
Novel Approach for the Search for Chemical Scaffolds with Activity at Both Acetylcholinesterase and the α7 Nicotinic Acetylcholine Receptor: A Perspective on Scaffolds with Dual Activity for the Treatment of Neurodegenerative Disorders

 ,
,
Abstract
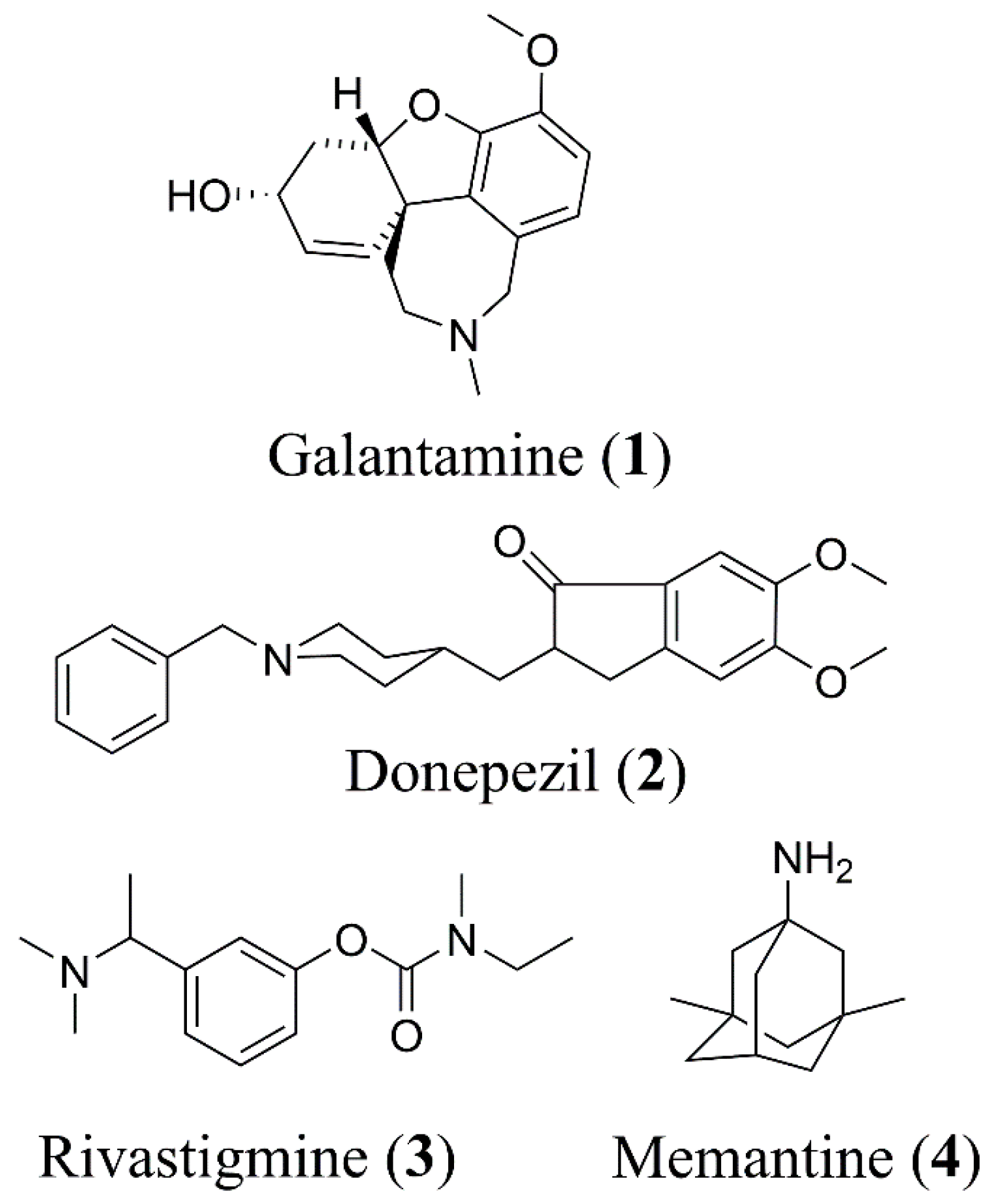
:1. Introduction
2. Results
2.1. Virtual Screening
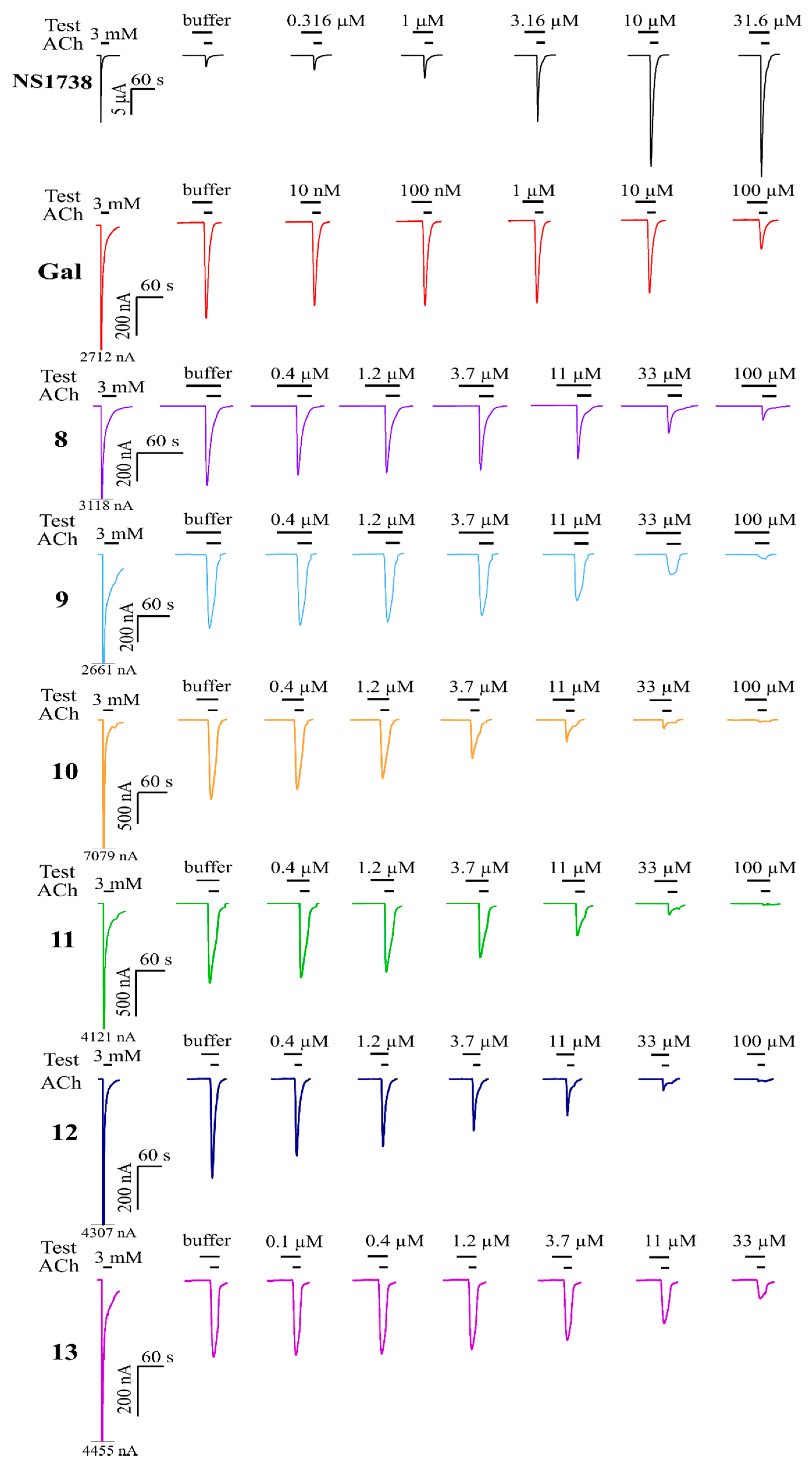
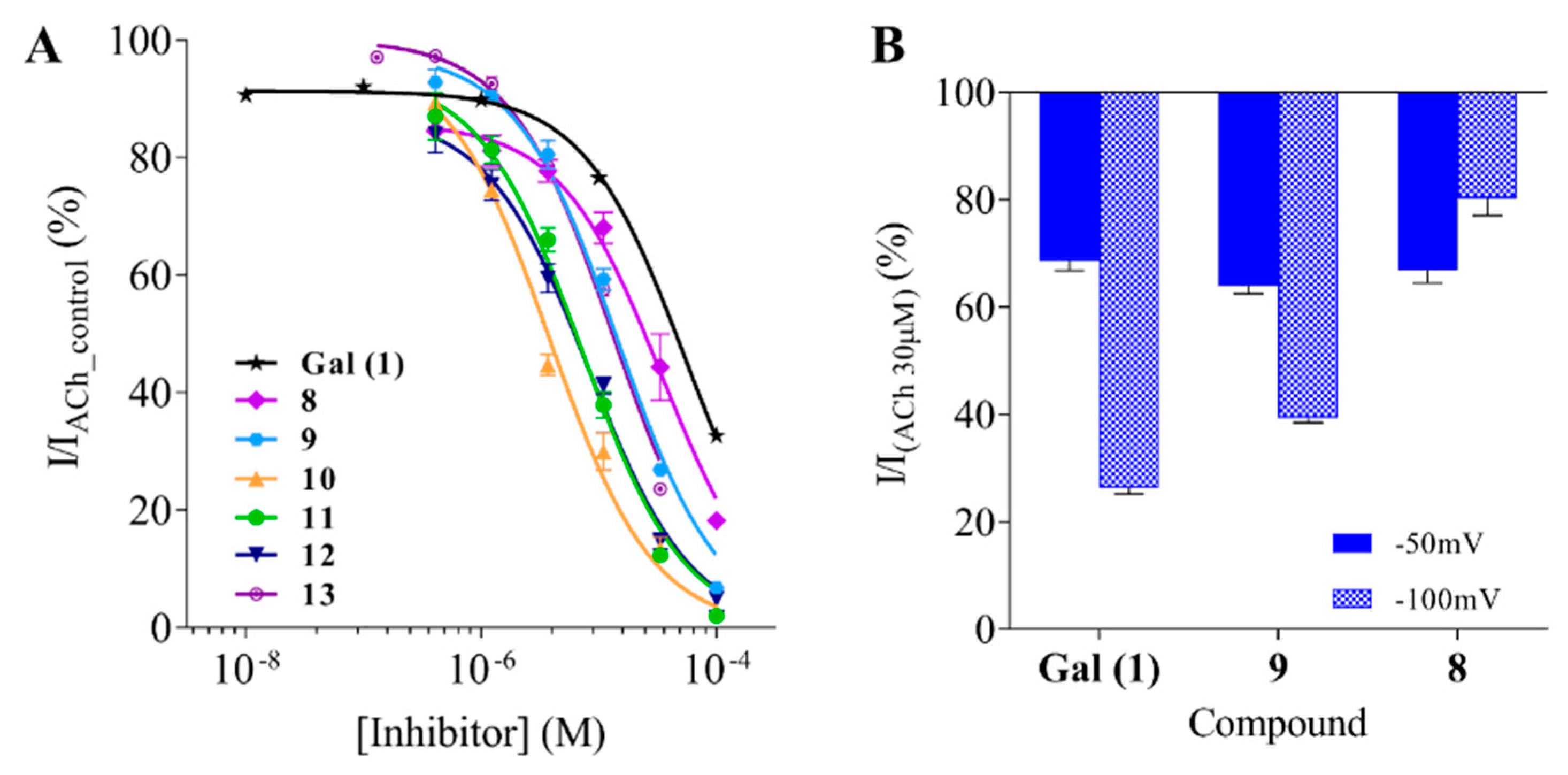
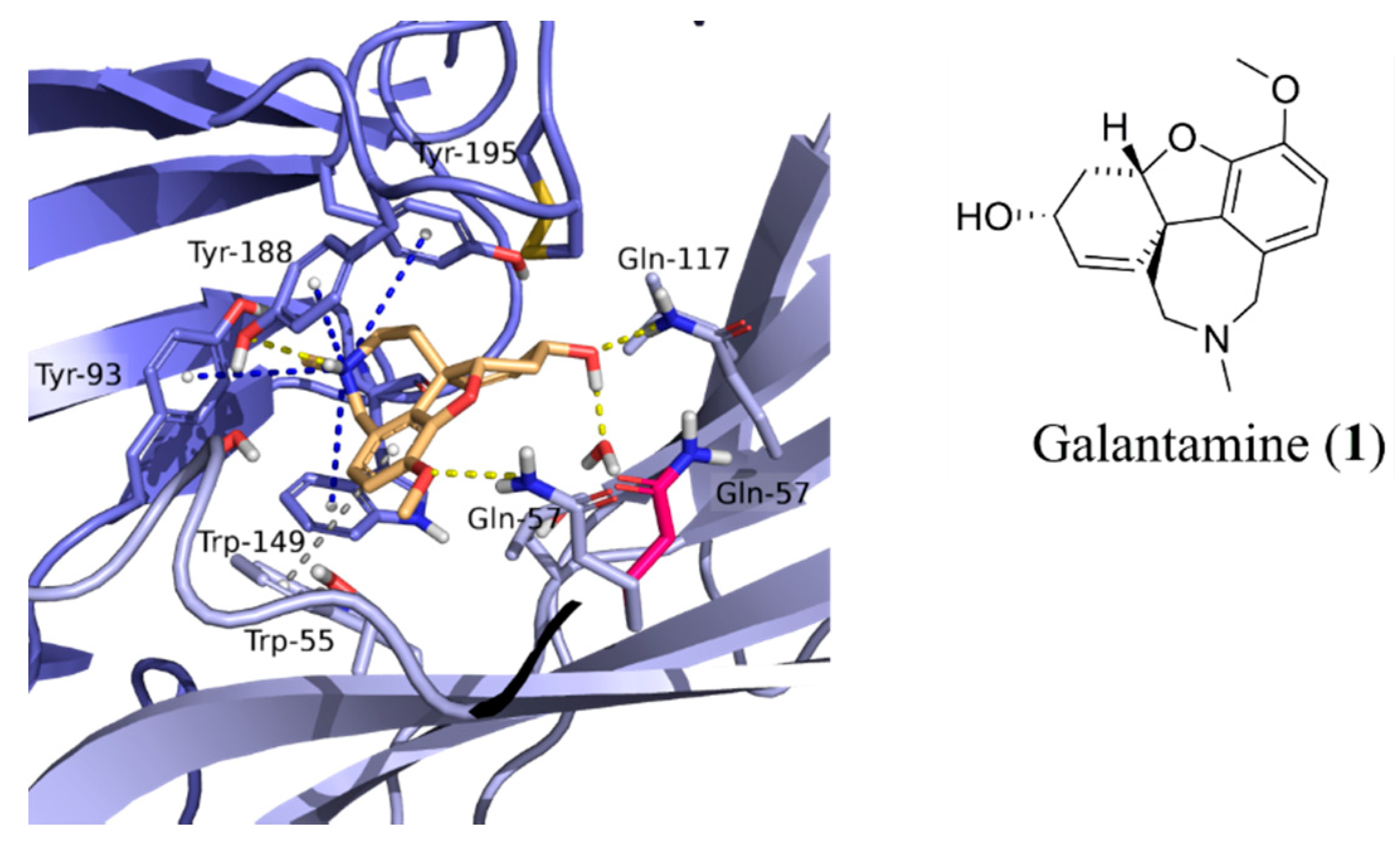
2.2. Assessment of Activity with AChE and α7 nAChRs
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Homology Modeling and Preparation of Protein Structures
4.3. Preparation of Databases
4.4. HTVS and Hits Selection
4.5. Validation of Activity at nAChRs
4.5.1. Molecular Biology
4.5.2. Expression of nAChR in Xenopus laevis Oocytes
4.5.3. Oocyte Electrophysiology
4.6. Validation of Activity at AChE
4.7. Data Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reiman, E.M.; Quiroz, Y.T.; Fleisher, A.S.; Chen, K.; Velez-Pardo, C.; Jimenez-Del-Rio, M.; Fagan, A.M.; Shah, A.R.; Alvarez, S.; Arbelaez, A.; et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: A case-control study. Lancet Neurol. 2012, 11, 1048–1056. [Google Scholar] [CrossRef]
- Grossberg, G.T.; Edwards, K.R.; Zhao, Q. Rationale for combination therapy with galantamine and memantine in Alzheimer’s disease. J. Clin. Pharm. 2006, 46, 17s–26s. [Google Scholar] [CrossRef]
- Zhao, X.; Marszalec, W.; Toth, P.T.; Huang, J.; Yeh, J.Z.; Narahashi, T. In vitro galantamine-memantine co-application: Mechanism of beneficial action. Neuropharmacology 2006, 51, 1181–1191. [Google Scholar] [CrossRef]
