
Determination of Sulfonamides in Feeds by High-Performance Liquid Chromatography after Fluorescamine Precolumn Derivatization


Abstract
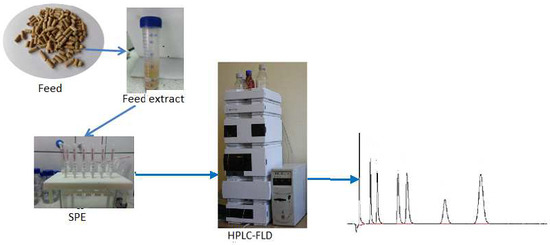
:
1. Introduction
2. Results and Discussion
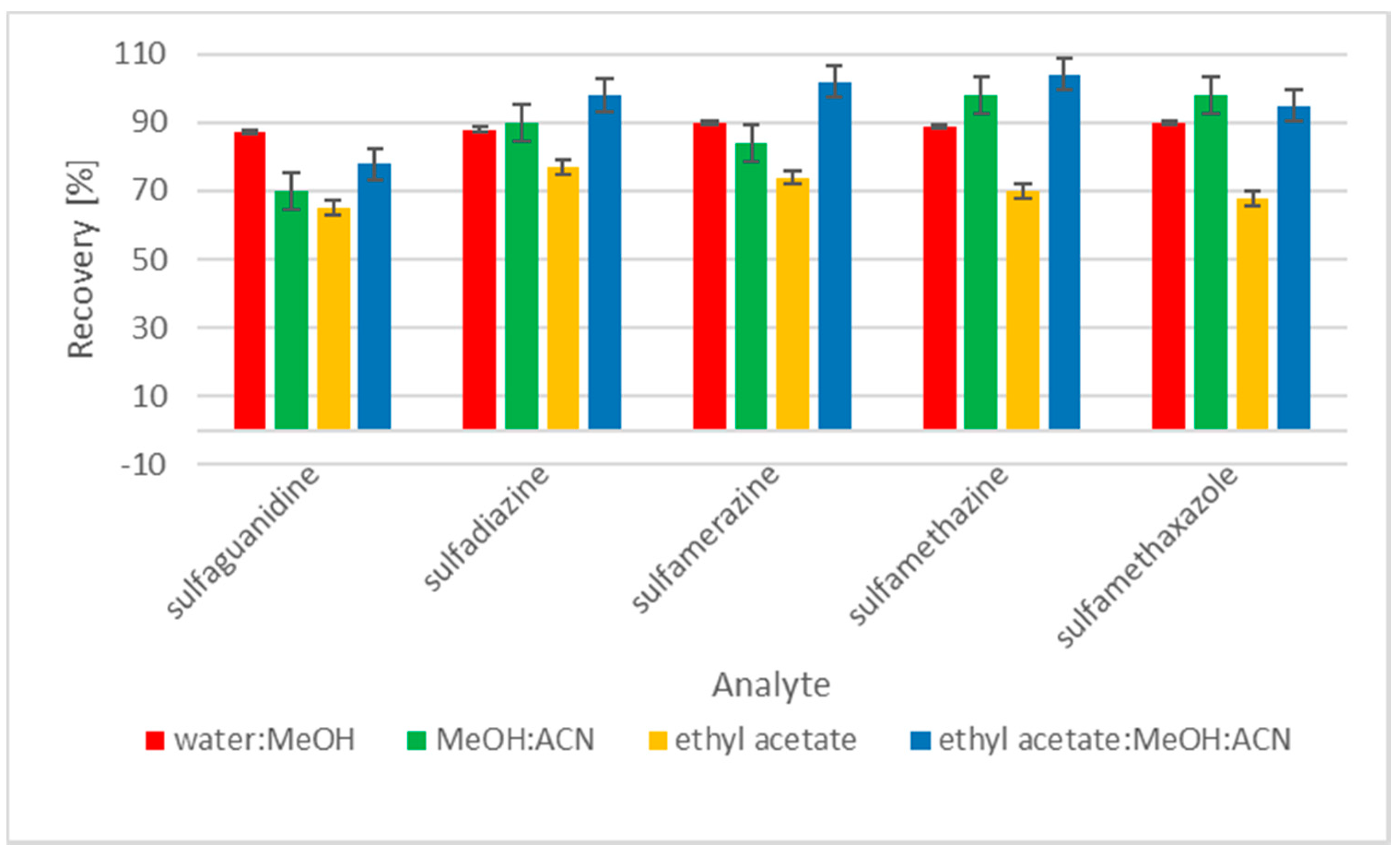
