
Huntington’s Disease: A Review of the Known PET Imaging Biomarkers and Targeting Radiotracers


Abstract
:1. Introduction
1.1. Positron Emission Tomography
1.2. Huntington’s Disease
2. Indicators of Huntington’s Disease: From A to Z
2.1. Adenosine Receptors
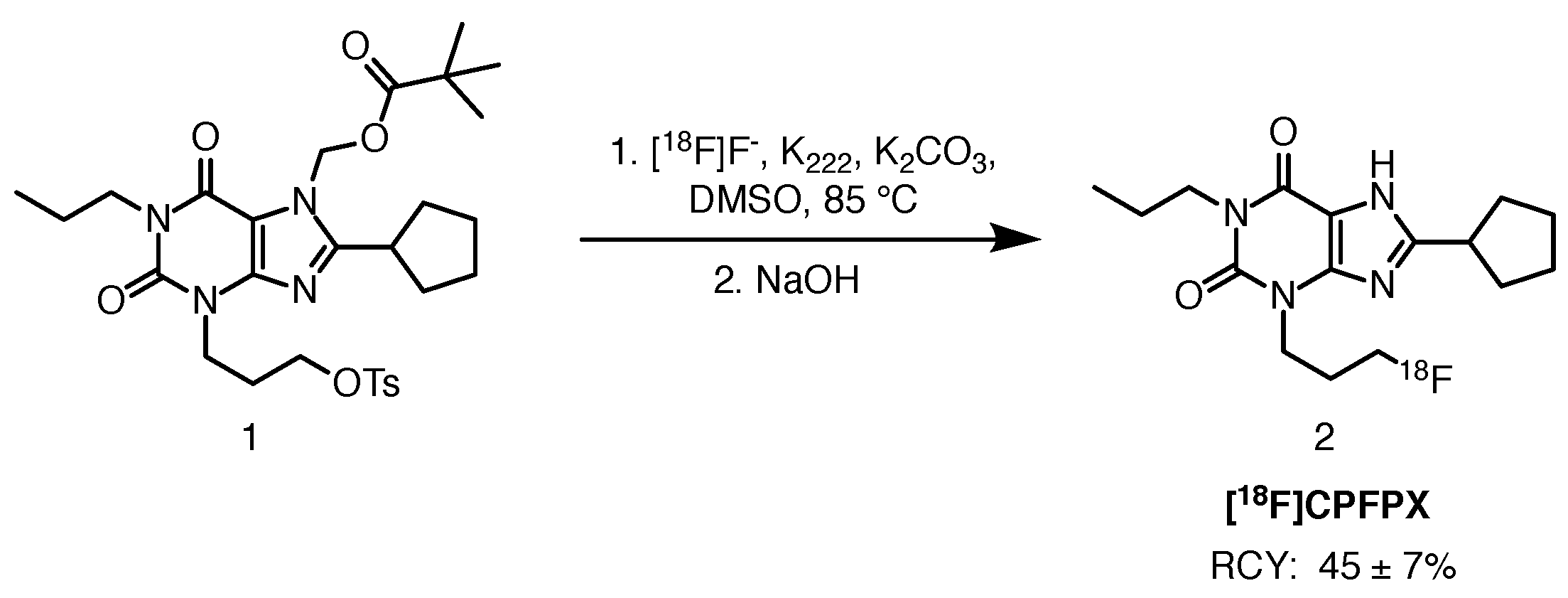
2.1.1. [18F]CPFPX
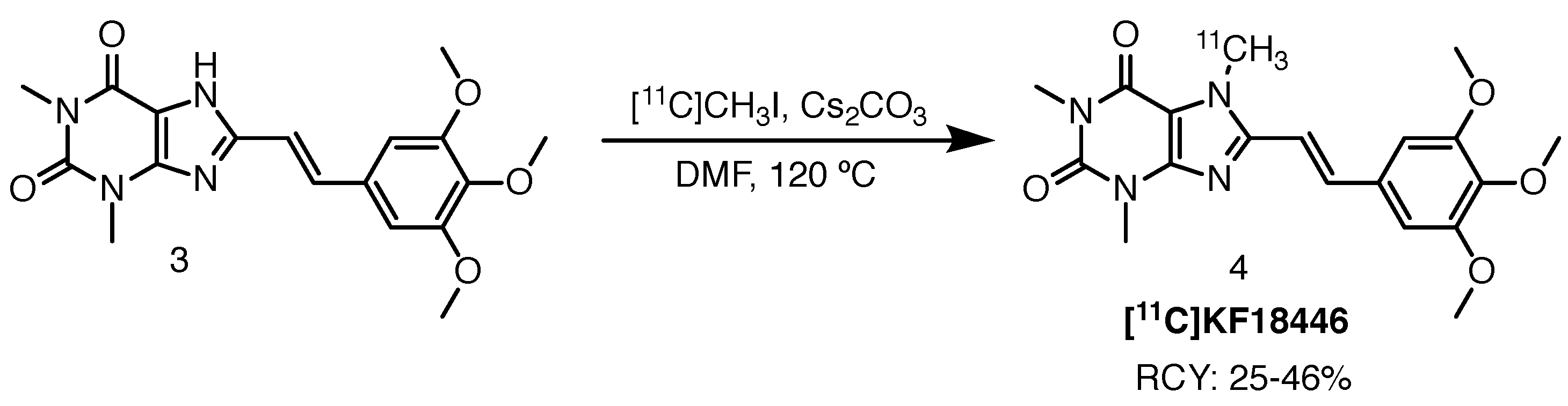
2.1.2. [11C]KF18446
2.2. Cannabinoid Receptors
2.2.1. 18F]MK-9470
2.3. Dopaminergic Receptors
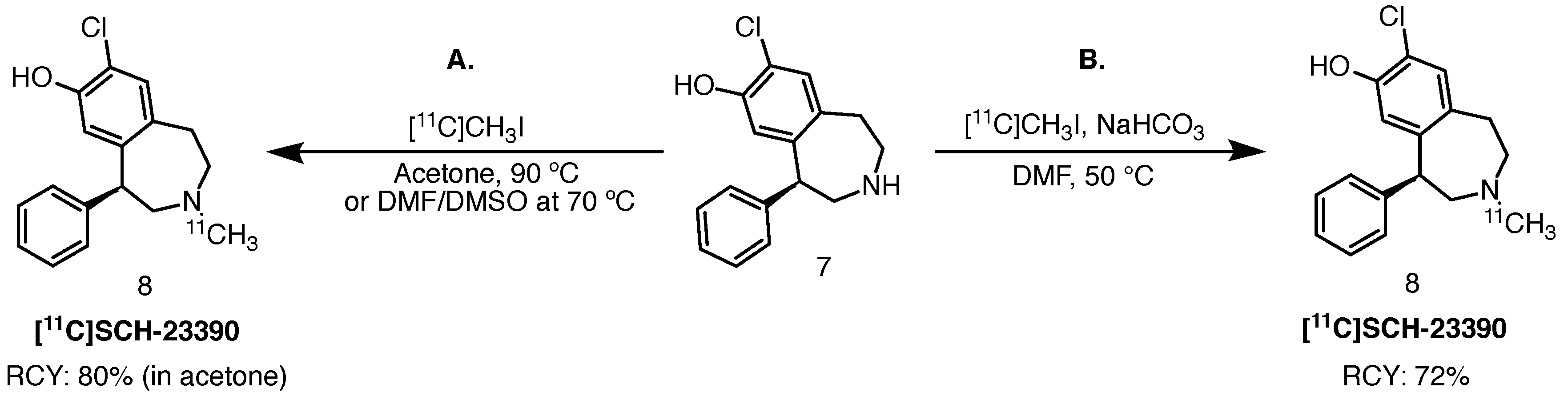
2.3.1. [11C]SCH-23390
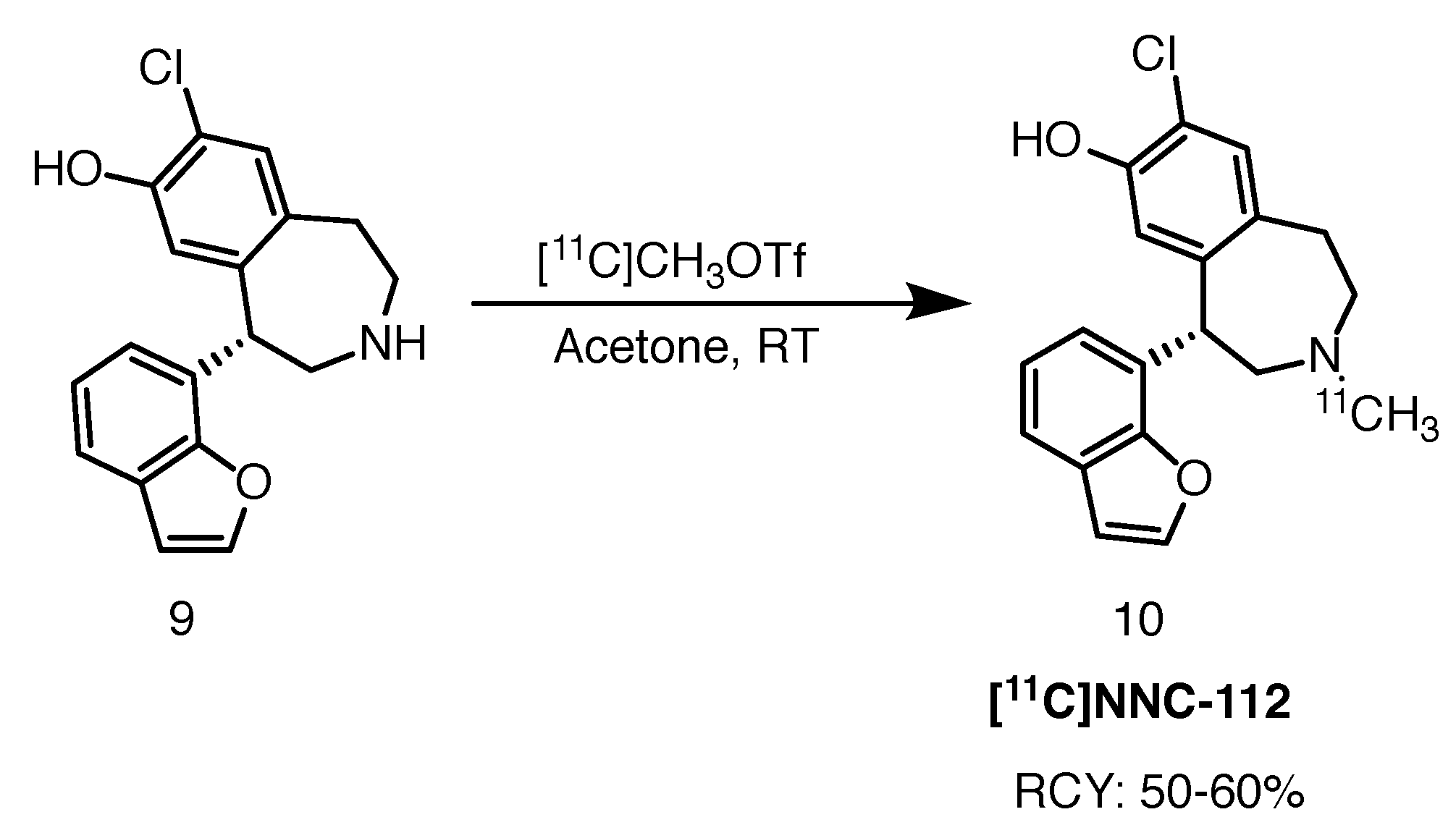
2.3.2. [11C]NNC-112
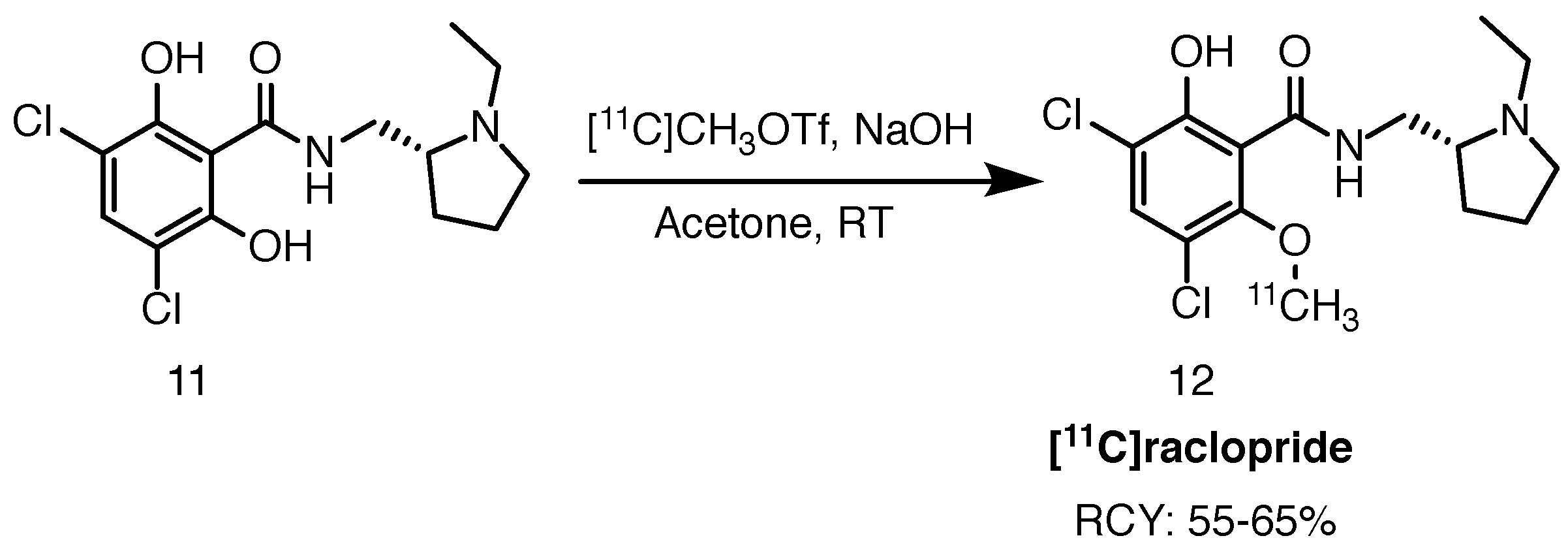
2.3.3. [11C]Raclopride
2.4. GABA Receptors
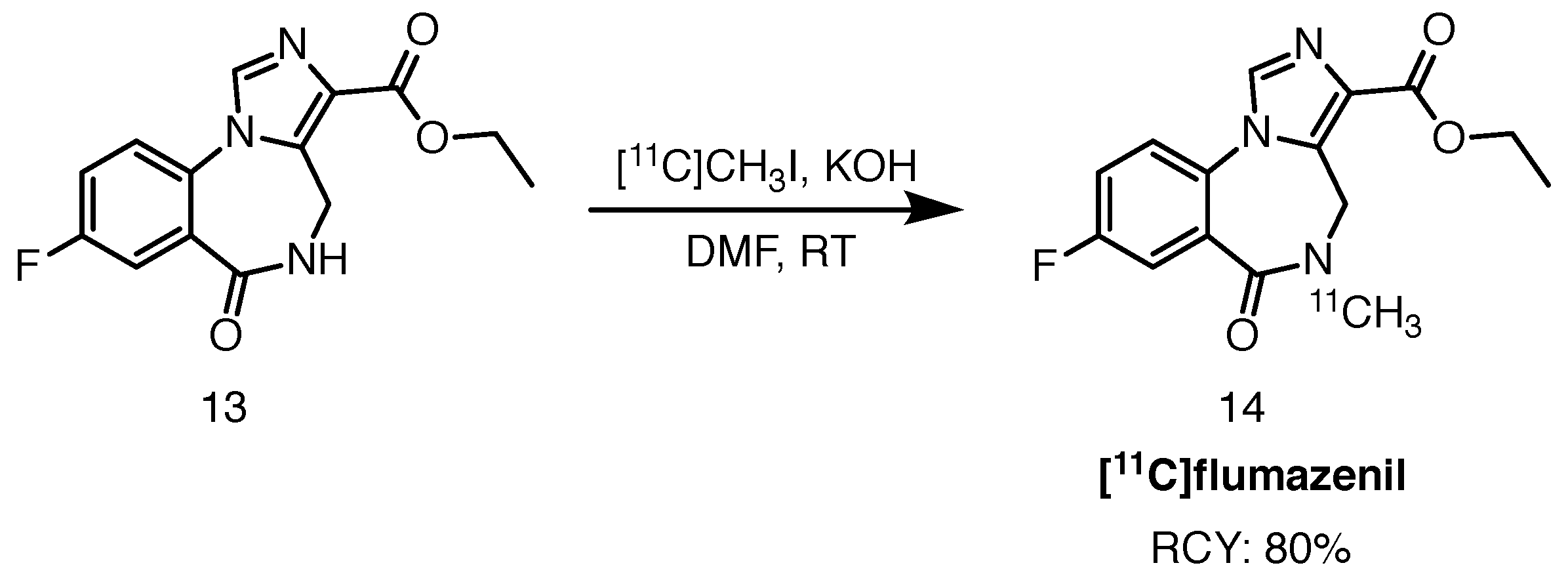
2.4.1. [11C]Flumazenil
2.5. Glucose Metabolism
2.6. Glutamatergic Receptors
2.6.1. [11C]ABP-688
2.7. Microglia
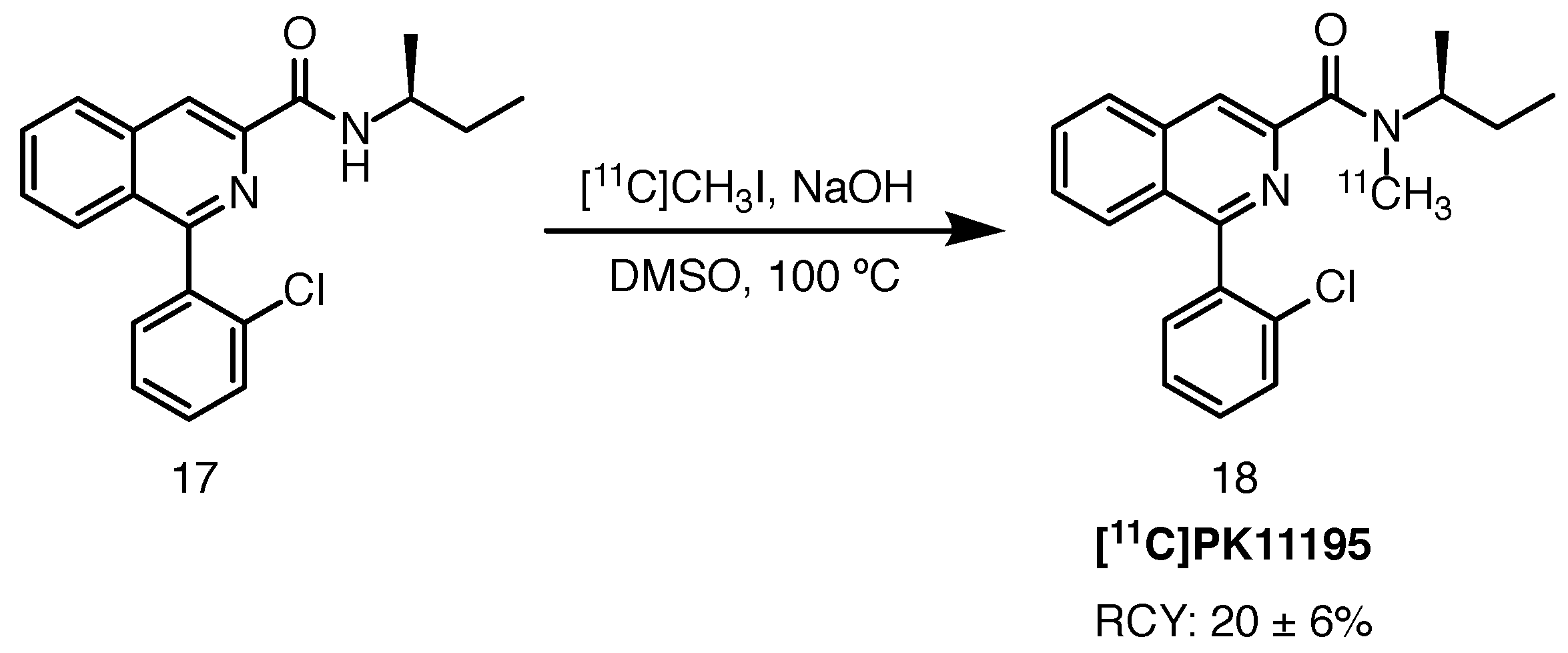
2.7.1. [11C]PK11195
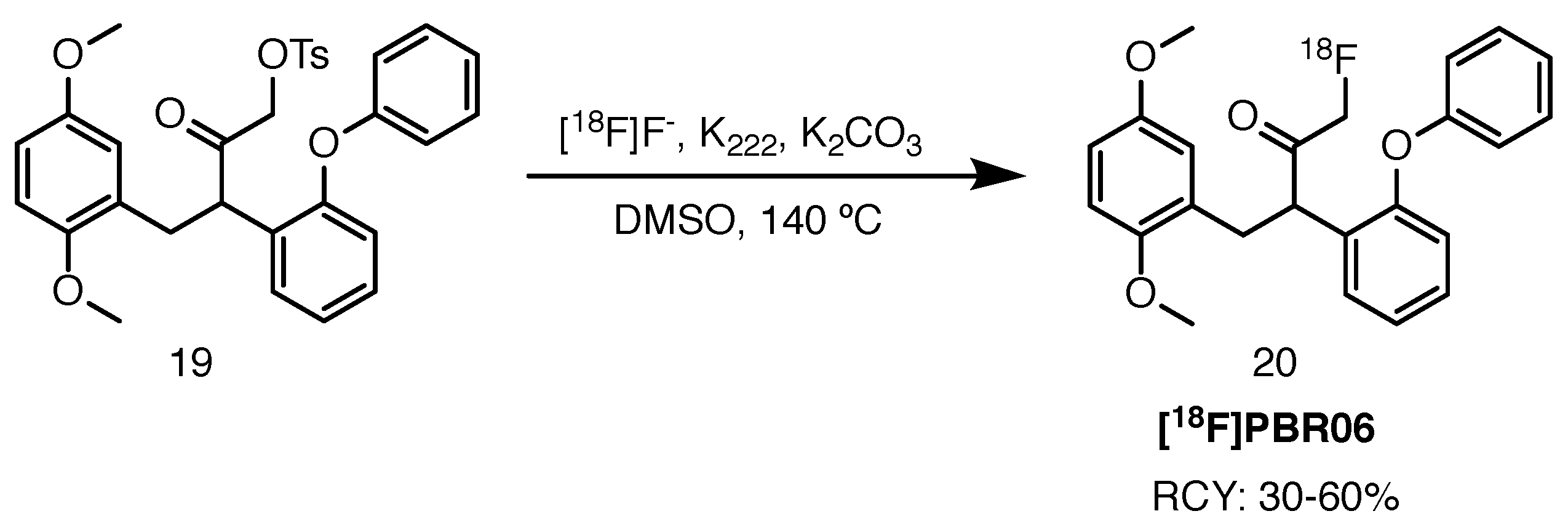
2.7.2. [18F]PBR06
2.8. Phosphodiesterase 10A
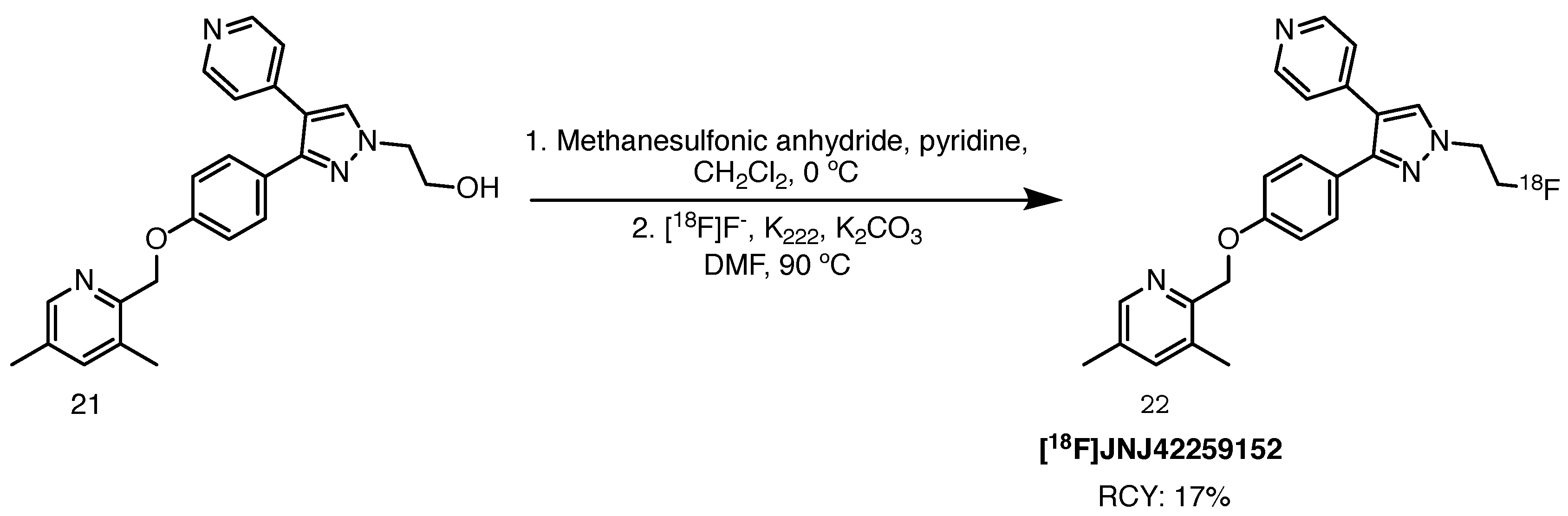
2.8.1. [18F]JNJ42249152
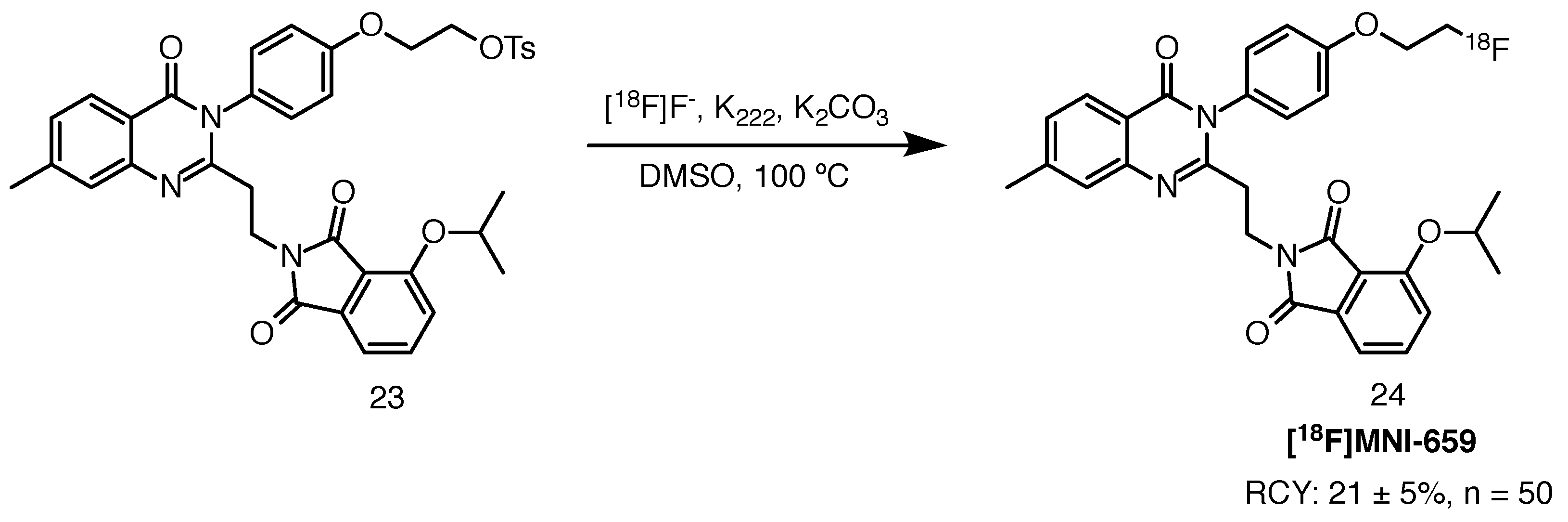
2.8.2. [18F]MNI-659
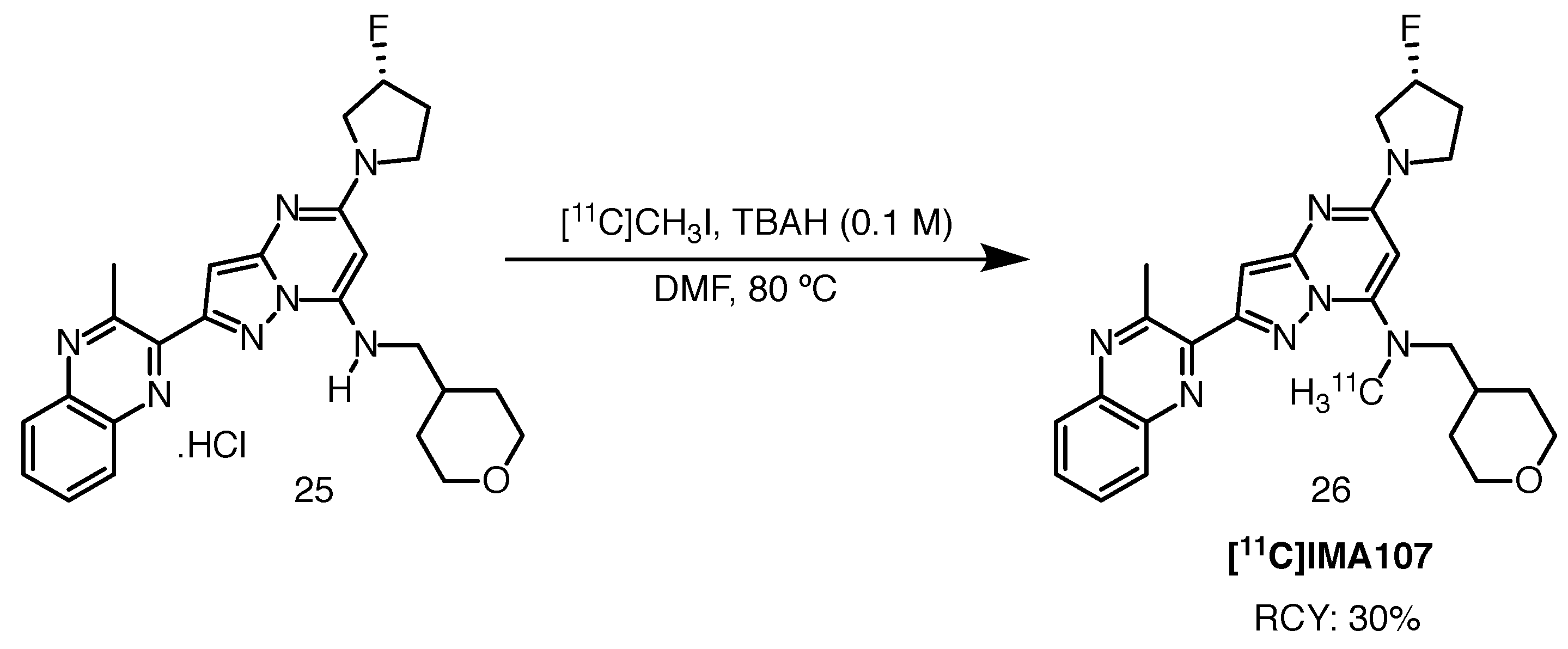
2.8.3. [11C]IMA-107
2.9. Synaptic Vesicle Protein 2A
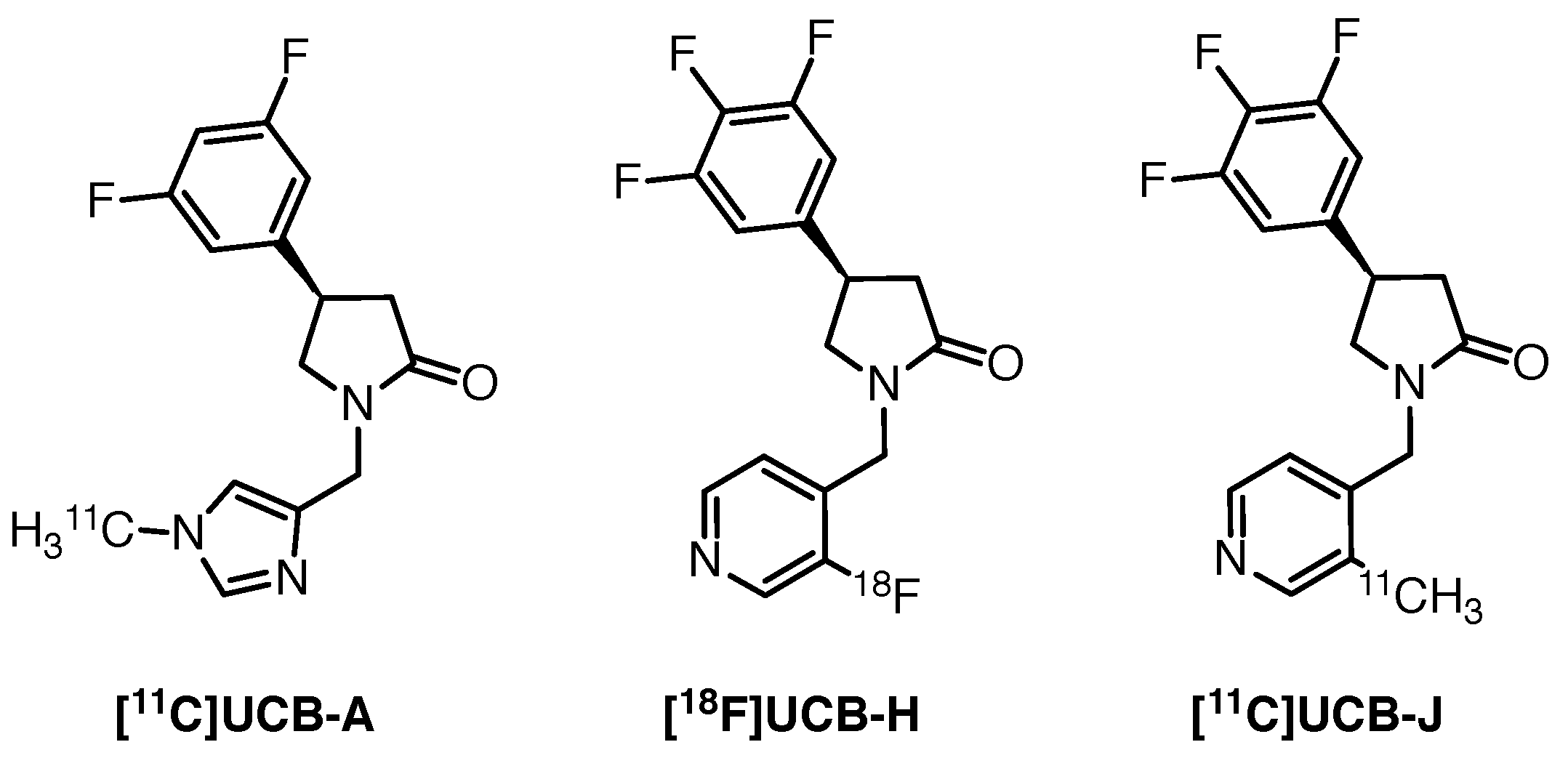
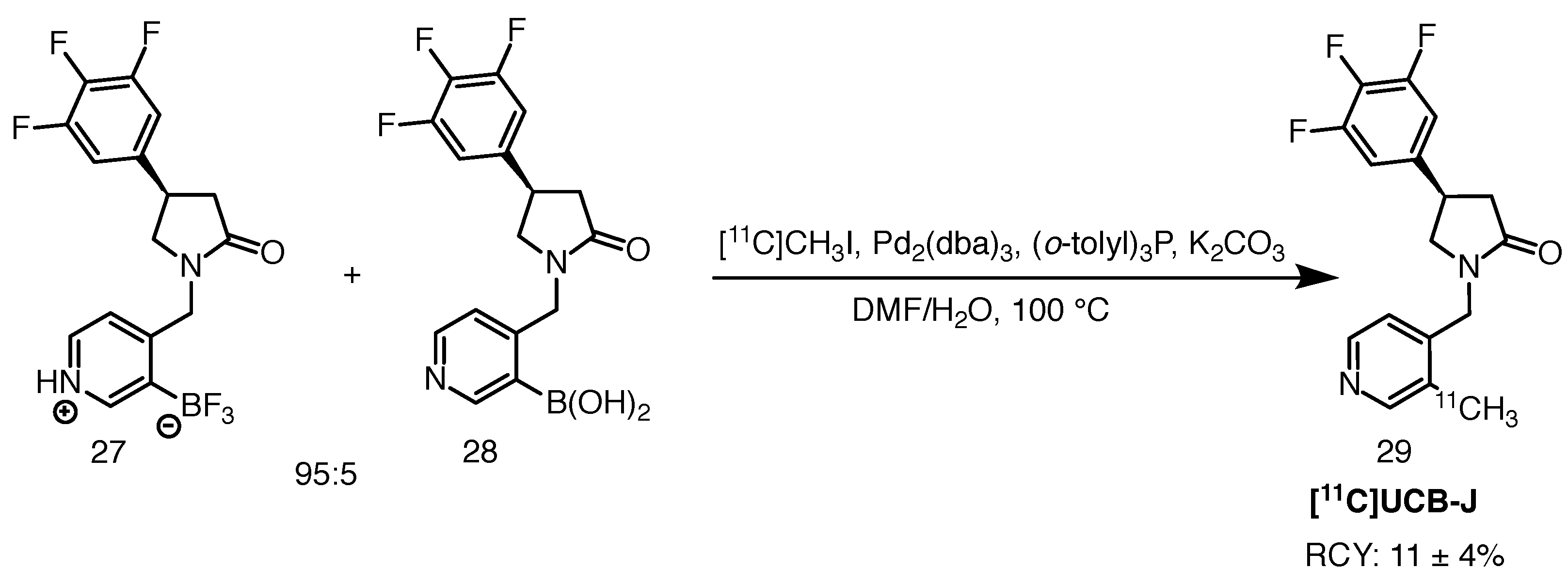
2.9.1. [11C]UCB-J
3. Conclusions
Funding
Conflicts of Interest
Abbreviations
| A1 and A2A | Adenosine receptors |
| AD | Alzheimer’s disease |
| BPND | Binding potential |
| Bq | Becquerel |
| CAG | Cytosine-adenine-guanine |
| CB1 | Cannabinoid receptor type 1 |
| CNS | Central nervous system |
| D1, D2 and D3 | Dopamine receptors |
| DMF | Dimethyl formamide |
| DMSO | Dimethyl sulfoxide |
| GABA | gamma-Aminobutyric acid |
| GMP | Good manufacturing practice |
| HD | Huntington’s disease |
| HPLC | High performance liquid chromatography |
| IB | Inclusion body |
| K222 | Kryptofix 222 |
| MeCN | Acetonitrile |
| mHTT | Mutant huntingtin |
| MRI | Magnetic Resonance Imaging |
| MSN | Medium spiny neuron |
| OTf | Trifluoromethanesulfonate, triflate |
| PET | Positron Emission Tomography |
| PD | Parkinson’s disease |
| RCP | Radiochemical purity; defined as the absence of other radiochemical compounds/species |
| RCY | Radiochemical yield; defined as the amount of activity in the product expressed as the percentage (%) of starting activity used in the considered process, all RCYs presented in this review are decay corrected |
| RT | Room temperature |
| SUV | Standardised uptake value |
| WT | Wildtype |
References
- Schapira, A.H.V.; Olanow, C.W.; Greenamyre, J.T.; Bezard, E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: Future therapeutic perspectives. Lancet 2014, 384, 545–555. [Google Scholar] [CrossRef]
- Wilson, H.; De Micco, R.; Niccolini, F.; Politis, M. Molecular Imaging Markers to Track Huntington’s Disease Pathology. Front. Neurol. 2017, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Pagano, G.; Niccolini, F.; Politis, M. Current status of PET imaging in Huntington’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1171–1182. [Google Scholar] [CrossRef] [Green Version]
- Fazio, P.; Paucar, M.; Svenningsson, P.; Varrone, A. Novel Imaging Biomarkers for Huntington’s Disease and Other Hereditary Choreas. Curr. Neurol. Neurosci. Rep. 2018, 18, 85. [Google Scholar] [CrossRef] [Green Version]
