
Discovery and Computational Analyses of Novel Small Molecule Zika Virus Inhibitors

Abstract
:1. Introduction
2. Results
2.1. Structure-Based Virtual Screening of a Diverse Library
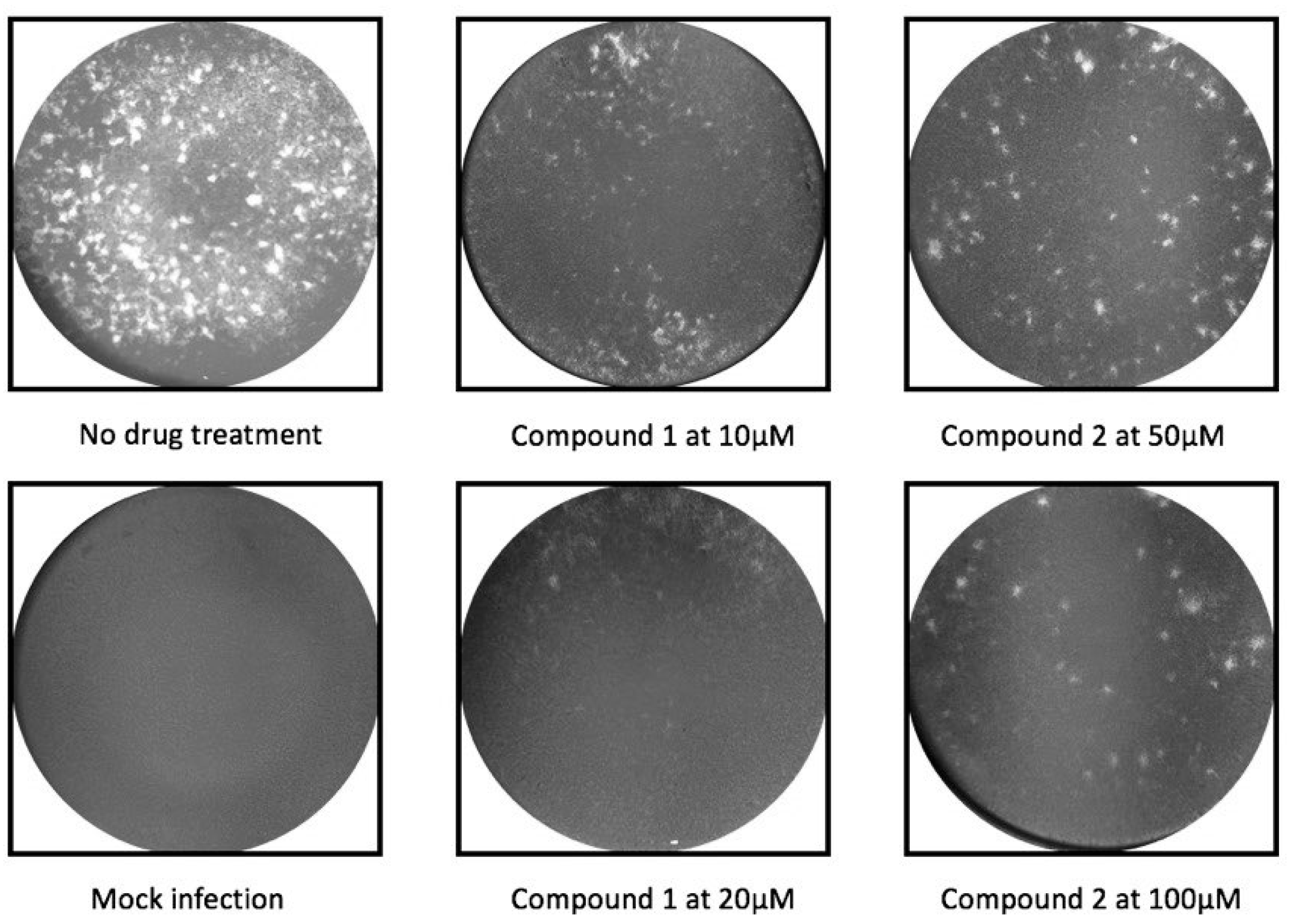