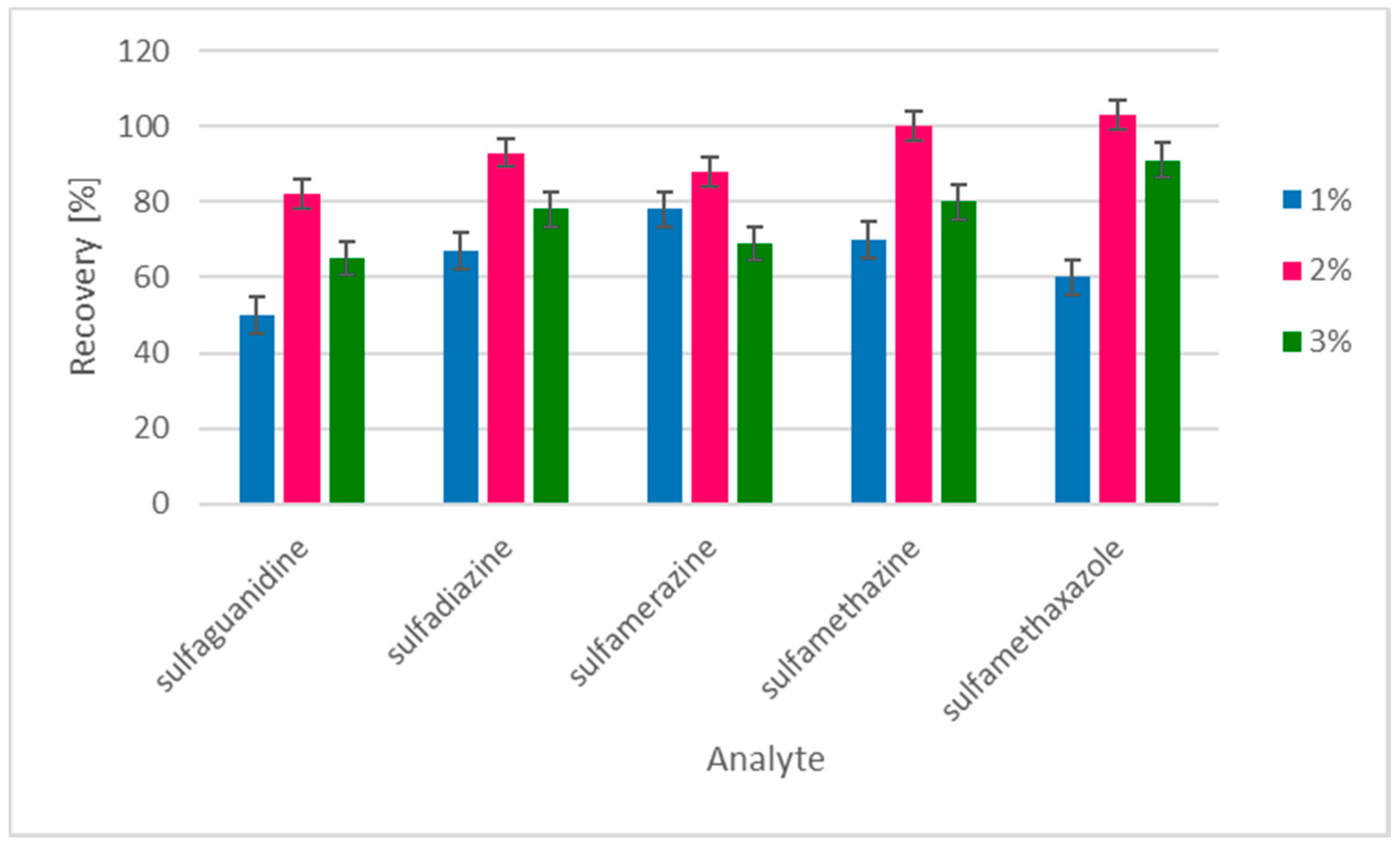
2.1. Sample Preparation
2.2. Chromatographic Conditions
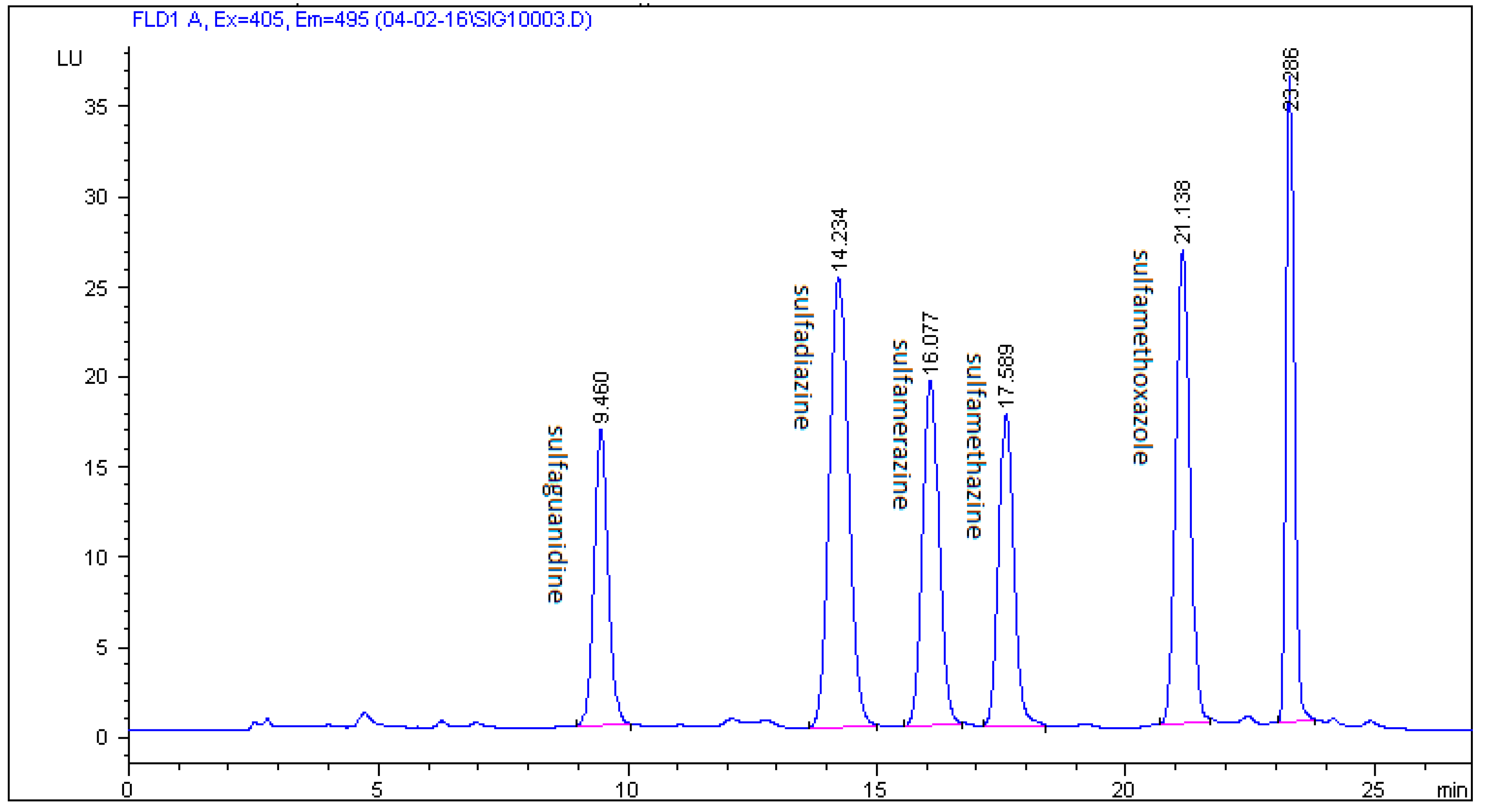
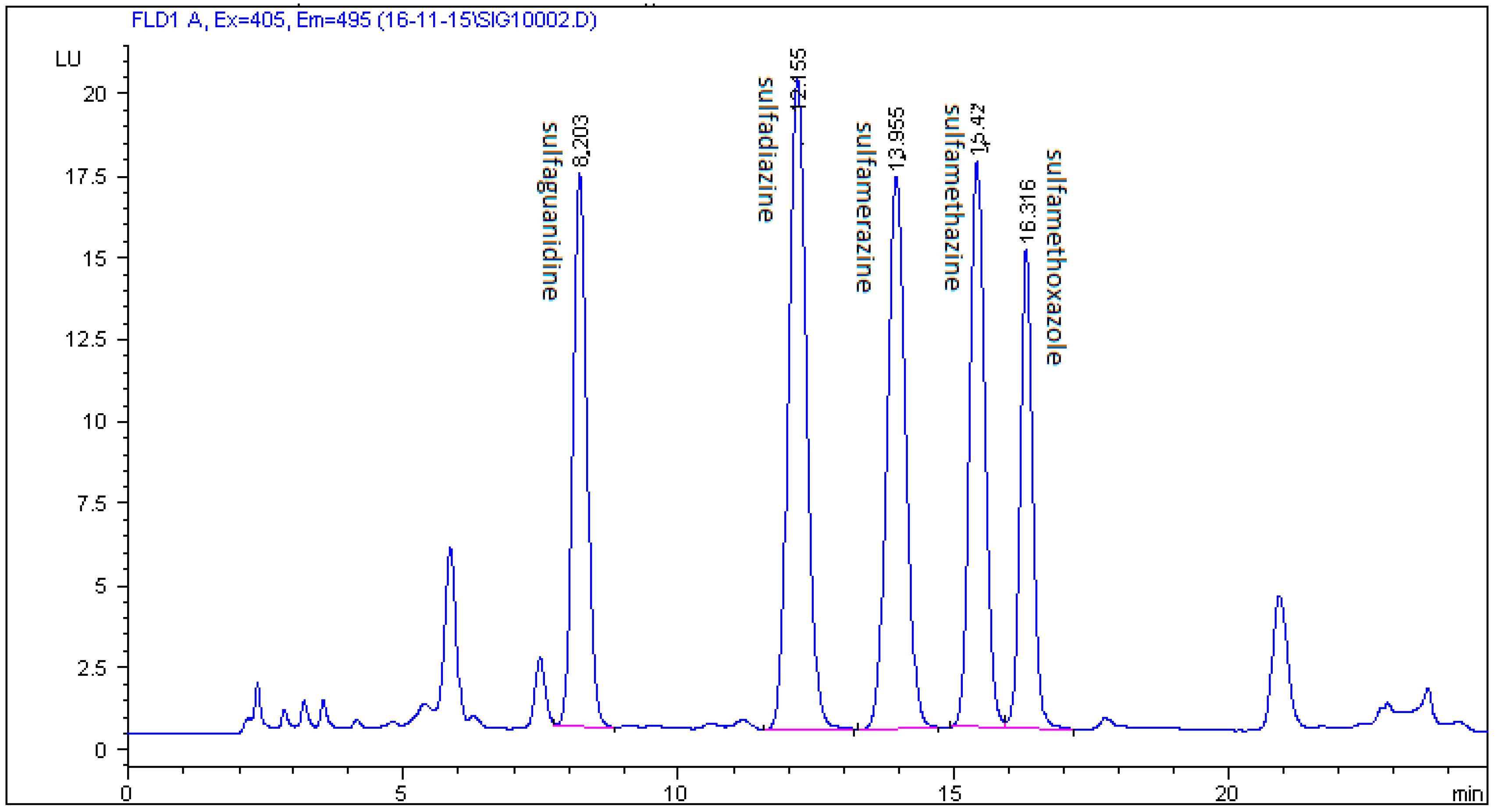
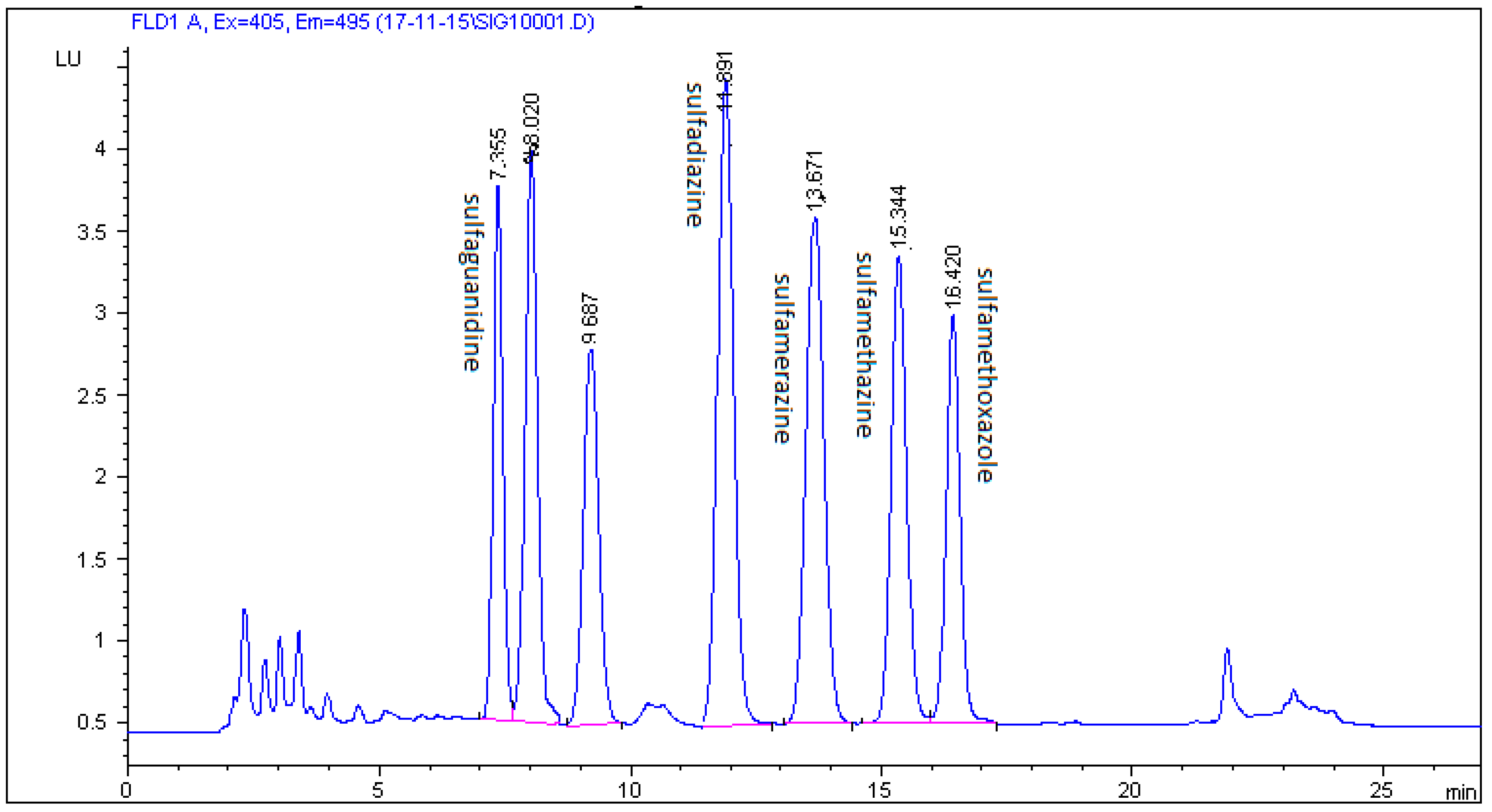
