New Poly(Ionic Liquid) Based Fiber for Determination of Oxytetracycline in Milk Samples by Application of SPME-CE Technique

 ,
, 
 , and
, and 
Abstract
:1. Introduction
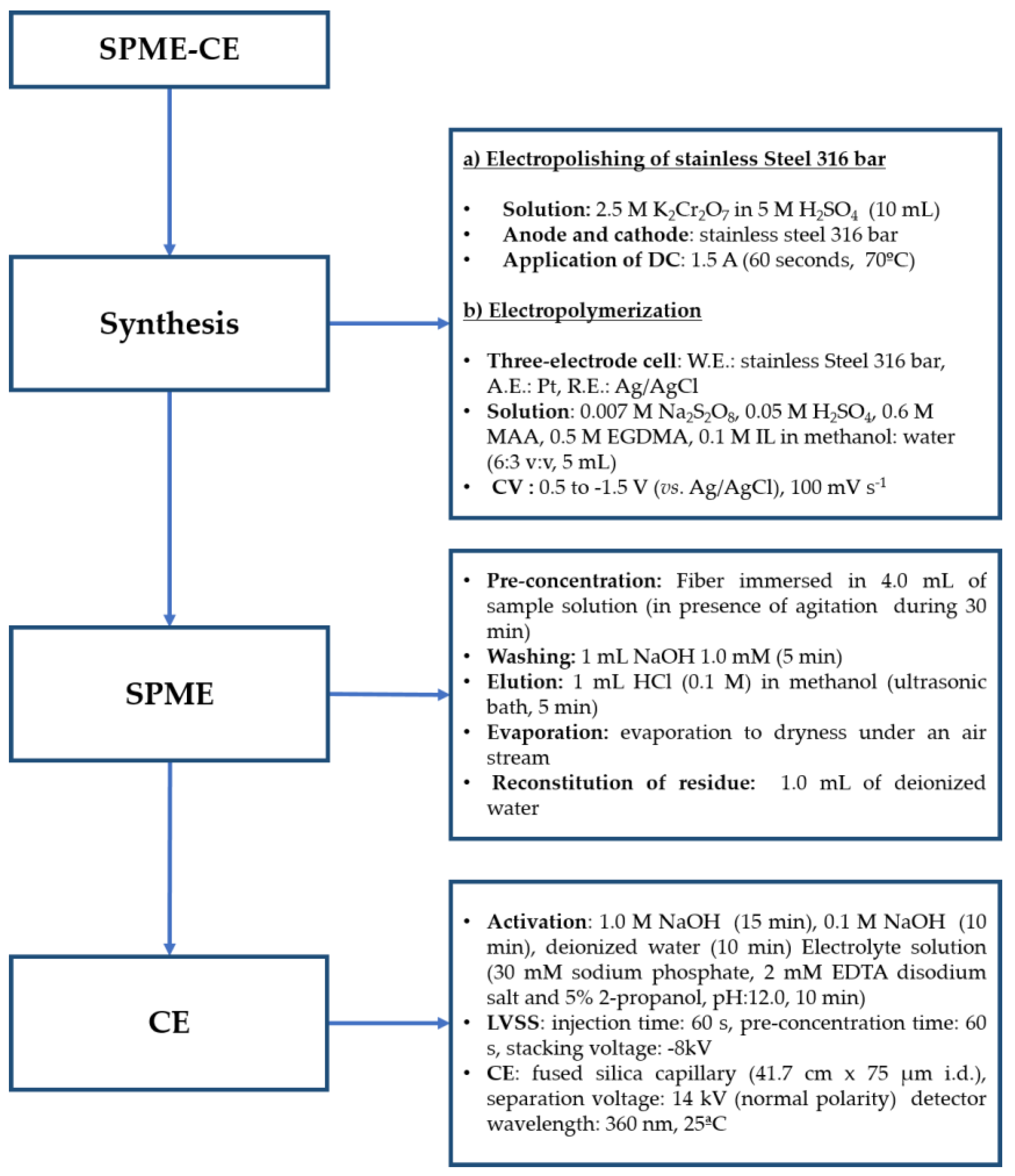
2. Results and Discussion
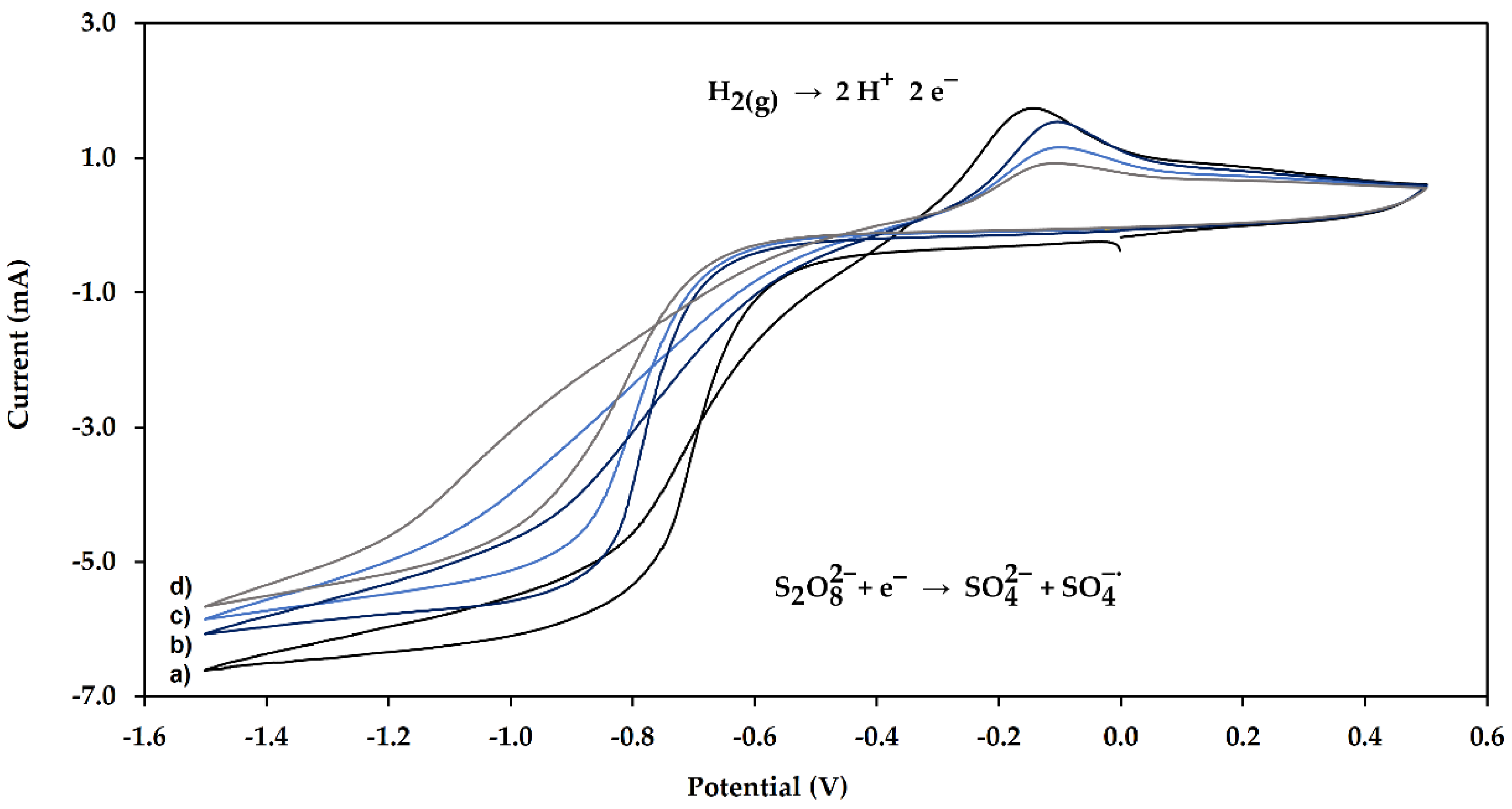
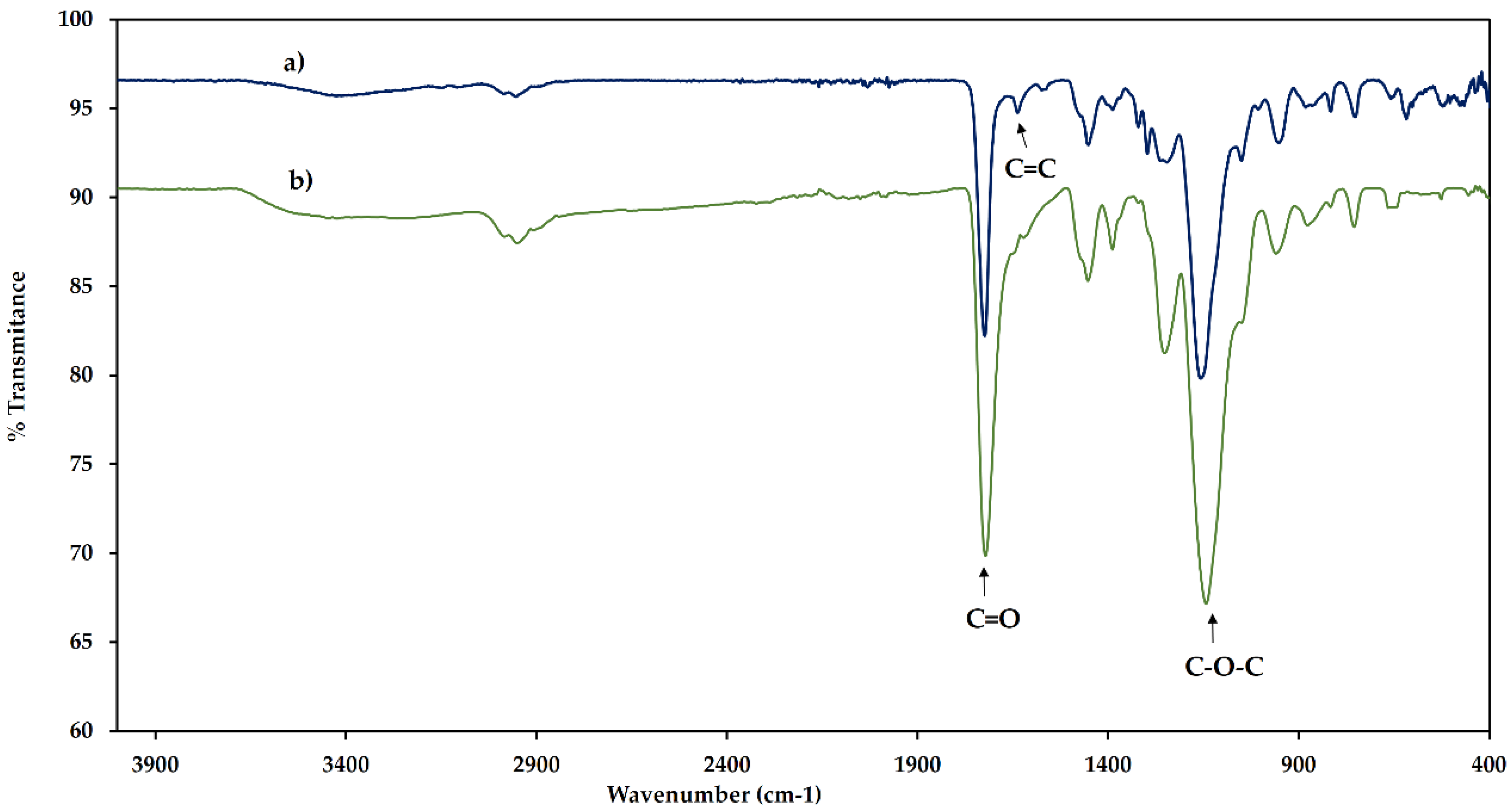