- Roussakis, A.A.; Piccini, P. PET Imaging in Huntington’s Disease. J. Huntingtons. Dis. 2015, 4, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Correia, K.; Loupe, J.; Kim, K.H.; Barker, D.; Hong, E.P.; Chao, M.J.; Long, J.D.; Lucente, D.; Vonsattel, J.P.G.; et al. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 2019, 178, 887–900.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalingam, S.; Levy, L. Genetics of Huntington Disease. Am. J. Neuroradiol. 2014, 35, 1070–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019, 49, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Neto, J.L.; Lee, J.M.; Afridi, A.; Gillis, T.; Guide, J.R.; Dempsey, S.; Lager, B.; Alonso, I.; Wheeler, V.C.; Pinto, R.M. Genetic contributors to intergenerational CAG repeat instability in Huntington’s disease knock-in mice. Genetics 2017, 205, 503–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherzinger, E.; Sittler, A.; Schweiger, K.; Heiser, V.; Lurz, R.; Hasenbank, R.; Bates, G.P.; Lehrach, H.; Wanker, E.E. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: Implications for Huntington’s disease pathology. Proc. Natl. Acad. Sci. USA 1999, 96, 4604–4609. [Google Scholar] [CrossRef] [Green Version]
- Chugani, H.T. Positron Emission Tomography in Pediatric Neurodegenerative Disorders. Pediatr. Neurol. 2019, 100, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.E. Huntington’s Disease and the Striatal Medium Spiny Neuron: Cell-Autonomous and Non-Cell-Autonomous Mechanisms of Disease. Neurotherapeutics 2012, 9, 270–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuorimaa, A.; Rissanen, E.; Airas, L. In Vivo PET Imaging of Adenosine 2A Receptors in Neuroinflammatory and Neurodegenerative Disease. Contrast Media Mol. Imaging 2017, 2017, 6975841. [Google Scholar] [CrossRef]
- Blum, D.; Chern, Y.; Domenici, M.R.; Buée, L.; Lin, C.Y.; Rea, W.; Ferré, S.; Popoli, P. The Role of Adenosine Tone and Adenosine Receptors in Huntington’s Disease. J. Caffeine Adenosine Res. 2018, 8, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Morigaki, R.; Goto, S. Striatal Vulnerability in Huntington’s Disease: Neuroprotection Versus Neurotoxicity. Brain Sci. 2017, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.V.; Kaster, M.P.; Tomé, A.R.; Agostinho, P.M.; Cunha, R.A. Adenosine receptors and brain diseases: Neuroprotection and neurodegeneration. Biochim. Biophys. Acta Biomembr. 2011, 1808, 1380–1399. [Google Scholar] [CrossRef] [Green Version]
- Blum, D.; Hourez, R.; Galas, M.C.; Popoli, P.; Schiffmann, S.N. Adenosine receptors and Huntington’s disease: implications for pathogenesis and therapeutics. Lancet Neurol. 2003, 2, 366–374. [Google Scholar] [CrossRef]
- Holschbach, M.H.; Olsson, R.A.; Bier, D.; Wutz, W.; Sihver, W.; Schüller, M.; Palm, B.; Coenen, H.H. Synthesis and Evaluation of No-Carrier-Added 8-Cyclopentyl-3-(3-[18F]fluoropropyl)-1-propylxanthine ([18F]CPFPX): A Potent and Selective A1-Adenosine Receptor Antagonist for in Vivo Imaging. J. Med. Chem. 2002, 45, 5150–5156. [Google Scholar] [CrossRef]
- Coenen, H.H.; Gee, A.D.; Adam, M.; Antoni, G.; Cutler, C.S.; Fujibayashi, Y.; Jeong, J.M.; Mach, R.H.; Mindt, T.L.; Pike, V.W.; et al. Consensus nomenclature rules for radiopharmaceutical chemistry—Setting the record straight. Nucl. Med. Biol. 2017, 55, v–xi. [Google Scholar] [CrossRef] [Green Version]