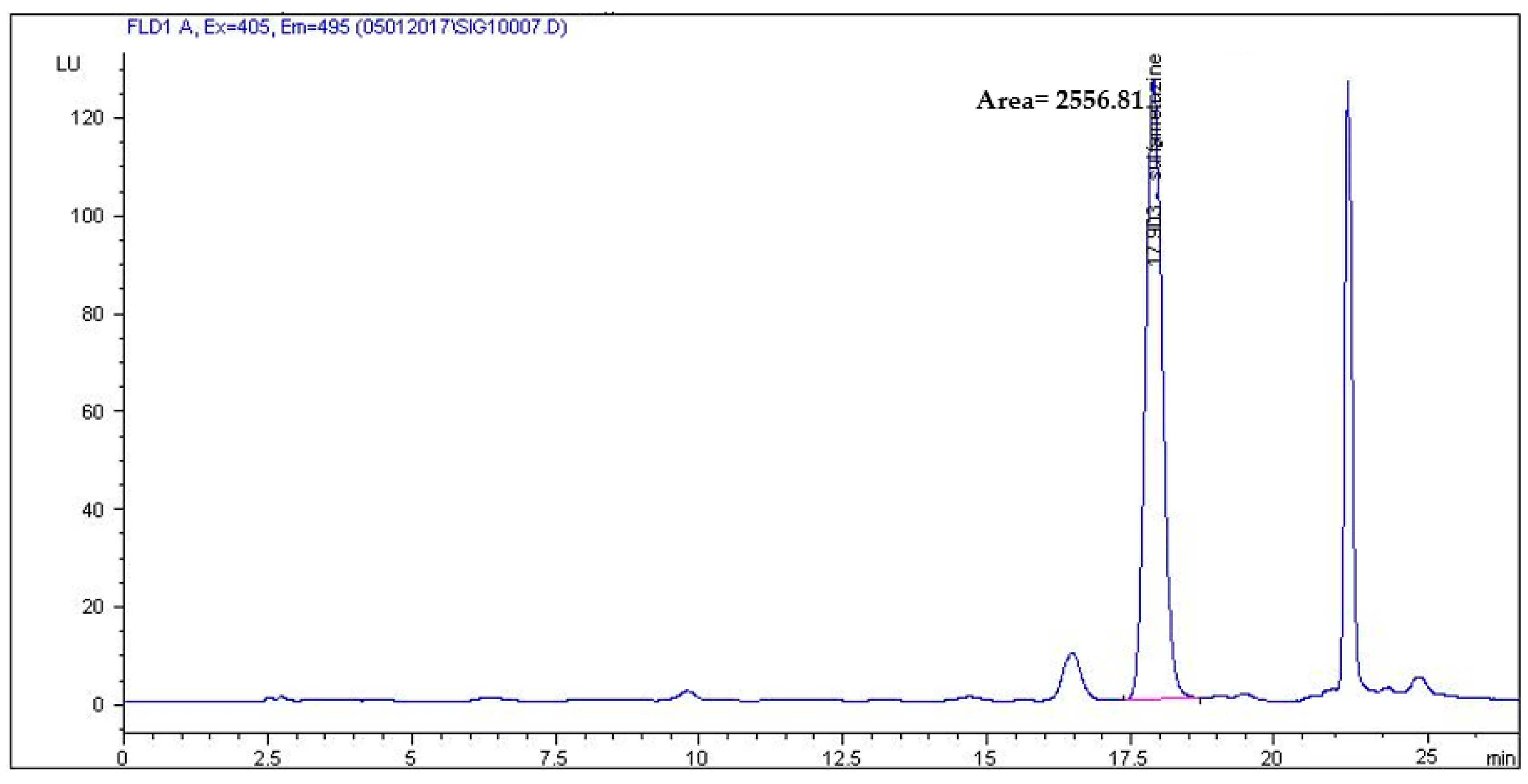
2.3. Method Validation
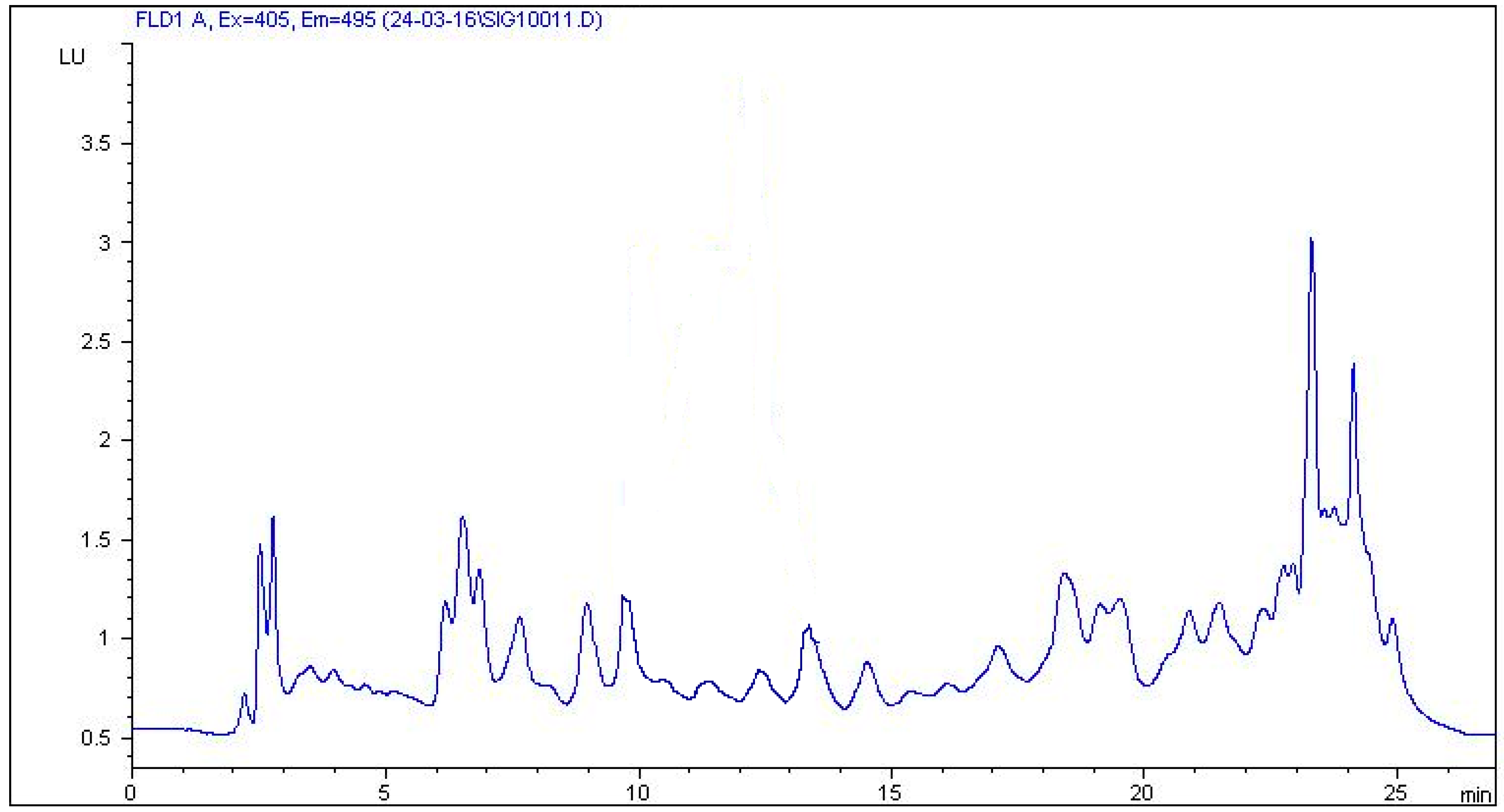
2.4. Real Sample Application
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Instrumentation
3.3. Chromatography
3.4. Standard Solutions
3.5. Sample Preparation
3.6. Clean-Up
3.7. Derivatization
3.8. Validation Procedure
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kim, H.J.; Jeong, M.H.; Park, H.J.; Kim, W.C.; Kim, J.E. Development of an immunoaffinity chromatography and HPLC-UV method for detection of 16 sulfonamides in feed. Food Chem. 2016, 196, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Hows, M.E.P.; Perrett, D.; Kay, J. Optimisation of a simultaneous separation of sulphonamides dihydrofolate reductase inhibitors and β-lactam antibiotics by capillary electrophoresis. J. Chromatogr. A 1997, 768, 97–104. [Google Scholar] [CrossRef]
- EC Directive 2002/32 (2002) Off J Eur Union L 140:10–21. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32002L0032 (accessed on 10 December 2018).
