trends on the Role of PET in Drug Development; World Scientific Pub Co Inc.: Singapore, 2012. [Google Scholar]
- Fernandes, E.; Barbosa, Z.; Clemente, G.; Alves, F.; Abrunhosa, A.J. Positron emitting tracers in pre-clinical drug development. Curr. Radiopharm. 2012, 5, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.W.; Long, N.J.; Vilar, R.; Gee, A.D. Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew. Chem. Int. Ed. 2008, 47, 8998–9033. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjugate Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Teare, H.; Robins, E.G.; Kirjavainen, A.; Forsback, S.; Sandford, G.; Solin, O.; Luthra, S.K.; Gouverneur, V. Radiosynthesis and Evaluation of [18F]Selectfluor bis(triflate). Angew. Chem. Int. Ed. 2010, 49, 6821–6824. [Google Scholar] [CrossRef]
- Pretze, M.; Große-Gehling, P.; Mamat, C. Cross-Coupling Reactions as Valuable Tool for the Preparation of PET Radiotracers. Molecules 2011, 16, 1129–1165. [Google Scholar] [CrossRef]
- Lee, E.; Kamlet, A.S.; Powers, D.C.; Neumann, C.N.; Boursalian, G.B.; Furuya, T.; Choi, D.C.; Hooker, J.M.; Ritter, T. A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 2011, 334, 639–642. [Google Scholar] [CrossRef]
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P.J.H. Copper-Catalyzed [18F]Fluorination of (Mesityl)(aryl)iodonium Salts. Org. Lett. 2014, 16, 3224–3227. [Google Scholar] [CrossRef]
- Neumann, C.N.; Hooker, J.M.; Ritter, T. Concerted nucleophilic aromatic substitution with 19F− and 18F−. Nature 2016, 534, 369–373. [Google Scholar] [CrossRef]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef]
- Beyzavi, M.H.; Mandal, D.; Strebl, M.G.; Neumann, C.N.; D.’Amato, E.M.; Chen, J.; Hooker, J.M.; Ritter, T. 18F-Deoxyfluorination of Phenols via Ru π-Complexes. Acs Cent. Sci. 2017, 3, 944–948. [Google Scholar] [CrossRef]
- Tredwell, M.; Preshlock, S.M.; Taylor, N.J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Génicot, C.; Gouverneur, V. A General Copper-Mediated Nucleophilic 18F Fluorination of Arenes. Angew. Chem. Int. Ed. 2014, 53, 7751–7755. [Google Scholar] [CrossRef] [PubMed]
- Coenen, H.H.; Ermert, J. 18F-labelling innovations and their potential for clinical application. Clin. Transl. Imaging 2018, 6, 169–193. [Google Scholar] [CrossRef]
- Deng, X.; Rong, J.; Wang, L.; Vasdev, N.; Zhang, L.; Josephson, L.; Liang, S.H. Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew. Chem. Int. Ed. 2019, 58, 2580–2605. [Google Scholar] [CrossRef] [PubMed]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Kordys, E.; Endepols, H.; Neumaier, B. Copper-Mediated Aromatic Radiofluorination Revisited: Efficient Production of PET Tracers on a Preparative Scale. Chem. A Eur. J. 2015, 21, 5972–5979. [Google Scholar] [CrossRef]
- Mossine, A.V.; Brooks, A.F.; Makaravage, K.J.; Miller, J.M.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Synthesis of [18F]Arenes via the Copper-Mediated [18F]Fluorination of Boronic Acids. Org. Lett. 2015, 17, 5780–5783. [Google Scholar] [CrossRef]
- Preshlock, S.; Calderwood, S.; Verhoog, S.; Tredwell, M.; Huiban, M.; Hienzsch, A.; Gruber, S.; Wilson, T.C.; Taylor, N.J.; Cailly, T.; et al. Enhanced copper-mediated 18F-fluorination of aryl boronic esters provides eight radiotracers for PET applications. Chem. Commun. 2016, 52, 8361–8364. [Google Scholar] [CrossRef]
- Schäfer, D.; Weiß, P.; Ermert, J.; Castillo Meleán, J.; Zarrad, F.; Neumaier, B. Preparation of No-Carrier-Added 6-[18F]Fluoro-l-tryptophan via Cu-Mediated Radiofluorination. Eur. J. Org. Chem. 2016, 2016, 4621–4628. [Google Scholar] [CrossRef]
- Giglio, B.C.; Fei, H.; Wang, M.; Wang, H.; He, L.; Feng, H.; Wu, Z.; Lu, H.; Li, Z. Synthesis of 5-[(18)F]Fluoro-α-methyl Tryptophan: New Trp Based PET Agents. Theranostics 2017, 7, 1524–1530. [Google Scholar] [CrossRef]