- Bauer, A.; Holschbach, M.H.; Cremer, M.; Weber, S.; Boy, C.; Shah, N.J.; Olsson, R.A.; Halling, H.; Coenen, H.H.; Zilles, K. Evaluation of 18F-CPFPX, a Novel Adenosine A1 Receptor Ligand: In Vitro Autoradiography and High-Resolution Small Animal PET. J. Nucl. Med. 2003, 44, 1682–1689. [Google Scholar]
- Matusch, A.; Saft, C.; Elmenhorst, D.; Kraus, P.H.; Gold, R.; Hartung, H.P.; Bauer, A. Cross sectional PET study of cerebral adenosine A1 receptors in premanifest and manifest Huntington’s disease. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, K.; Noguchi, J.; Wakabayashi, S.I.; Shimada, J.; Ogi, N.; Nariai, T.; Tanaka, A.; Endo, K.; Suzuki, F.; Senda, M. 11C-Labeled KF18446: A Potential Central Nervous System Adenosine A2a Receptor Ligand. J. Nucl. Med. 2000, 41, 345–354. [Google Scholar] [PubMed]
- Ishiwata, K.; Ogi, N.; Hayakawa, N.; Oda, K.; Nagaoka, T.; Toyama, H.; Suzuki, F.; Endo, K.; Tanaka, A.; Senda, M. Adenosine A2A receptor imaging with [11C]KF18446 PET in the rat brain after quinolinic acid lesion: Comparison with the dopamine receptor imaging. Ann. Nucl. Med. 2002, 16, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Kendall, D.A.; Yudowski, G.A. Cannabinoid Receptors in the Central Nervous System: Their Signaling and Roles in Disease. Front. Cell. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, M.; Faull, R.; Dragunow, M. Loss of cannabinoid receptors in the substantia nigra in Huntington’s disease. Neuroscience 1993, 56, 523–527. [Google Scholar] [CrossRef]
- Glass, M.; Dragunow, M.; Faull, R. The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABAA receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience 2000, 97, 505–519. [Google Scholar] [CrossRef]
- Van Laere, K.; Casteels, C.; Dhollander, I.; Goffin, K.; Grachev, I.; Bormans, G.; Vandenberghe, W. Widespread Decrease of Type 1 Cannabinoid Receptor Availability in Huntington Disease In Vivo. J. Nucl. Med. 2010, 51, 1413–1417. [Google Scholar] [CrossRef] [Green Version]
- Thomae, D.; Morley, T.J.; Hamill, T.; Carroll, V.M.; Papin, C.; Twardy, N.M.; Lee, H.S.; Hargreaves, R.; Baldwin, R.M.; Tamagnan, G.; et al. Automated one-step radiosynthesis of the CB1 receptor imaging agent [18F]MK-9470. J. Label. Compd. Radiopharm. 2014, 57, 611–614. [Google Scholar] [CrossRef]
- Gagnon, D.; Petryszyn, S.; Sanchez, M.G.; Bories, C.; Beaulieu, J.M.; De Koninck, Y.; Parent, A.; Parent, M. Striatal Neurons Expressing D1 and D2 Receptors are Morphologically Distinct and Differently Affected by Dopamine Denervation in Mice. Sci. Rep. 2017, 7, 41432. [Google Scholar] [CrossRef] [Green Version]
- Bourne, J.A. SCH 23390: The First Selective Dopamine D1-Like Receptor Antagonist. CNS Drug Rev. 2006, 7, 399–414. [Google Scholar] [CrossRef]
- Friedman, A.M.; DeJesus, O.T.; Woolverton, W.L.; Moffaert, G.V.; Goldberg, L.I.; Prasad, A.; Barnett, A.; Dinerstein, R.J. Positron tomography of a radio-brominated analog of the D1/DA1 antagonist, SCH 23390. Eur. J. Pharmacol. 1985, 108, 327–328. [Google Scholar] [CrossRef]
- Dejesus, O.; Moffaert, G.V.; Friedman, A. Evaluation of positron-emitting SCH 23390 analogs as tracers for CNS dopamine D1 receptors. Nucl. Med. Biol. 1989, 16, 47–50. [Google Scholar] [CrossRef]