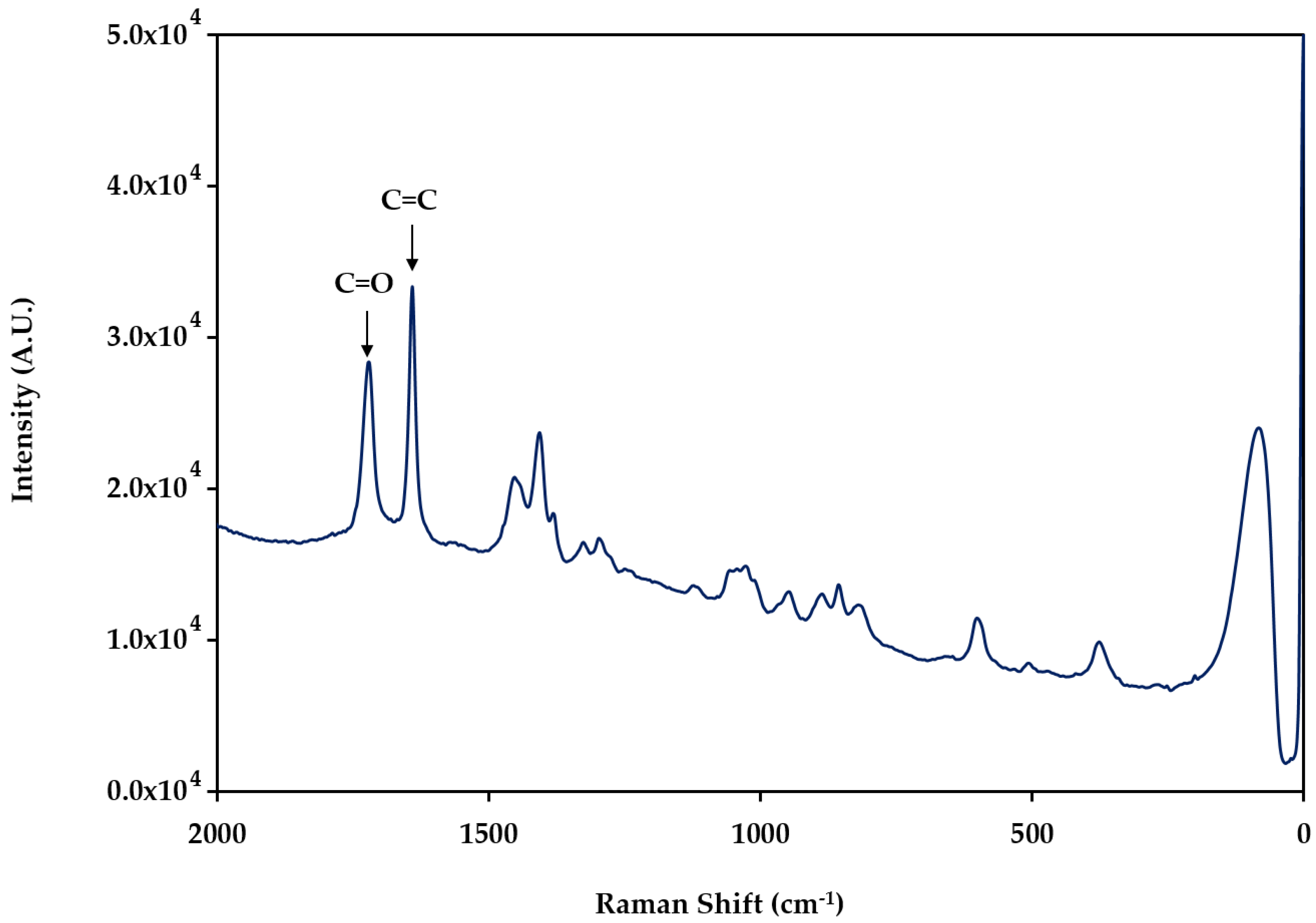
2.1. Electropolymerization
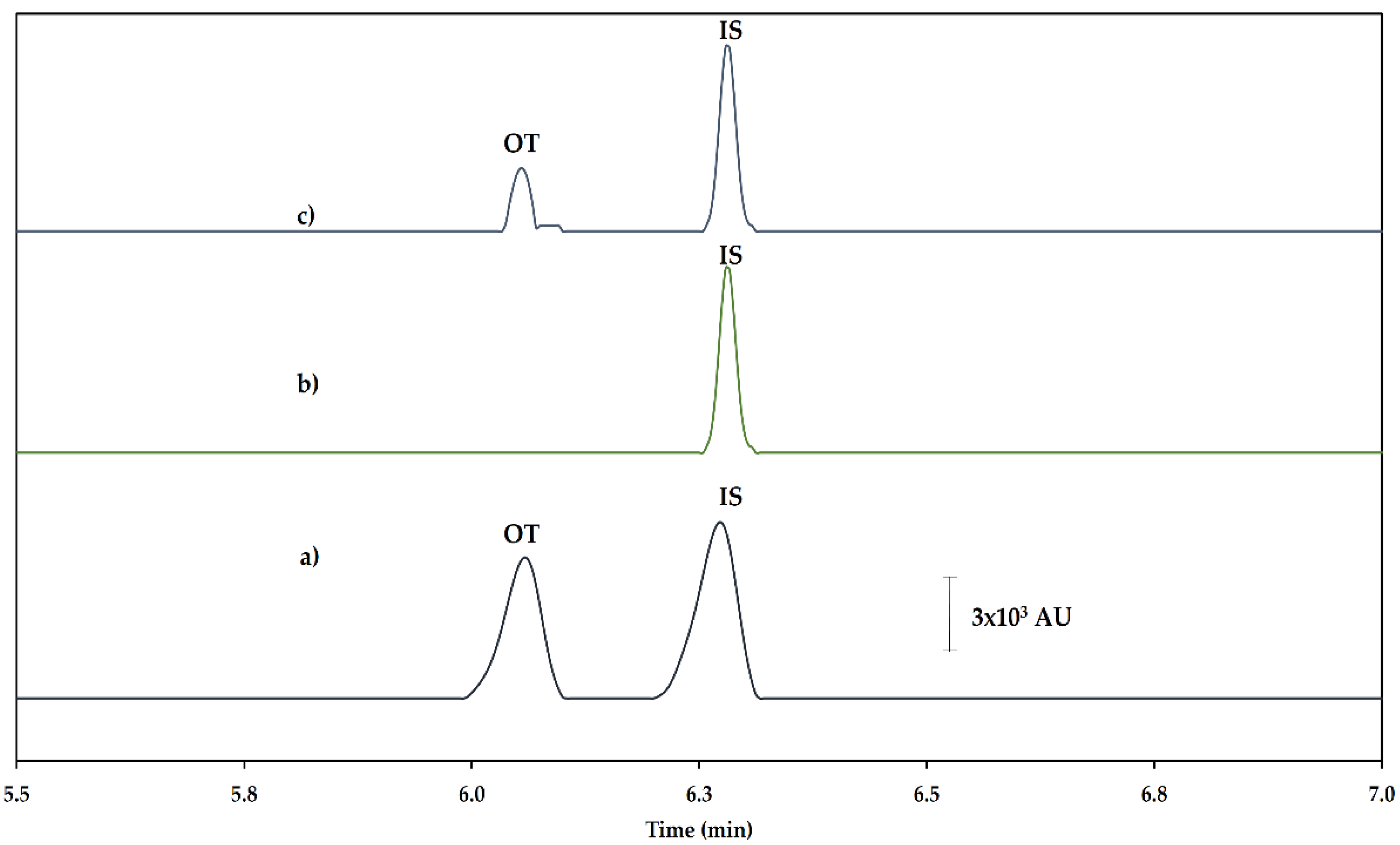
2.2. Optimization of the SPME Procedure
2.3. Method Validation
2.4. The Environmental Impact of the Methodology
3. Experimental Section
3.1. Reagents and Chemicals
3.2. Equipment
3.3. Electropolymerization
3.4. Sample Preparation
3.5. Method Validation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Berruga, I.M.; Molina, A.; Althaus, L.R.; Molina, P.M. Control and prevention of antibiotic residues and contaminants in sheep and goat’s milk. Small Rumin. Res. 2016, 142, 38–43. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, M.P.; Pezza, H.R.; Pezza, L. Simple and clean determination of tetracyclines by flow injection analysis. Spectrochim. Acta Part A 2016, 153, 386–392. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, S.; Aga, D.S. Analysis of tetracycline antibiotics in solid: Advances in extraction, cleanup, and quantification. TrAC Trends Anal. Chem. 2007, 26, 456–465. [Google Scholar] [CrossRef]
- United States Food and Drug Administration (FDA). Code of Federal Regulations, Tolerances for Residues of New Animal Drugs in Food; FDA: Silver Spring, MD, USA, 2018; p. 6. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfcfr/CFRSearchcfm?fr=556.500 (accessed on 10 January 2019).