- Lopes, J.P.; Tarozzo, G.; Reggiani, A.; Piomelli, D.; Cavalli, A. Galantamine potentiates the neuroprotective effect of memantine against NMDA-induced excitotoxicity. Brain Behav. 2013, 3, 67–74. [Google Scholar] [CrossRef]
- Atri, A.; Molinuevo, J.L.; Lemming, O.; Wirth, Y.; Pulte, I.; Wilkinson, D. Memantine in patients with Alzheimer’s disease receiving donepezil: New analyses of efficacy and safety for combination therapy. Alzheimer’s Res. 2013, 5, 6. [Google Scholar] [CrossRef]
- Grossberg, G.T.; Manes, F.; Allegri, R.F.; Gutierrez-Robledo, L.M.; Gloger, S.; Xie, L.; Jia, X.D.; Pejovic, V.; Miller, M.L.; Perhach, J.L.; et al. The safety, tolerability, and efficacy of once-daily memantine (28 mg): A multinational, randomized, double-blind, placebo-controlled trial in patients with moderate-to-severe Alzheimer’s disease taking cholinesterase inhibitors. CNS Drugs 2013, 27, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Exp. Rev. Clin. Pharm. 2013, 6. [Google Scholar] [CrossRef]
- Hughes, R.E.; Nikolic, K.; Ramsay, R.R. One for all? Hitting multiple Alzheimer’s Disease targets with one drug. Front. Neurosci. 2016, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, K.; Mavridis, L.; Djikic, T.; Vucicevic, J.; Agbaba, D.; Yelekci, K.; Mitchell, J.B.O. Drug design for cns diseases: polypharmacological profiling of compounds using cheminformatic, 3D-QSAR and Virtual screening methodologies. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef]
- Zhu, T.; Cao, S.; Su, P.C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef] [PubMed]
- Moser, D.; Wisniewska, J.M.; Hahn, S.; Achenbach, J.; Buscató, E.L.; Klingler, F.-M.; Hofmann, B.; Steinhilber, D.; Proschak, E. Dual-target virtual screening by pharmacophore elucidation and molecular shape filtering. ACS Med. Chem. Lett. 2012, 3, 155–158. [Google Scholar] [CrossRef]