- EC Directive 2009/8 (2009) Off J Eur Union L 40:19–25. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2009:274:0025:0027:EN:PDF (accessed on 10 December 2018).
- EC Regulation No 183/2005 of the European Parlament and of the Council of 12 January 2005 laying down requirements of feed hygiene. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32005R0183&from=EN (accessed on 10 December 2018).
- Przeniosło-Siwczyńska, M.; Kwiatek, K. Evaluation of multi—Plate microbial assay for the screening of antibacterial substances in animal feedingstuffs. Bull. Vet. Inst. Pulawy. 2007, 57, 599–602. [Google Scholar]
- Reeves, V.B. Confirmation of multiple sulfonamide residues in bovine milk by gas chromatography-positive chemical ionization mass spectrometry. J. Chromatogr. B 1999, 723, 127–137. [Google Scholar] [CrossRef]
- Abdallah, H.; Arnaudguilhem, C.; Jaber, F.; Lobinski, R. Multiresidue analysis of 22 sulfonamides and their metabolites in animal tissues using quick, easy, cheap, effective, rugged, and safe extraction and high resolution mass spectrometry (hybrid linear ion trap-Orbitrap. J. Chromatogr. A 2014, 1355, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Bogialli, S.; Curini, R.; di Corcia, A.; Nazzari, M.; Polci, M.L. Rapid confirmatory assay for detrmining 12 sulfonamide antimicrobials in milka and eggs by matrix solid-phase dispersion and liquid chromatography-mass spectrometry. J. Agr. Food Chem. 2003, 51, 4225–4232. [Google Scholar] [CrossRef] [PubMed]
- Galarini, R.; Diana, F.; Moretti, S.; Puppini, B.; Saluti, G.; Persic, L. Development and validation of a new qualitative ELISA screening for multiresidue detection of sulfonamides in food and feed. Food Control. 2014, 35, 300–310. [Google Scholar] [CrossRef]
- Posyniak, A.; Jażdżewski, K.; Pietruszka, K.; Mitrowska, K.; Gajda, A. Improved analytical procedure for the determination of sulfonamides in honey. Bull. Vet. Inst. Pulawy. 2008, 52, 87–91. [Google Scholar]
- Croubels, S.; Wassink, P.; De Backer, P. Simultaneous determination of sulfadiazine and trimethoprim in animal feed by liquid chromatography with UV and tandem mass spectrometric detection. Anal. Chim. Acta 2002, 473, 183–194. [Google Scholar] [CrossRef]
- Hela, W.; Brandtner, M.; Widek, R.; Schuh, R. Determiantion of sulfonamides in animal tissues using cation exchange reserved phase sorbent for sample cleanup and HPLC-DAD for detection. Food Chem. 2003, 83, 601–608. [Google Scholar] [CrossRef]
- Iammarino, M.; Palermo, C.; Nardiello, D.; Muscarella, M. Optimization and validation of a confirmatory method for determination of ten sulfonamides in feeds by LC and UV-diode array detection. Chromatographia 2011, 73, 75–82. [Google Scholar] [CrossRef]
- Pietruszka, K.; Posyniak, A. Simple method to determine of sulfonamides in muscles by high performance liquid chromatography. Bull. Vet. Inst. Pulawy. 2010, 54, 393–396. [Google Scholar]
- Blasco, C.; Corcia, A.D.; Picó, Y. Determination of tetracyclines in multi-specie animal tissues by pressurized liquid extraction and liquid chromatography–tandem mass spectrometry. Food Chem. 2009, 116, 1005–1012. [Google Scholar] [CrossRef]
- Chafer-Pericas, C.; Maquieira, A.; Puchades, R.; Miralles, J.; Moreno, A. Multiresidue determination of antibiotics in feed and fish samples for food safety evaluation. Comparison of immunoassay vs LC-MS/MS. Food Control. 2011, 22, 993–999. [Google Scholar] [CrossRef]
- Gavilán, R.E.; Nebot, C.; Patyra, E.; Miranda, J.M.; Franco, C.M.; Cepeda, A. Simultaneous analysis of coccidiostats and sulphonamides in non-target feed by HPLC-MS/MS and validation following the Commission Decision 2002/657/EC. Food Additiv. Contam. 2018, 35, 1093–1106. [Google Scholar]
- Kaklamanos, G.; Vincent, U.; von Holst, C. Analysis of antimicrobial agents in pig feed by liquid chromatography coupled to orbitrap mass spectrometry. J. Chromatogr. B 2013, 1293, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; He, P.; Li, Z.; Li, R. Simultaneous determination of 16 sulfonamides in animal feeds by UHPLC-MS-MS. J. Chromatogr. Sci. 2011, 49, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Patyra, E.; Nebot, C.; Gavilán, R.E.; Cepeda, A.; Kwiatek, K. Development and validation of multi-residue and multi-class method for antibacterial substances analysis in non-target feed by liquid chromatography—Tandem mass spectrometry. Food Additiv. Contam. 2018, 35, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Pereira Lopes, R.; De Freitas Passos, E.E.; de Alkimim Filho, J.F.; Vargas Azevedo, E.; Vasconcellos Augusti, D.; Augusti, R. Development and validation of a method for the determination of sulfonamides in animal feed by modified QuEChERS and LC-MS/MS analysis. Food Control. 2012, 28, 192–198. [Google Scholar] [CrossRef]
- Anon: Commission Decision 2002/657/EC. 2002. of 12August implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. OJ. L 221:8–36. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2002:221:0008:0036:EN:PDF (accessed on 10 December 2018).