- Stenkrona, P.; Matheson, G.J.; Cervenka, S.; Sigray, P.P.; Halldin, C.; Farde, L. [11C]SCH23390 binding to the D1-dopamine receptor in the human brain—A comparison of manual and automated methods for image analysis. EJNMMI Res. 2018, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- DeJesus, O.; Moffaert, G.V.; Friedman, A. Synthesis of [11C]SCH 23390 for dopamine D1 receptor studies. Int. J. Rad. Appl. Instr. A 1987, 38, 345–348. [Google Scholar] [CrossRef]
- Halldin, C.; Stone-Elander, S.; Farde, L.; Ehrin, E.; Fasth, K.J.; Långström, B.; Sedvall, G. Preparation of 11C-labelled SCH 23390 for the in vivo study of dopamine D-1 receptors using positron emission tomography. Appl. Radiat. Isot. 1986, 37, 1039–1043. [Google Scholar] [CrossRef]
- Sedvall, G.; Karlsson, P.; Lundin, A.; Anvret, M.; Suhara, T.; Halldin, C.; Farde, L. Dopamine D1 receptor number—A sensitive PET marker for early brain degeneration in Huntington’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1994, 243, 249–255. [Google Scholar]
- Andrews, T.C.; Weeks, R.A.; Turjanski, N.; Gunn, R.N.; Watkins, L.H.A.; Sahakian, B.; Hodges, J.R.; Rosser, A.E.; Wood, N.W.; Brooks, D.J. Huntington’s disease progression: PET and clinical observations. Brain 1999, 122, 2353–2363. [Google Scholar] [CrossRef] [Green Version]
- Häggkvist, J.; Tóth, M.; Tari, L.; Varnäs, K.; Svedberg, M.; Forsberg, A.; Nag, S.; Dominguez, C.; Munoz-Sanjuan, I.; Bard, J.; et al. Longitudinal Small-Animal PET Imaging of the zQ175 Mouse Model of Huntington Disease Shows In Vivo Changes of Molecular Targets in the Striatum and Cerebral Cortex. J. Nucl. Med. 2017, 58, 617–622. [Google Scholar] [CrossRef] [Green Version]
- Halldin, C.; Foged, C.; Chou, Y.H.; Karlsson, P.; Swahn, C.G.; Johan, S.; Sedvall, G.; Farde, L. Carbon-11-NNC 112: A Radioligand for PET Examination of Striatal and Neocortical D1-Dopamine Receptors. J. Nucl. Med. 1998, 39, 2061–2068. [Google Scholar]
- Esmaeilzadeh, M.; Farde, L.; Karlsson, P.; Varrone, A.; Halldin, C.; Waters, S.; Tedroff, J. Extrastriatal dopamine D2 receptor binding in Huntington’s disease. Hum. Brain Mapp. 2011, 32, 1626–1636. [Google Scholar] [CrossRef]
- Ehrin, E.; Farde, L.; de Paulis, T.; Eriksson, L.; Greitz, T.; Johnström, P.; Litton, J.E.; Nilsson, J.G.; Sedvall, G.; Stone-Elander, S.; et al. Preparation of 11C-labelled raclopride, a new potent dopamine receptor antagonist: Preliminary PET studies of cerebral dopamine receptors in the monkey. Int. J. Appl. Radiat. Isot. 1985, 36, 269–273. [Google Scholar] [CrossRef]
- Langer, O.; Någren, K.; Dolle, F.; Lundkvist, C.; Sandell, J.; Swahn, C.G.; Vaufrey, F.; Crouzel, C.; Maziere, B.; Halldin, C. Precursor synthesis and radiolabelling of the dopamine D2 receptor ligand [11C]raclopride from [11C]methyl triflate. J. Label. Compd. Radiopharm. 1999, 42, 1183–1193. [Google Scholar] [CrossRef]
- Lee, Y.S.; Jeong, J.M.; Cho, Y.H.; Lee, J.H.; Lee, H.J.; Kim, J.E.; Lee, Y.S.; Kang, K.W. Evaluation of base-free 11C-Raclopride synthesis with various solvents. J. Nucl. Med. 2015, 56, 2501. [Google Scholar]
- Haroun, S.; Sanei, Z.; Jivan, S.; Schaffer, P.; Ruth, T.J.; Li, P.C. Continuous-flow synthesis of [11C]raclopride, a positron emission tomography radiotracer, on a microfluidic chip. Can. J. Chem. 2013, 91, 326–332. [Google Scholar] [CrossRef]
- Gómez-Vallejo, V.; Llop, J. Fully automated and reproducible radiosynthesis of high specific activity [11C]raclopride and [11C]Pittsburgh compound-B using the combination of two commercial synthesizers. Nucl. Med. Commun. 2011, 32, 1011–1017. [Google Scholar] [CrossRef]