- Zhou, S.; Li, Y.; Hou, T. Feasibility of using molecular docking-based virtual screening for searching dual target kinase inhibitors. J. Chem. Inf. Model. 2013, 53, 982–996. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Talebi, M.; Mahmoudi, J.; Babri, S.; Shanehbandi, D. Selective activation of alpha7 nicotinic acetylcholine receptor by PHA-543613 improves Abeta25-35-mediated cognitive deficits in mice. Neuroscience 2015, 298, 81–93. [Google Scholar] [CrossRef]
- Gotti, C.; Zoli, M.; Clementi, F. Brain nicotinic acetylcholine receptors: Native subtypes and their relevance. Trends Pharm. Sci. 2006, 27, 482–491. [Google Scholar] [CrossRef]
- Rubboli, F.; Court, J.A.; Sala, C.; Morris, C.; Chini, B.; Perry, E.; Clementi, F. Distribution of Nicotinic Receptors in the Human Hippocampus and Thalamus. Eur. J. Neurosci. 1994, 6, 1596–1604. [Google Scholar] [CrossRef]
- Timmermann, D.B.; Gronlien, J.H.; Kohlhaas, K.L.; Nielsen, E.O.; Dam, E.; Jorgensen, T.D.; Ahring, P.K.; Peters, D.; Holst, D.; Christensen, J.K.; et al. An allosteric modulator of the α7 nicotinic acetylcholine receptor possessing cognition-enhancing properties in vivo. J. Pharm. Exper. 2007, 323, 294–307. [Google Scholar] [CrossRef]
- Prickaerts, J.; van Goethem, N.P.; Chesworth, R.; Shapiro, G.; Boess, F.G.; Methfessel, C.; Reneerkens, O.A.H.; Flood, D.G.; Hilt, D.; Gawryl, M.; et al. EVP-6124, a novel and selective α7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of α7 nicotinic acetylcholine receptors. Neuropharmacology 2012, 62, 1099–1110. [Google Scholar] [CrossRef]