- European Union Commission Regulation No. 37/2010, Official Journal of the European Union L15, 2010, 1–72. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32010R0037 (accessed on 10 January 2019).
- Furusawa, N. Rapid liquid chromatographic determination of oxytetracycline in milk. J. Chromatogr. A 1999, 839, 247–251. [Google Scholar] [CrossRef]
- Pena, A.; Pelantova, N.; Lino, C.M.; Silveira, M.I.N.; Solich, P. Validation of an analytical methodology for determination of oxytetracycline and tetracycline residues in honey by HPLC with fluorescence detection. J. Agric. Food Chem. 2005, 53, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Rajput, Y.S.; Singh, G.; Sharma, R. Synthesis and characterization of oxytetracycline imprinted polymer for application in food. Appl. Nanosci. 2016, 6, 209–214. [Google Scholar] [CrossRef]
- Miranda, J.M.; Rodríguez, J.A.; Galán-Vidal, C.A. Simultaneous determination of tetracyclines in poultry muscle by capillary zone electrophoresis. J. Chromatogr. A 2009, 1216, 3366–3371. [Google Scholar] [CrossRef]
- Spietelun, A.; Kloskowski, A.; Chrzanowski, W.; Namiesńik, J. Understanding solid-phase microextraction: Key factors influencing the extraction process and trends in improving the technique. Chem. Rev. 2013, 113, 1667–1685. [Google Scholar] [CrossRef]
- Joshi, M.D.; Ho, T.D.; Cole, W.T.S.; Anderson, J.L. Determination of polychlorinated biphenyls in ocean water and bovine milk using crosslinked polymeric ionic liquid sorbent coatings by solid-phase microextraction. Talanta 2014, 118, 172–179. [Google Scholar] [CrossRef]
- Chen, W.; Zeng, J.; Chen, J.; Huang, X.; Jiang, Y.; Wang, Y.; Chen, X. High extraction efficiency for polar aromatic compounds in natural water samples using multiwalled carbon nanotubes/Nafion solid-phase microextraction coating. J. Chromatogr. A 2009, 1216, 9143–9148. [Google Scholar] [CrossRef]
- Fontanals, N.; Borrull, F.; Marcé, R.M. Ionic liquids in solid-phase extraction. Trends Anal. Chem. 2012, 41, 15–26. [Google Scholar] [CrossRef]
- Zhao, F.; Meng, Y.; Anderson, J.L. Polymeric ionic liquids as selective coatings for the extraction of esters using solid-phase microextraction. J. Chromatogr. A 2008, 1208, 1–9. [Google Scholar] [CrossRef]
- Meng, Y.; Anderson, J.L. Tuning the selectivity of polymeric ionic liquid sorbent coatings for the extraction of polycyclic aromatic hydrocarbons using solid-phase microextraction. J. Chromatogr. A 2010, 1217, 6143–6152. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.D.; Canestraro, A.J.; Anderson, J.L. Ionic liquids in solid-phase microextraction: A review. Anal. Chim. Acta 2011, 695, 18–43. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.A.; Rodríguez, J.A.; Páez-Hernández, M.E.; Guevara-Lara, A.; Barrado, E.; Hernández, P. Chromium(VI) removal from aqueous solution by magnetite coated by a polymeric ionic liquid-based adsorbent. Materials 2017, 10, 502. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Antonietti, M. Poly(ionic liquid)s: Polymers expanding classical property profiles. Polymer 2011, 52, 1469–1482. [Google Scholar] [CrossRef] [Green Version]