- Antonini, A.; Leenders, K.L.; Eidelberg, D. [11C]Raclopride-PET studies of the Huntington’s disease rate of progression: Relevance of the trinucleotide repeat length. Ann. Neurol. 1998, 43, 253–255. [Google Scholar] [CrossRef]
- Pavese, N.; Politis, M.; Tai, Y.F.; Barker, R.A.; Tabrizi, S.J.; Mason, S.L.; Brooks, D.J.; Piccini, P. Cortical dopamine dysfunction in symptomatic and premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 2010, 37, 356–361. [Google Scholar] [CrossRef]
- Hsu, Y.T.; Chang, Y.G.; Chern, Y. Insights into GABAAergic system alteration in Huntington’s disease. Open Biol. 2018, 8, 180165. [Google Scholar] [CrossRef] [Green Version]
- Garret, M.; Du, Z.; Chazalon, M.; Cho, Y.H.; Baufreton, J. Alteration of GABAergic neurotransmission in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 292–300. [Google Scholar] [CrossRef]
- Künig, G.; Leenders, K.L.; Sanchez-Pernaute, R.; Antonini, A.; Vontobel, P.; Verhagen, A.; Günther, I. Benzodiazepine receptor binding in Huntington’s disease: [11C]Flumazenil uptake measured using positron emission tomography. Ann. Neurol. 2000, 47, 644–648. [Google Scholar] [CrossRef]
- Cleij, M.C.; Clark, J.C.; Baron, J.C.; Aigbirhio, F.I. Rapid preparation of [11C]flumazenil: captive solvent synthesis combined with purification by analytical sized columns. J. Label. Compd. Radiopharm. 2007, 50, 19–24. [Google Scholar] [CrossRef]
- Abd-Elrahman, K.S.; Hamilton, A.; Hutchinson, S.R.; Liu, F.; Russell, R.C.; Ferguson, S.S.G. mGluR5 antagonism increases autophagy and prevents disease progression in the zQ175 mouse model of Huntington’s disease. Sci. Signal. 2017, 10, eaan6387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, F.M.; Hamilton, A.; Doria, J.G.; Guimaraes, I.M.; Cregan, S.P.; Ferguson, S.S. Metabotropic glutamate receptor 5 as a potential therapeutic target in Huntington’s disease. Expert Opin. Ther. Targets 2014, 18, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.M.; Paquet, M.; Ferreira, L.T.; Cregan, T.; Swan, P.; Cregan, S.P.; Ferguson, S.S.G. Metabotropic Glutamate Receptor-Mediated Cell Signaling Pathways Are Altered in a Mouse Model of Huntington’s Disease. J. Neurosci. 2010, 30, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Ametamey, S.M.; Kessler, L.J.; Honer, M.; Wyss, M.T.; Buck, A.; Hintermann, S.; Auberson, Y.P.; Gasparini, F.; Schubiger, P.A. Radiosynthesis and Preclinical Evaluation of 11C-ABP688 as a Probe for Imaging the Metabotropic Glutamate Receptor Subtype 5. J. Nucl. Med. 2006, 47, 698–705. [Google Scholar]
- Ametamey, S.M.; Treyer, V.; Streffer, J.; Wyss, M.T.; Schmidt, M.; Blagoev, M.; Hintermann, S.; Auberson, Y.; Gasparini, F.; Fischer, U.C.; et al. Human PET Studies of Metabotropic Glutamate Receptor Subtype 5 with 11C-ABP688. J. Nucl. Med. 2007, 48, 247–252. [Google Scholar]
- DeLorenzo, C.; Milak, M.S.; Brennan, K.G.; Kumar, J.S.D.; Mann, J.J.; Parsey, R.V. In vivo positron emission tomography imaging with [11C]ABP688: binding variability and specificity for the metabotropic glutamate receptor subtype 5 in baboons. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1083–1094. [Google Scholar] [CrossRef] [Green Version]
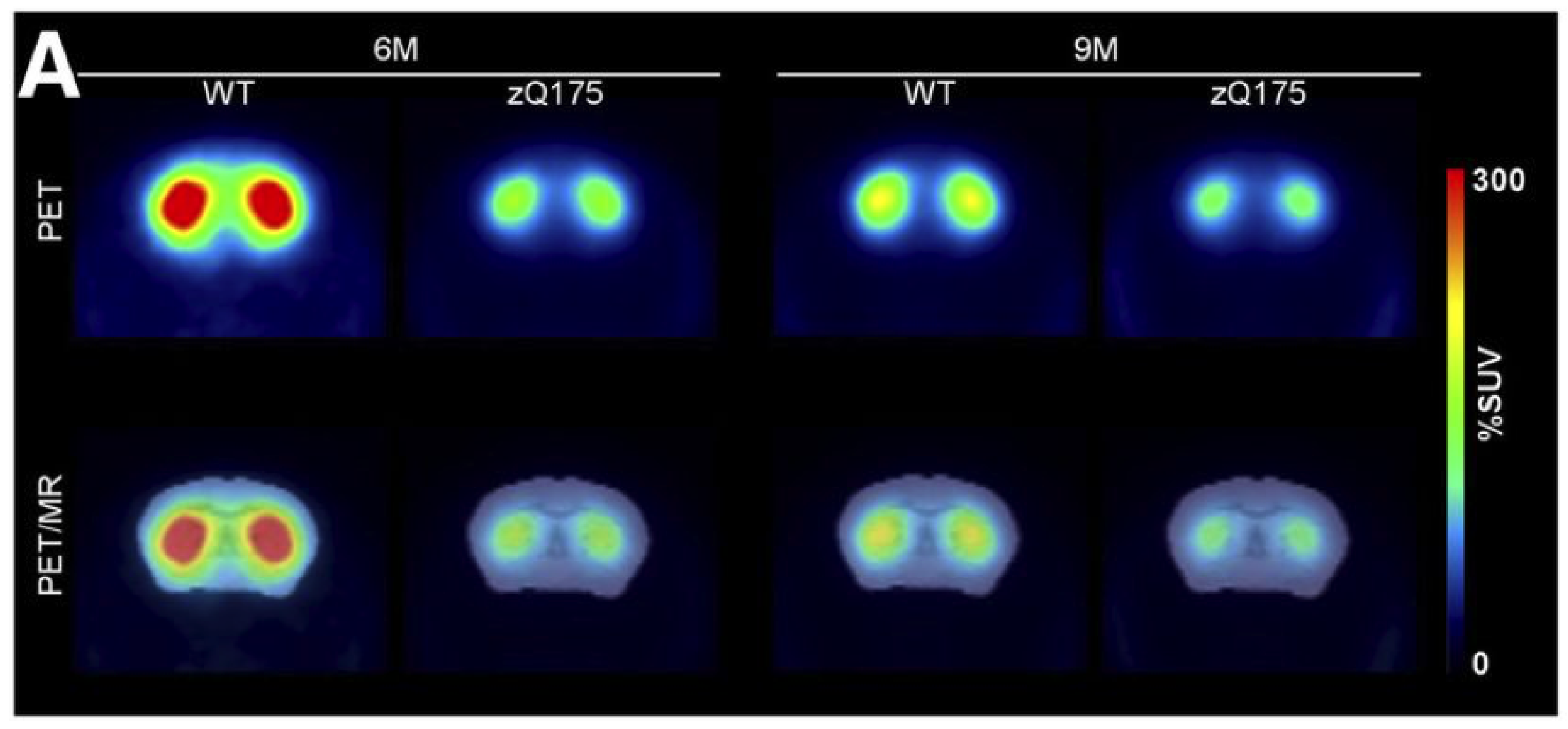
- Bertoglio, D.; Kosten, L.; Verhaeghe, J.; Thomae, D.; Wyffels, L.; Stroobants, S.; Wityak, J.; Dominguez, C.; Mrzljak, L.; Staelens, S. Longitudinal Characterization of mGluR5 Using 11C-ABP688 PET Imaging in the Q175 Mouse Model of Huntington Disease. J. Nucl. Med. 2018, 59, 1722–1727. [Google Scholar] [CrossRef] [Green Version]