- Samochocki, M.; Hoffle, A.; Fehrenbacher, A.; Jostock, R.; Ludwig, J.; Christner, C.; Radina, M.; Zerlin, M.; Ullmer, C.; Pereira, E.F.; et al. Galantamine is an allosterically potentiating ligand of neuronal nicotinic but not of muscarinic acetylcholine receptors. J. Pharm. Exper. 2003, 305, 1024–1036. [Google Scholar] [CrossRef]
- Texido, L.; Ros, E.; Martin-Satue, M.; Lopez, S.; Aleu, J.; Marsal, J.; Solsona, C. Effect of galantamine on the human alpha7 neuronal nicotinic acetylcholine receptor, the Torpedo nicotinic acetylcholine receptor and spontaneous cholinergic synaptic activity. Br. J. Pharm. 2005, 145, 672–678. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Hansen, S.B.; Taylor, P. Galanthamine and non-competitive inhibitor binding to ACh-binding protein: Evidence for a binding site on non-alpha-subunit interfaces of heteromeric neuronal nicotinic receptors. J. Mol. Biol. 2007, 369, 895–901. [Google Scholar] [CrossRef]
- Shahsavar, A.; Gajhede, M.; Kastrup, J.S.; Balle, T. Structural studies of nicotinic acetylcholine receptors: using acetylcholine-binding protein as a structural surrogate. Basic Clin. Pharm. Toxicol. 2016, 118, 399–407. [Google Scholar] [CrossRef]
- Kowal, N.M.; Ahring, P.K.; Liao, V.W.Y.; Indurti, D.C.; Harvey, B.S.; O’Connor, S.M.; Chebib, M.; Olafsdottir, E.S.; Balle, T. Galantamine is not a positive allosteric modulator of human α4β2 or α7 nicotinic acetylcholine receptors. Br. J. Pharm. 2018, 175, 2911–2925. [Google Scholar] [CrossRef]
- Han, S.Y.; Sweeney, J.E.; Bachman, E.S.; Schweiger, E.J.; Forloni, G.; Coyle, J.T.; Davis, B.M.; Joullié, M.M. Chemical and pharmacological characterization of galanthamine, an acetylcholinesterase inhibitor, and its derivatives. A potential application in Alzheimer’s disease? Eur. J. Med. Chem. 1992, 27, 673–687. [Google Scholar] [CrossRef]