- Zischler, J.; Kolks, N.; Modemann, D.; Neumaier, B.; Zlatopolskiy, B.D. Alcohol-Enhanced Cu-Mediated Radiofluorination. Chem. A Eur. J. 2017, 23, 3251–3256. [Google Scholar] [CrossRef]
- Mossine, A.V.; Brooks, A.F.; Ichiishi, N.; Makaravage, K.J.; Sanford, M.S.; Scott, P.J.H. Development of Customized [18F]Fluoride Elution Techniques for the Enhancement of Copper-Mediated Late-Stage Radiofluorination. Sci. Rep. 2017, 7, 233. [Google Scholar] [CrossRef] [PubMed]
- Blevins, D.W.; Kabalka, G.W.; Osborne, D.R.; Akula, M.R. Effect of Added Cu(OTf)2 on the Cu(OTf)2(Py)4-Mediated Radiofluorination of Benzoyl and Phthaloylglycinates. Nat. Sci. 2018, 10, 125–133. [Google Scholar] [CrossRef]
- Zhou, D.; Chu, W.; Katzenellenbogen, J. Exploration of alcohol-enhanced Cu-mediated radiofluorination towards practical labeling. J. Nucl. Med. 2018, 59, 187. [Google Scholar]
- Mossine, A.V.; Brooks, A.F.; Bernard-Gauthier, V.; Bailey, J.J.; Ichiishi, N.; Schirrmacher, R.; Sanford, M.S.; Scott, P.J.H. Automated synthesis of PET radiotracers by copper-mediated 18F-fluorination of organoborons: Importance of the order of addition and competing protodeborylation. J. Label. Compd. Radiopharm. 2018, 61, 228–236. [Google Scholar] [CrossRef]
- Lahdenpohja, S.; Keller, T.; Rajander, J.; Kirjavainen, A.K. Radiosynthesis of the norepinephrine transporter tracer [18F]NS12137 via copper-mediated 18F-labelling. J. Label. Compd. Radiopharm. 2019, 62, 259–264. [Google Scholar] [CrossRef]
- Antuganov, D.; Zykov, M.; Timofeev, V.; Timofeeva, K.; Antuganova, Y.; Orlovskaya, V.; Fedorova, O.; Krasikova, R. Copper-Mediated Radiofluorination of Aryl Pinacolboronate Esters: A Straightforward Protocol by Using Pyridinium Sulfonates. Eur. J. Org. Chem. 2019, 2019, 918–922. [Google Scholar] [CrossRef]
- Zhang, B.; Fraser, B.H.; Klenner, M.A.; Chen, Z.; Liang, S.H.; Massi, M.; Robinson, A.J.; Pascali, G. [18F]Ethenesulfonyl Fluoride as a Practical Radiofluoride Relay Reagent. Chem. A Eur. J. 2019, 25, 7613–7617. [Google Scholar] [CrossRef]
- Zhang, X.; Basuli, F.; Swenson, R.E. An azeotropic drying-free approach for copper-mediated radiofluorination without addition of base. J. Label. Compd. Radiopharm. 2019, 62, 139–145. [Google Scholar] [CrossRef]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Richarz, R.; Lauchner, K.; Neumaier, B. Minimalist approach meets green chemistry: Synthesis of 18F- labeled (hetero)aromatics in pure ethanol. J. Label. Compd. Radiopharm. 2019, 62, 404–410. [Google Scholar] [CrossRef]
- Mossine, A.V.; Tanzey, S.S.; Brooks, A.F.; Makaravage, K.J.; Ichiishi, N.; Miller, J.M.; Henderson, B.D.; Skaddan, M.B.; Sanford, M.S.; Scott, P.J.H. One-pot synthesis of high molar activity 6-[18F]fluoro-l-DOPA by Cu-mediated fluorination of a BPin precursor. Org. Biomol. Chem. 2019. [Google Scholar] [CrossRef]
- Lahdenpohja, S.O.; Rajala, N.A.; Rajander, J.; Kirjavainen, A.K. Fast and efficient copper-mediated 18F-fluorination of arylstannanes, aryl boronic acids, and aryl boronic esters without azeotropic drying. Ejnmmi Radiopharm. Chem. 2019, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.J.; Emer, E.; Preshlock, S.; Schedler, M.; Tredwell, M.; Verhoog, S.; Mercier, J.; Genicot, C.; Gouverneur, V. Derisking the Cu-Mediated 18F-Fluorination of Heterocyclic Positron Emission Tomography Radioligands. J. Am. Chem. Soc. 2017, 139, 8267–8276. [Google Scholar] [CrossRef] [PubMed]
- Drerup, C.; Ermert, J.; Coenen, H.H. Synthesis of a Potent Aminopyridine-Based nNOS-Inhibitor by Two Recent No-Carrier-Added 18F-Labelling Methods. Molecules 2016, 21, 1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, V.T.; Klaveness, J.; Olberg, D.E. One-step synthesis of [18F]cabozantinib for use in positron emission tomography imaging of c-Met. J. Label. Compd. Radiopharm. 2018, 61, 11–17. [Google Scholar] [CrossRef]