- SANTE/11945/2015. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed. Available online: https://www.accredia.it/en/documento/guidance-sante-119452015-guidance-document-on-analytical-quality-control-and-method-validation-procedures-for-pesticides-residues-analysis-in-food-and-feed/ (accessed on 10 December 2018).
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Recovery [%] | Repeatability [%] | Whitin-Laboratory Reproducibility [%] | CCα [µg/kg] | CCβ [µg/kg] | LOD [µg/kg] | LOQ [µg/kg] | U [%] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Concentration Levels [µg/kg] | Concentration Levels [µg/kg] | Concentration Levels [µg/kg] | ||||||||||||
| 200 | 1000 | 2000 | 200 | 1000 | 2000 | 200 | 1000 | 2000 | ||||||
| Sulfaguanidine | 79.3 | 102.1 | 89.9 | 7.8 | 8.7 | 3.0 | 11.8 | 11.5 | 11.1 | 265.2 | 315.2 | 34.5 | 41.3 | 24.4 |
| Sulfadiazine | 97.4 | 114.0 | 90.4 | 3.8 | 7.7 | 2.7 | 11.8 | 7.4 | 9.5 | 197.7 | 239.2 | 52.4 | 58.9 | 19.8 |
| Sulfamerazine | 103.4 | 103.0 | 93.5 | 5.7 | 8.0 | 5.9 | 6.1 | 10.0 | 9.7 | 224.5 | 263.2 | 63.5 | 68.4 | 21.2 |
| Sulfametazine | 103.9 | 103.5 | 91.7 | 4.6 | 6.5 | 6.1 | 5.9 | 14.9 | 10.9 | 266.7 | 326.9 | 79.5 | 89.9 | 20.3 |
| sulfamethoxazole | 94.8 | 102.2 | 100.9 | 5.9 | 7.9 | 9.1 | 8.3 | 14.2 | 10.1 | 274.6 | 337.9 | 39.7 | 43.1 | 23.3 |
| Time (min) | 0.08% Acetic Acid in Milli-Q Water (A) (%) | Acetonitrile (B) (%) | Methanol (C) (%) |
|---|---|---|---|
| 0–10 | 48 | 10 | 42 |
| 10–15 | 41 | 10 | 49 |
| 15–17 | 41 | 10 | 49 |
| 17–20 | 18 | 40 | 42 |
| 20–22 | 48 | 10 | 42 |
| 22–27 | 48 | 10 | 42 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patyra, E.; Przeniosło-Siwczyńska, M.; Kwiatek, K. Determination of Sulfonamides in Feeds by High-Performance Liquid Chromatography after Fluorescamine Precolumn Derivatization. Molecules 2019, 24, 452. https://doi.org/10.3390/molecules24030452
Patyra E, Przeniosło-Siwczyńska M, Kwiatek K. Determination of Sulfonamides in Feeds by High-Performance Liquid Chromatography after Fluorescamine Precolumn Derivatization. Molecules. 2019; 24(3):452. https://doi.org/10.3390/molecules24030452
Chicago/Turabian StylePatyra, Ewelina, Monika Przeniosło-Siwczyńska, and Krzysztof Kwiatek. 2019. "Determination of Sulfonamides in Feeds by High-Performance Liquid Chromatography after Fluorescamine Precolumn Derivatization" Molecules 24, no. 3: 452. https://doi.org/10.3390/molecules24030452
APA StylePatyra, E., Przeniosło-Siwczyńska, M., & Kwiatek, K. (2019). Determination of Sulfonamides in Feeds by High-Performance Liquid Chromatography after Fluorescamine Precolumn Derivatization. Molecules, 24(3), 452. https://doi.org/10.3390/molecules24030452