- Bores, G.M.; Huger, F.P.; Petko, W.; Mutlib, A.E.; Camacho, F.; Rush, D.K.; Selk, D.E.; Wolf, V.; Kosley, R.W., Jr.; Davis, L.; et al. Pharmacological evaluation of novel Alzheimer’s disease therapeutics: Acetylcholinesterase inhibitors related to galanthamine. J. Pharm. Exper. 1996, 277, 728–738. [Google Scholar]
- Triggle, D.J.; Mitchell, J.M.; Filler, R. The pharmacology of physostigmine. Cns Drug Rev. 2006, 4, 87–136. [Google Scholar] [CrossRef]
- Jia, P.; Sheng, R.; Zhang, J.; Fang, L.; He, Q.; Yang, B.; Hu, Y. Design, synthesis and evaluation of galanthamine derivatives as acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2009, 44, 772–784. [Google Scholar] [CrossRef]
- Weed, M.R.; Polino, J.; Signor, L.; Bookbinder, M.; Keavy, D.; Benitex, Y.; Morgan, D.G.; King, D.; Macor, J.E.; Zaczek, R.; et al. Nicotinic alpha 7 receptor agonists EVP-6124 and BMS-933043, attenuate scopolamine-induced deficits in visuo-spatial paired associates learning. PLoS ONE 2017, 12, e0187609. [Google Scholar] [CrossRef]
- Dominguez, J.L.; Fernandez-Nieto, F.; Castro, M.; Catto, M.; Paleo, M.R.; Porto, S.; Sardina, F.J.; Brea, J.M.; Carotti, A.; Villaverde, M.C.; et al. Computer-aided structure-based design of multitarget leads for Alzheimer’s disease. J. Chem. Inf. Model. 2015, 55, 135–148. [Google Scholar] [CrossRef]
- Lepailleur, A.; Freret, T.; Lemaitre, S.; Boulouard, M.; Dauphin, F.; Hinschberger, A.; Dulin, F.; Lesnard, A.; Bureau, R.; Rault, S. Dual histamine H3R/serotonin 5-HT4R ligands with antiamnesic properties: Pharmacophore-based virtual screening and polypharmacology. J. Chem. Inf. Model. 2014, 54, 1773–1784. [Google Scholar] [CrossRef]