- Politis, M.; Pavese, N.; Tai, Y.F.; Kiferle, L.; Mason, S.L.; Brooks, D.J.; Tabrizi, S.J.; Barker, R.A.; Piccini, P. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: A multimodal imaging study. Hum. Brain Mapp. 2011, 32, 258–270. [Google Scholar] [CrossRef]
- Dupont, A.C.; Largeau, B.; Santiago Ribeiro, M.; Guilloteau, D.; Tronel, C.; Arlicot, N. Translocator Protein-18 kDa (TSPO) Positron Emission Tomography (PET) Imaging and Its Clinical Impact in Neurodegenerative Diseases. Int. J. Mol. Sci. 2017, 18, 785. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Toyama, H.; Hatano, K.; Suzuki, H.; Ichise, M.; Momosaki, S.; Kudo, G.; Ito, F.; Kato, T.; Yamaguchi, H.; Katada, K.; et al. In vivo imaging of microglial activation using a peripheral benzodiazepine receptor ligand: [11C]PK-11195 and animal PET following ethanol injury in rat striatum. Ann. Nucl. Med. 2008, 22, 417–424. [Google Scholar] [CrossRef]
- Alves, V.H.; Abrunhosa, A.J.; Castelo-Branco, M. Optimisation of synthesis, purification and reformulation of (R)-[N-Methyl-11C]PK11195 for in vivo PET imaging studies. In Proceedings of the 2013 IEEE 3rd Portuguese Meeting in Bioengineering (ENBENG), Braga, Portugal, 20–23 February 2013; pp. 1–5. [Google Scholar]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 2007, 130, 1759–1766. [Google Scholar] [CrossRef] [Green Version]
- Simmons, D.A.; James, M.L.; Belichenko, N.P.; Semaan, S.; Condon, C.; Kuan, J.; Shuhendler, A.J.; Miao, Z.; Chin, F.T.; Longo, F.M. TSPO-PET imaging using [18F]PBR06 is a potential translatable biomarker for treatment response in Huntington’s disease: preclinical evidence with the p75NTR ligand LM11A-31. Hum. Mol. Genet. 2018, 27, 2893–2912. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, Y.; Kimura, Y.; Siméon, F.G.; Dickstein, L.P.; Pike, V.W.; Innis, R.B.; Fujita, M. Biodistribution and Radiation Dosimetry in Humans of a New PET Ligand, 18F-PBR06, to Image Translocator Protein (18 kDa). J. Nucl. Med. 2010, 51, 145–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imaizumi, M.; Briard, E.; Zoghbi, S.S.; Gourley, J.P.; Hong, J.; Musachio, J.L.; Gladding, R.; Pike, V.W.; Innis, R.B.; Fujita, M. Kinetic evaluation in nonhuman primates of two new PET ligands for peripheral benzodiazepine receptors in brain. Synapse 2007, 61, 595–605. [Google Scholar] [CrossRef]
- Wang, M.; Gao, M.; Miller, K.D.; Zheng, Q.H. Synthesis of [11C]PBR06 and [18F]PBR06 as agents for positron emission tomographic (PET) imaging of the translocator protein (TSPO). Steroids 2011, 76, 1331–1340. [Google Scholar] [CrossRef]
- Briard, E.; Zoghbi, S.S.; Siméon, F.G.; Imaizumi, M.; Gourley, J.P.; Shetty, H.U.; Lu, S.; Fujita, M.; Innis, R.B.; Pike, V.W. Single-Step High-Yield Radiosynthesis and Evaluation of a Sensitive 18F-Labeled Ligand for Imaging Brain Peripheral Benzodiazepine Receptors with PET. J. Med. Chem. 2009, 52, 688–699. [Google Scholar] [CrossRef] [Green Version]
- Ooms, M.; Attili, B.; Celen, S.; Koole, M.; Verbruggen, A.; Van Laere, K.; Bormans, G. [18F]JNJ42259152 binding to phosphodiesterase 10A, a key regulator of medium spiny neuron excitability, is altered in the presence of cyclic AMP. J. Neurochem. 2016, 139, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Hebb, A.; Robertson, H.; Denovan-Wright, E. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington’s disease transgenic mice prior to the onset of motor symptoms. Neuroscience 2004, 123, 967–981. [Google Scholar] [CrossRef]