- Devasurendra, A.M.; Zhang, C.; Young, J.A.; Viranga Tillekeratne, L.M.; Anderson, J.L.; Kirchhoff, J.R. Electropolymerized pyrrole-based conductive polymeric ionic liquids and their application for solid-phase microextraction. Appl. Mater. Interfaces 2017, 9, 24955–24963. [Google Scholar] [CrossRef] [PubMed]
- Galuszka, A.; Migaszewski, Z.; Namiesnik, J. The 12 principles of Green analytical chemistry and the SIGNIFICANCE mnemonic of green analytical practices. TrAC 2013, 50, 78–84. [Google Scholar] [CrossRef]
- Cram, S.L.; Spinks, G.M.; Wallace, G.G.; Brown, H.R. Mechanism of electropolymerisation of methyl methacrylate and glycidyl acrylate on stainless steel. Electrochim. Acta 2002, 47, 1935–1948. [Google Scholar] [CrossRef]
- Cram, S.L.; Spinks, G.M.; Wallace, G.G.; Brown, H.R. Electrochemical polymerization of acrylics on stainless steel cathodes. J. Appl. Polym. Sci. 2003, 87, 765–773. [Google Scholar] [CrossRef]
- Bi, W.; Tian, M.; Row, K.H. Separation of phenolic acids from natural plant extracts using molecularly imprinted anion-exchange polymer confined ionic liquids. J. Chromatogr. A 2012, 1232, 37–42. [Google Scholar] [CrossRef] [PubMed]
- García-Campaña, A.M.; Gámiz-Gracia, L.; Lara, F.J.; Del Olmo Iruela, M.; Cruces-Blanco, C. Applications of capillary electrophoresis to the determination of antibiotics in food and environmental samples. Anal. Bioanal. Chem. 2009, 395, 967–986. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.R.; Rupp, H.S.; Wu, W.H. Complexities in tetracycline analysis-chemistry, matrix extraction, cleanup, and liquid chromatography. J. Chromatogr. A 2005, 1075, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Ben, W.; Qiang, Z.; Adams, C.; Zhang, H.; Chen, L. Simultaneous determination of sulfonamides, tetracyclines and tiamulin in swine wastewater by solid-phase extraction and liquid chromatography-mass spectrometry. J. Chromatogr. A 2008, 1202, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Konieczka, P.; Namieśnik, J. Estimating uncertainty in analytical procedures based on chromatographic techniques. J. Chromatogr. A 2010, 1217, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Saudi Arabian Standards Organization (SASO). Guide to the Expression of Uncertainty in Measurement, No. 13/2000; SASO: Riyadh, Saudi Arabian, 2000; Available online: https://www.bipm.org/utils/common/documents/jcgm/JCGM_100_2008_E.pdf (accessed on 10 January 2019).