- Hameg, A.; Bayle, F.; Nuss, P.; Dupuis, P.; Garay, R.P.; Dib, M. Affinity of cyamemazine, an anxiolytic antipsychotic drug, for human recombinant dopamine vs. serotonin receptor subtypes. Biochem. Pharm. 2003, 65, 435–440. [Google Scholar] [CrossRef]
- Martin, L.F.; Freedman, R. Schizophrenia and the alpha7 nicotinic acetylcholine receptor. Int. Rev. Neurobiol. 2007, 78, 225–246. [Google Scholar]
- Young, J.W.; Geyer, M.A. Evaluating the role of the alpha-7 nicotinic acetylcholine receptor in the pathophysiology and treatment of schizophrenia. Biochem. Pharm. 2013, 86, 1122–1132. [Google Scholar] [CrossRef] [Green Version]
- Durrieu, G.; Olivier, P.; Bagheri, H.; Montastruc, J.L. Overview of adverse reactions to nefopam: An analysis of the French Pharmacovigilance database. Fundam. Clin. Pharmacol. 2007, 21, 555–558. [Google Scholar] [CrossRef]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Levy, L.; McClure, D. The pharmacology of flazalone: A new class of anti-inflammatory agent. J. Pharm. Exper. 1976, 198, 473–480. [Google Scholar]
- Dellisanti, C.D.; Yao, Y.; Stroud, J.C.; Wang, Z.Z.; Chen, L. Crystal structure of the extracellular domain of nAChR alpha1 bound to alpha-bungarotoxin at 1.94 A resolution. Nat. Neurosci. 2007, 10, 953–962. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. Publ. Protein Soc. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-Scale Systematic Analysis of 2D Fingerprint Methods and Parameters to Improve Virtual Screening Enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [Google Scholar] [CrossRef]