- Boscutti, G.; A Rabiner, E.; Plisson, C. PET Radioligands for imaging of the PDE10A in human: Current status. Neurosci. Lett. 2019, 691, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Andrés, J.I.; De Angelis, M.; Alcázar, J.; Iturrino, L.; Langlois, X.; Dedeurwaerdere, S.; Lenaerts, I.; Vanhoof, G.; Celen, S.; Bormans, G. Synthesis, In Vivo Occupancy, and Radiolabeling of Potent Phosphodiesterase Subtype-10 Inhibitors as Candidates for Positron Emission Tomography Imaging. J. Med. Chem. 2011, 54, 5820–5835. [Google Scholar] [CrossRef] [PubMed]
- Van Laere, K.; Ahmad, R.U.; Hudyana, H.; Dubois, K.; Schmidt, M.E.; Celen, S.; Bormans, G.; Koole, M. Quantification of 18F-JNJ-42259152, a Novel Phosphodiesterase 10A PET Tracer: Kinetic Modeling and Test-Retest Study in Human Brain. J. Nucl. Med. 2013, 54, 1285–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Laere, K.; Ahmad, R.U.; Hudyana, H.; Celen, S.; Dubois, K.; Schmidt, M.E.; Bormans, G.; Koole, M. Human biodistribution and dosimetry of 18F-JNJ42259152, a radioligand for phosphodiesterase 10A imaging. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Bourgeois, S.; Postnov, A.; Schmidt, M.E.; Bormans, G.; Van Laere, K.; Vandenberghe, W. PET imaging shows loss of striatal PDE10A in patients with Huntington disease. Neurology 2014, 82, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Barret, O.; Thomae, D.; Tavares, A.; Alagille, D.; Papin, C.; Waterhouse, R.; McCarthy, T.; Jennings, D.; Marek, K.; Russell, D.; et al. In Vivo Assessment and Dosimetry of 2 Novel PDE10A PET Radiotracers in Humans: 18F-MNI-659 and 18F-MNI-654. J. Nucl. Med. 2014, 55, 1297–1304. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.S.; Barret, O.; Jennings, D.L.; Friedman, J.H.; Tamagnan, G.D.; Thomae, D.; Alagille, D.; Morley, T.J.; Papin, C.; Papapetropoulos, S.; et al. The Phosphodiesterase 10 Positron Emission Tomography Tracer, [18F]MNI-659, as a Novel Biomarker for Early Huntington Disease. JAMA Neurol. 2014, 71, 1520. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.S.; Jennings, D.L.; Barret, O.; Tamagnan, G.D.; Carroll, V.M.; Caillé, F.; Alagille, D.; Morley, T.J.; Papin, C.; Seibyl, J.P.; et al. Change in PDE10 across early Huntington disease assessed by [18F]MNI-659 and PET imaging. Neurology 2016, 86, 748–754. [Google Scholar] [CrossRef]
- Plisson, C.; Weinzimmer, D.; Jakobsen, S.; Natesan, S.; Salinas, C.; Lin, S.F.; Labaree, D.; Zheng, M.Q.; Nabulsi, N.; Marques, T.R.; et al. Phosphodiesterase 10A PET Radioligand Development Program: From Pig to Human. J. Nucl. Med. 2014, 55, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Niccolini, F.; Haider, S.; Reis Marques, T.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Piccini, P.; Kapur, S.; Rabiner, E.A.; et al. Altered PDE10A expression detectable early before symptomatic onset in Huntington’s disease. Brain 2015, 138, 3016–3029. [Google Scholar] [CrossRef] [Green Version]