- Bernard-Gauthier, V.; Mossine, A.V.; Mahringer, A.; Aliaga, A.; Bailey, J.J.; Shao, X.; Stauff, J.; Arteaga, J.; Sherman, P.; Grand′Maison, M.; et al. Identification of [18F]TRACK, a Fluorine-18-Labeled Tropomyosin Receptor Kinase (Trk) Inhibitor for PET Imaging. J. Med. Chem. 2018, 61, 1737–1743. [Google Scholar] [CrossRef]
- Wilson, T.; Xavier, M.-A.; Knight, J.; Verhoog, S.; Torres, J.B.; Mosley, M.; Hopkins, S.; Wallington, S.; Allen, D.; Kersemans, V.; et al. PET imaging of PARP expression using [18F]olaparib. J. Nucl. Med. 2018, 60, 504–510. [Google Scholar] [CrossRef] [Green Version]
- Guibbal, F.; Meneyrol, V.; Ait-Arsa, I.; Diotel, N.; Patché, J.; Veeren, B.; Bénard, S.; Gimié, F.; Yong-Sang, J.; Khantalin, I.; et al. Synthesis and Automated Labeling of [18F]Darapladib, a Lp-PLA2 Ligand, as Potential PET Imaging Tool of Atherosclerosis. Acs. Med. Chem. Lett. 2019, 10, 743–748. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Clemente, G.S.; Elsinga, P.H.; Dömling, A. MCR Scaffolds Get Hotter with 18F-Labeling. Molecules 2019, 24, 1327. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, S.; Nguyen, H.; Nguyen, M.; Abdel-Rasoul, M.; Nguyen, V.; Nguyen, C.D.; Nguyen, K.T.; Li, L.; Kitzmiller, J.P. Pleiotropic Effects of Statins: Untapped Potential for Statin Pharmacotherapy. Curr. Vasc. Pharmacol. 2019, 17, 239–261. [Google Scholar] [CrossRef]
- Fatehi Hassanabad, A.; McBride, S.A. Statins as Potential Therapeutics for Lung Cancer: Molecular Mechanisms and Clinical Outcomes. Am. J. Clin. Oncol. 2019, 42, 732–736. [Google Scholar] [CrossRef]
- Fier, P.S.; Luo, J.; Hartwig, J.F. Copper-Mediated Fluorination of Arylboronate Esters. Identification of a Copper(III) Fluoride Complex. J. Am. Chem. Soc. 2013, 135, 2552–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refat, M.S.; Al-Saif, F.A. Synthesis, spectral, thermal, and antimicrobial studies of transition metal complexes of atorvastatin calcium as a lipid-lowering agent. J. Therm. Anal. Calorim. 2015, 120, 863–878. [Google Scholar] [CrossRef]
- Hatcher, L.Q.; Karlin, K.D. Ligand Influences in Copper-Dioxygen Complex-Formation and Substrate Oxidations. In Advances in Inorganic Chemistry; Academic Press: Cambridge, MA, USA, 2006; Volume 58, pp. 131–184. [Google Scholar]
- Novozhilov, Y.V.; Dorogov, M.V.; Blumina, M.V.; Smirnov, A.V.; Krasavin, M. An improved kilogram-scale preparation of atorvastatin calcium. Chem. Cent. J. 2015, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowden, G.D.; Pichler, B.J.; Maurer, A. A Design of Experiments (DoE) Approach Accelerates the Optimization of Copper-Mediated 18F-Fluorination Reactions of Arylstannanes. Sci. Rep. 2019, 9, 11370. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
S. Clemente, G.; Zarganes-Tzitzikas, T.; Dömling, A.; H. Elsinga, P. Late-Stage Copper-Catalyzed Radiofluorination of an Arylboronic Ester Derivative of Atorvastatin. Molecules 2019, 24, 4210. https://doi.org/10.3390/molecules24234210
S. Clemente G, Zarganes-Tzitzikas T, Dömling A, H. Elsinga P. Late-Stage Copper-Catalyzed Radiofluorination of an Arylboronic Ester Derivative of Atorvastatin. Molecules. 2019; 24(23):4210. https://doi.org/10.3390/molecules24234210
Chicago/Turabian StyleS. Clemente, Gonçalo, Tryfon Zarganes-Tzitzikas, Alexander Dömling, and Philip H. Elsinga. 2019. "Late-Stage Copper-Catalyzed Radiofluorination of an Arylboronic Ester Derivative of Atorvastatin" Molecules 24, no. 23: 4210. https://doi.org/10.3390/molecules24234210
APA StyleS. Clemente, G., Zarganes-Tzitzikas, T., Dömling, A., & H. Elsinga, P. (2019). Late-Stage Copper-Catalyzed Radiofluorination of an Arylboronic Ester Derivative of Atorvastatin. Molecules, 24(23), 4210. https://doi.org/10.3390/molecules24234210