- Mirza, N.R.; Larsen, J.S.; Mathiasen, C.; Jacobsen, T.A.; Munro, G.; Erichsen, H.K.; Nielsen, A.N.; Troelsen, K.B.; Nielsen, E.O.; Ahring, P.K. NS11394 [3′-[5-(1-hydroxy-1-methyl-ethyl)-benzoimidazol-1-yl]-biphenyl-2-carbonitrile], a unique subtype-selective GABAA receptor positive allosteric modulator: In vitro actions, pharmacokinetic properties and in vivo anxiolytic efficacy. J. Pharm. Exper. 2008, 327, 954–968. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Structure | AChE G-Score a % Inhibition b IC50 (µM) (pIC50 ± SEM) | α7 nAChR G-Score a % Inhibition b IC50 (µM) (pIC50 ± SEM) | No | Structure | AChE G-Score a % Inhibition b IC50 (µM) (pIC50 ± SEM) | α7 nAChR G-Score a % Inhibition b IC50 (µM) (pIC50 ± SEM) |
|---|---|---|---|---|---|---|---|
| 5 |  Physostigmine | - 91.5 ± 0.3% c IC50 = 0.78 (6.13 ± 0.02) | - - - - | 12 |  | −11.28 38.3 ± 4.2% - - | −14.33 95.2 ± 2.0% IC50 = 8.3 (5.08 ± 0.04) |
| 1 |  | −11.03 91.5 ± 0.1% d IC50 = 0.68 (6.14 ± 0.01) | −15.10 67.3 ± 0.5% IC50 = 54.8 (4.26 ± 0.04) | 13 |  Nefopam | −10.30 0% - - | −13.39 76.3 ± 0.5% e IC50 = 13.1 (4.88 ± 0.02) |
| 6 |  | −11.51 96.4 ± 0.3% IC50 = 0.28 (6.55 ± 0.02) | −15.10 45.4 ± 2.3% - - | 14 |  | −12.07 6.5 ± 1.7% - - | −13.87 87.4 ± 1.4% - - |
| 7 |  | −10.28 96.5 ± 0.1% IC50 = 0.23 (6.13 ± 0.02) | −13.05 64.8 ± 0.7% - - | 15 |  | −11.88 30.3 ± 4.7% - - | −13.03 75.8 ± 0.4% - - |
| 8 |  | −13.99 78.5 ± 0.6% IC50 = 10.6 (4.96 ± 0.03) | −13.03 81.8 ± 1.3% IC50 = 34.3 (4.46 ± 0.05) | 16 |  | −10.64 56.7 ± 4.2% - - | −13.11 79.0 ± 1.6% - - |
| 9 |  | −10.71 88.1 ± 0.8% IC50 = 5.04 (5.29 ± 0.03) | −13.11 93.2 ± 0.3% IC50 = 14.5 (4.84 ± 0.04) | 17 |  | −12.92 23.9 ± 5.7% - - | −14.94 84.8 ± 0.7% - - |
| 10 |  Cyamemazine | −10.35 32.7 ± 4.2% - - | −13.27 97.2 ± 0.5% IC50 = 3.9 (5.44 ± 0.05) | 18 |  | −11.23 18.0 ± 8.9% - - | −15.05 2.5 ± 2.5% - - |