- Heurling, K.; Ashton, N.J.; Leuzy, A.; Zimmer, E.R.; Blennow, K.; Zetterberg, H.; Eriksson, J.; Lubberink, M.; Schöll, M. Synaptic vesicle protein 2A as a potential biomarker in synaptopathies. Mol. Cell. Neurosci. 2019, 97, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Vogl, C.; Mochida, S.; Wolff, C.; Whalley, B.J.; Stephens, G.J. The Synaptic Vesicle Glycoprotein 2A Ligand Levetiracetam Inhibits Presynaptic Ca2+ Channels through an Intracellular Pathway. Mol. Pharmacol. 2012, 82, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercier, J.; Provins, L.; Valade, A. Discovery and development of SV2A PET tracers: Potential for imaging synaptic density and clinical applications. Drug Discov. Today Technol. 2017, 25, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Mercier, J.; Archen, L.; Bollu, V.; Carré, S.; Evrard, Y.; Jnoff, E.; Kenda, B.; Lallemand, B.; Michel, P.; Montel, F.; et al. Discovery of Heterocyclic Nonacetamide Synaptic Vesicle Protein 2A (SV2A) Ligands with Single-Digit Nanomolar Potency: Opening Avenues towards the First SV2A Positron Emission Tomography (PET) Ligands. ChemMedChem 2014, 9, 693–698. [Google Scholar] [CrossRef]
- Bahri, M.A.; Plenevaux, A.; Aerts, J.; Bastin, C.; Becker, G.; Mercier, J.; Valade, A.; Buchanan, T.; Mestdagh, N.; Ledoux, D.; et al. Measuring brain synaptic vesicle protein 2A with positron emission tomography and [18F]UCB-H. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 481–486. [Google Scholar] [CrossRef]
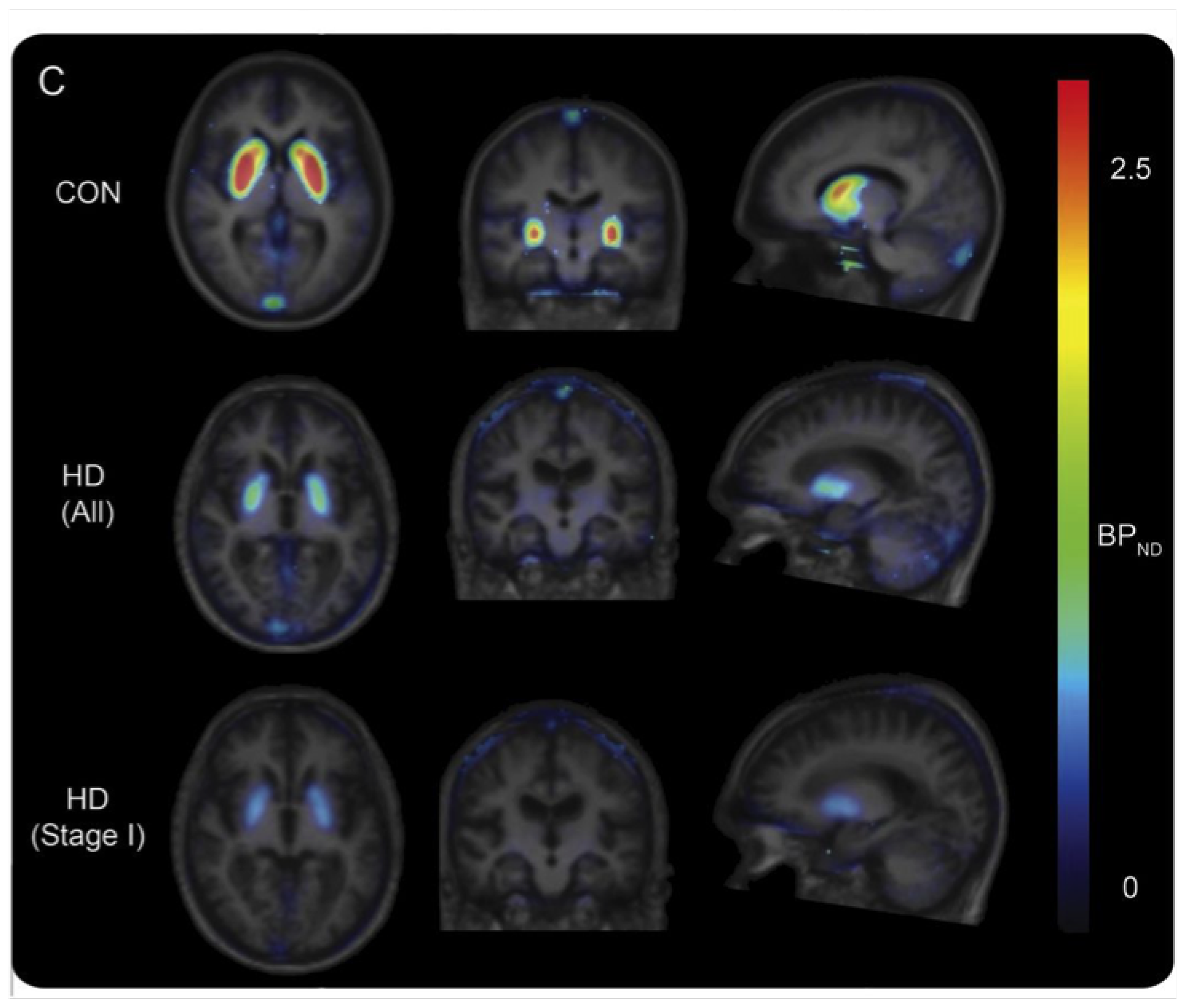
- Nabulsi, N.B.; Mercier, J.; Holden, D.; Carre, S.; Najafzadeh, S.; Vandergeten, M.C.; Lin, S.f.; Deo, A.; Price, N.; Wood, M.; et al. Synthesis and Preclinical Evaluation of 11C-UCB-J as a PET Tracer for Imaging the Synaptic Vesicle Glycoprotein 2A in the Brain. J. Nucl. Med. 2016, 57, 777–784. [Google Scholar] [CrossRef] [Green Version]
- DiFilippo, A.; Murali, D.; Ellison, P.; Barnhart, T.; Engle, J.; Christian, B. Improved synthesis of [11C]UCB-J for PET imaging of synaptic density. J. Nucl. Med. 2019, 60, 1624. [Google Scholar]
- Bertoglio, D.; Verhaeghe, J.; Miranda, A.; Kertesz, I.; Cybulska, K.; Korat, Š.; Wyffels, L.; Stroobants, S.; Mrzljak, L.; Dominguez, C.; et al. Validation and noninvasive kinetic modeling of [11C]UCB-J PET imaging in mice. J. Cereb. Blood Flow Metab. 2019. [Google Scholar] [CrossRef] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
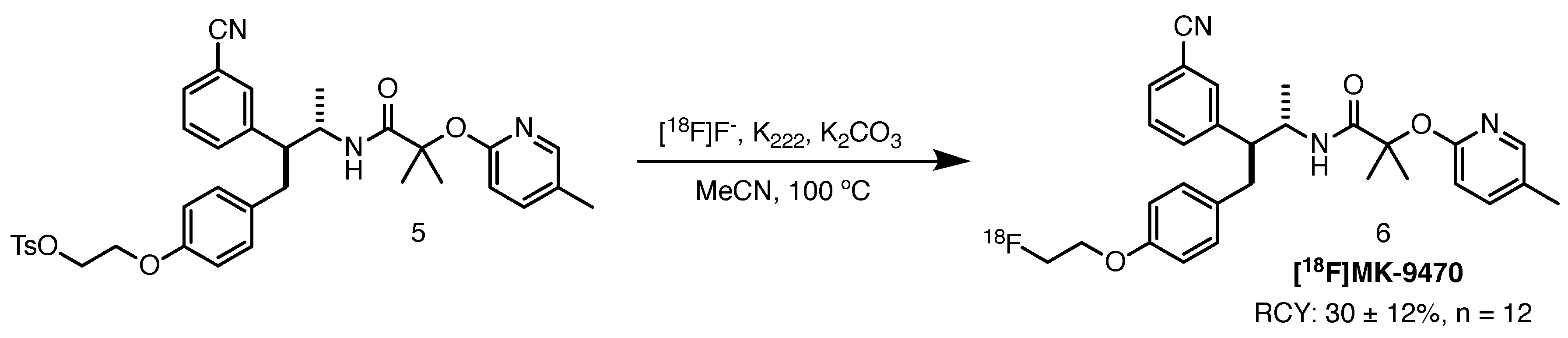{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
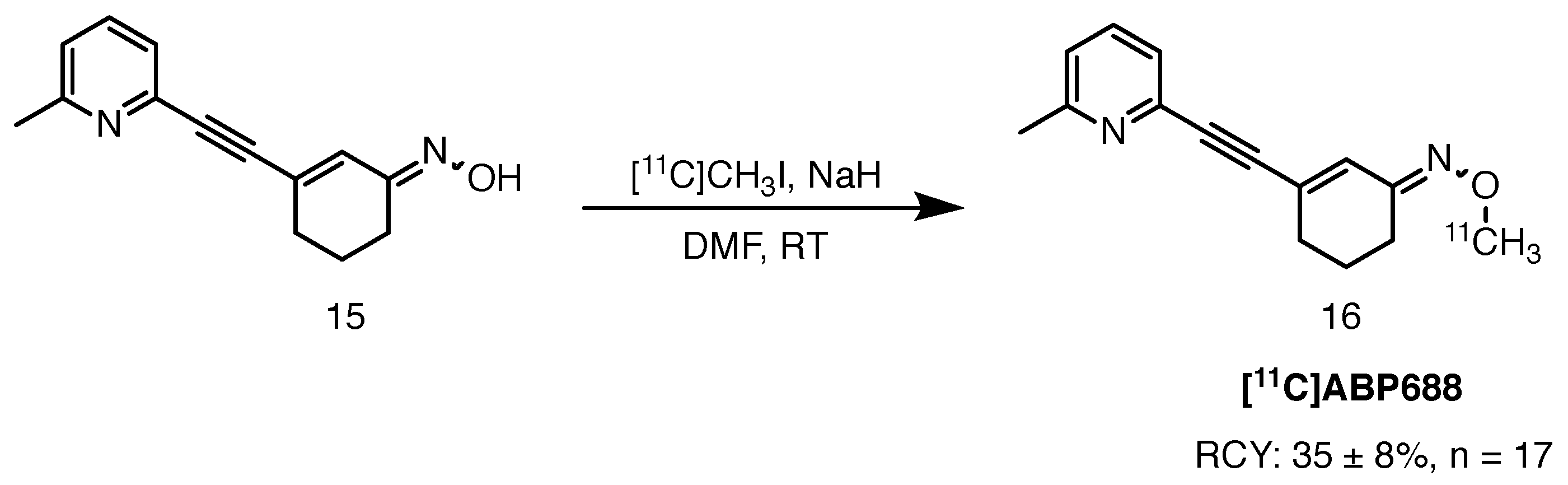{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cybulska, K.; Perk, L.; Booij, J.; Laverman, P.; Rijpkema, M. Huntington’s Disease: A Review of the Known PET Imaging Biomarkers and Targeting Radiotracers. Molecules 2020, 25, 482. https://doi.org/10.3390/molecules25030482
Cybulska K, Perk L, Booij J, Laverman P, Rijpkema M. Huntington’s Disease: A Review of the Known PET Imaging Biomarkers and Targeting Radiotracers. Molecules. 2020; 25(3):482. https://doi.org/10.3390/molecules25030482
Chicago/Turabian StyleCybulska, Klaudia, Lars Perk, Jan Booij, Peter Laverman, and Mark Rijpkema. 2020. "Huntington’s Disease: A Review of the Known PET Imaging Biomarkers and Targeting Radiotracers" Molecules 25, no. 3: 482. https://doi.org/10.3390/molecules25030482
APA StyleCybulska, K., Perk, L., Booij, J., Laverman, P., & Rijpkema, M. (2020). Huntington’s Disease: A Review of the Known PET Imaging Biomarkers and Targeting Radiotracers. Molecules, 25(3), 482. https://doi.org/10.3390/molecules25030482