- Cinquina, A.L.; Longo, F.; Anastasi, G.; Giannetti, L.; Cozzani, R. Validation of a high-performance liquid chromatography method for the determination of oxytetracycline, tetracycline, chlortetracycline and doxycycline in bovine milk and muscle. J. Chromatogr. A 2003, 987, 227–233. [Google Scholar] [CrossRef]
- Yang, X.-Q.; Yang, C.-X.; Yan, X.P. Zeolit imidazolate framework-8 as sorbent for on-line solid-phase extraction coupled with high-performance liquid chromatography for the determination of tetracyclines in water and milk samples. J. Chromatogr. A 2013, 1304, 28–33. [Google Scholar] [CrossRef]
- Islas, G.; Rodríguez, J.A.; Pérez-Silva, I.; Miranda, J.M.; Ibarra, I.S. Solid-Phase extraction and large volume sample stacking-capillary electrophoresis for determination of tetracycline residues in milk. J. Anal. Methods Chem. 2018, 2018, 1–7. [Google Scholar] [CrossRef]
- Ibarra, I.S.; Rodríguez, J.A.; Miranda, J.M.; Vega, M.; Barrado, E. Magnetic solid phase extraction based on phenyl silica adsorbent for the determination of tetracyclines in milk samples by capillary electrophoresis. J. Chromatogr. A 2011, 1218, 2196–2202. [Google Scholar] [CrossRef]
- Gałuszka, A.; Konieczka, P.; Migaszewski, Z.M.; Namiesnik, J. Analytical eco-scale for assessing the greenness of analytical procedures. TrAC Trends Anal. Chem. 2012, 37, 61–72. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| Concentration µg L−1 | 250 | Uncertainty | |
| Calibration equation | Sample (usample) | 0.001 | |
| Analytical sensitivity ± Sb1 | 0.691 ± 0.024 | Calibration (ucal) | 0.028 |
| Intercept ± Sb0 | −0.028 ± 0.024 | Recovery (utrue) | 0.899 |
| R2 | 0.9962 | Repeatability (urep) | 2.248 |
| LOD µg L−1 | 70.36 | LOD (uLOD) | 0.280 |
| Combined uncertainty | 0.98% | ||
| RSD (n = 3) | 1.57 | Expanded uncertainty | 1.96% |
| Result | |||
| Concentration ± U(*k = 2) µg L−1 | 250 ± 4.9 |
| Methodology | LOD (µg L−1) | %RSD (Intra day/Inter day) | Reference |
|---|---|---|---|
| HPLC-DAD | 50 | 2.3/4.1 | [6] |
| SPE-HPLC-DAD | 113(µg kg−1) | 4.70 (inter day) | [29] |
| SPE-HPLC-PDA | 1.5 | 1.6/3.6 | [30] |
| SPE-LVSS-CE | 19 | 9.2/7.0 | [31] |
| MSPE-CE | 2 | 1.7/2.2 | [32] |
| SPME-LVSS-CE-DAD | 70 (68 µg kg−1) | 3.5/4.5 | This work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, T.A.; Flores-Aguilar, J.F.; Santos, E.M.; Rodriguez, J.A.; Ibarra, I.S. New Poly(Ionic Liquid) Based Fiber for Determination of Oxytetracycline in Milk Samples by Application of SPME-CE Technique. Molecules 2019, 24, 430. https://doi.org/10.3390/molecules24030430
Ferreira TA, Flores-Aguilar JF, Santos EM, Rodriguez JA, Ibarra IS. New Poly(Ionic Liquid) Based Fiber for Determination of Oxytetracycline in Milk Samples by Application of SPME-CE Technique. Molecules. 2019; 24(3):430. https://doi.org/10.3390/molecules24030430
Chicago/Turabian StyleFerreira, T. Alexandra, J. Francisco Flores-Aguilar, Eva M. Santos, Jose A. Rodriguez, and Israel S. Ibarra. 2019. "New Poly(Ionic Liquid) Based Fiber for Determination of Oxytetracycline in Milk Samples by Application of SPME-CE Technique" Molecules 24, no. 3: 430. https://doi.org/10.3390/molecules24030430
APA StyleFerreira, T. A., Flores-Aguilar, J. F., Santos, E. M., Rodriguez, J. A., & Ibarra, I. S. (2019). New Poly(Ionic Liquid) Based Fiber for Determination of Oxytetracycline in Milk Samples by Application of SPME-CE Technique. Molecules, 24(3), 430. https://doi.org/10.3390/molecules24030430