| 11 |  Flazalone | −11.29 14.0 ± 9.3% - - | −14.60 98.0 ± 0.1% IC50 = 7.2 (5.14 ± 0.05) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowal, N.M.; Indurthi, D.C.; Ahring, P.K.; Chebib, M.; Olafsdottir, E.S.; Balle, T. Novel Approach for the Search for Chemical Scaffolds with Activity at Both Acetylcholinesterase and the α7 Nicotinic Acetylcholine Receptor: A Perspective on Scaffolds with Dual Activity for the Treatment of Neurodegenerative Disorders. Molecules 2019, 24, 446. https://doi.org/10.3390/molecules24030446
Kowal NM, Indurthi DC, Ahring PK, Chebib M, Olafsdottir ES, Balle T. Novel Approach for the Search for Chemical Scaffolds with Activity at Both Acetylcholinesterase and the α7 Nicotinic Acetylcholine Receptor: A Perspective on Scaffolds with Dual Activity for the Treatment of Neurodegenerative Disorders. Molecules. 2019; 24(3):446. https://doi.org/10.3390/molecules24030446
Chicago/Turabian StyleKowal, Natalia M., Dinesh C. Indurthi, Philip K. Ahring, Mary Chebib, Elin S. Olafsdottir, and Thomas Balle. 2019. "Novel Approach for the Search for Chemical Scaffolds with Activity at Both Acetylcholinesterase and the α7 Nicotinic Acetylcholine Receptor: A Perspective on Scaffolds with Dual Activity for the Treatment of Neurodegenerative Disorders" Molecules 24, no. 3: 446. https://doi.org/10.3390/molecules24030446
APA StyleKowal, N. M., Indurthi, D. C., Ahring, P. K., Chebib, M., Olafsdottir, E. S., & Balle, T. (2019). Novel Approach for the Search for Chemical Scaffolds with Activity at Both Acetylcholinesterase and the α7 Nicotinic Acetylcholine Receptor: A Perspective on Scaffolds with Dual Activity for the Treatment of Neurodegenerative Disorders. Molecules, 24(3), 446. https://doi.org/10.3390/molecules24030446