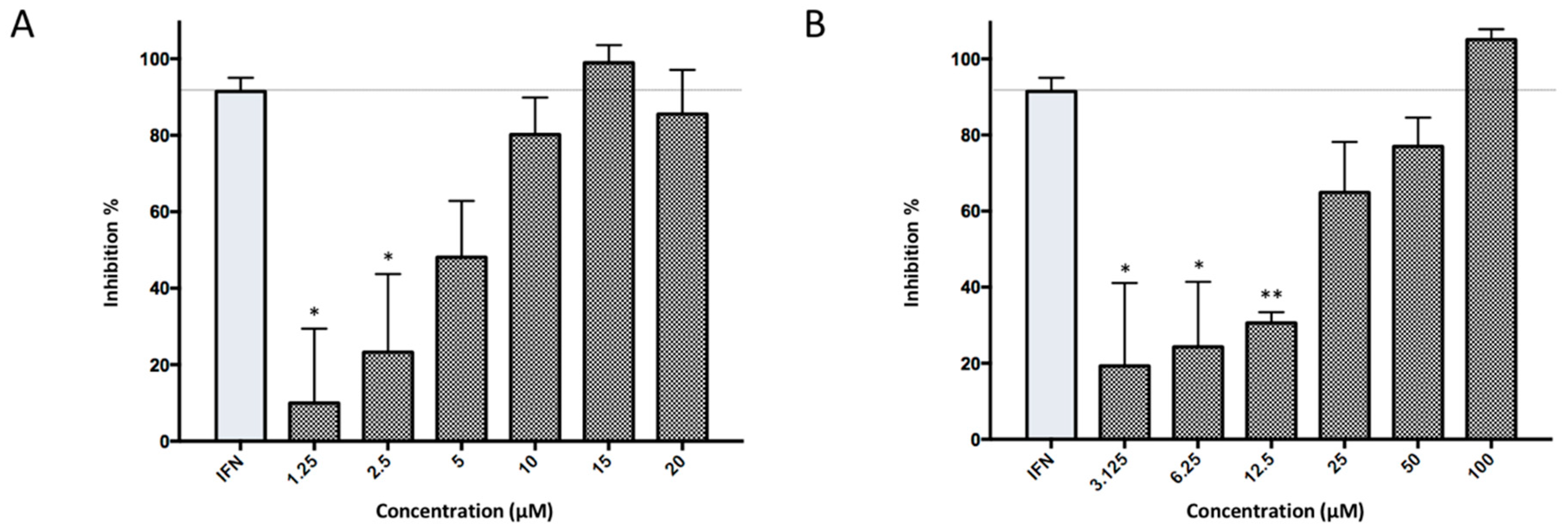
2.2. Antiviral Activity Against in Vitro ZIKV Infection
2.3. Potential Inhibition on Viral Replication Process
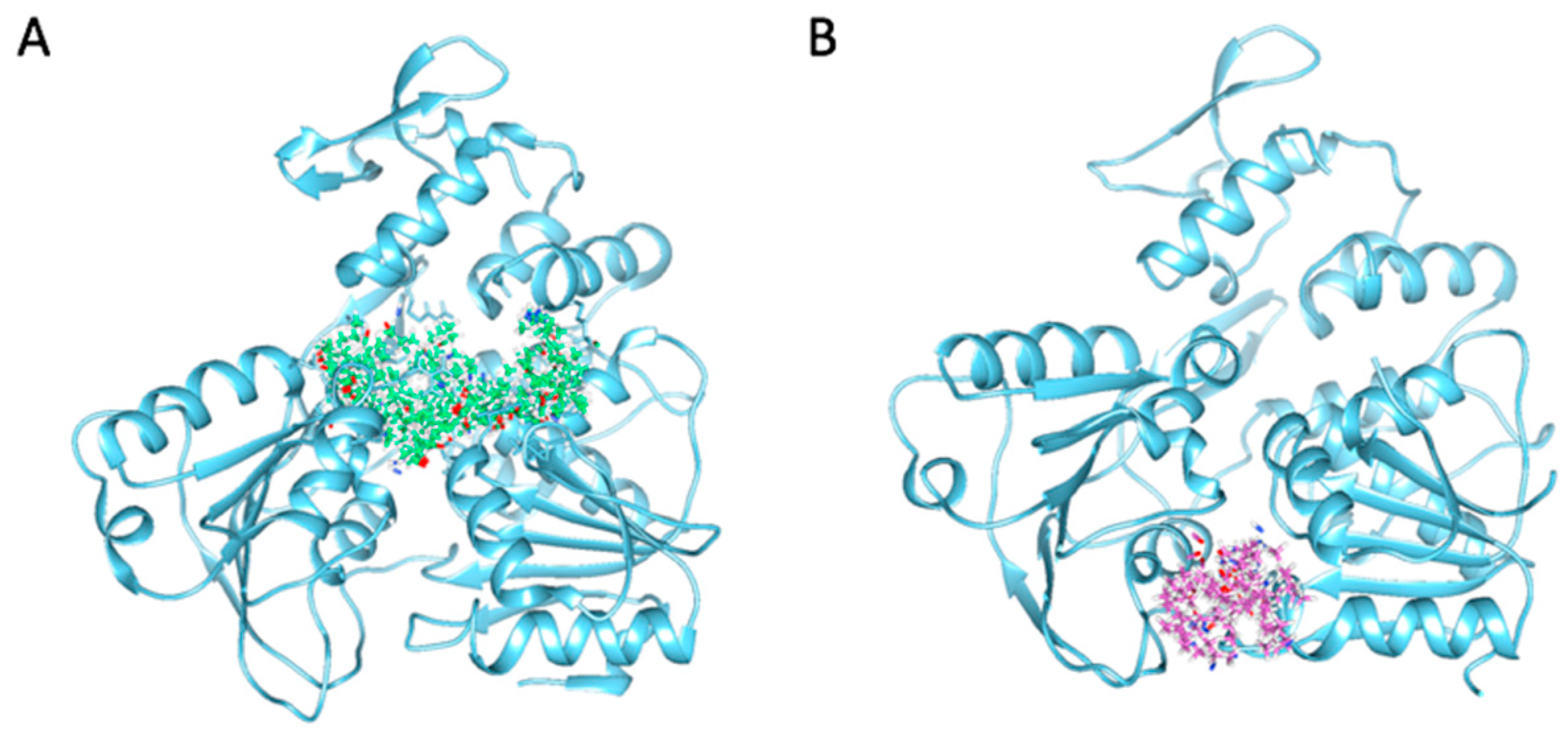
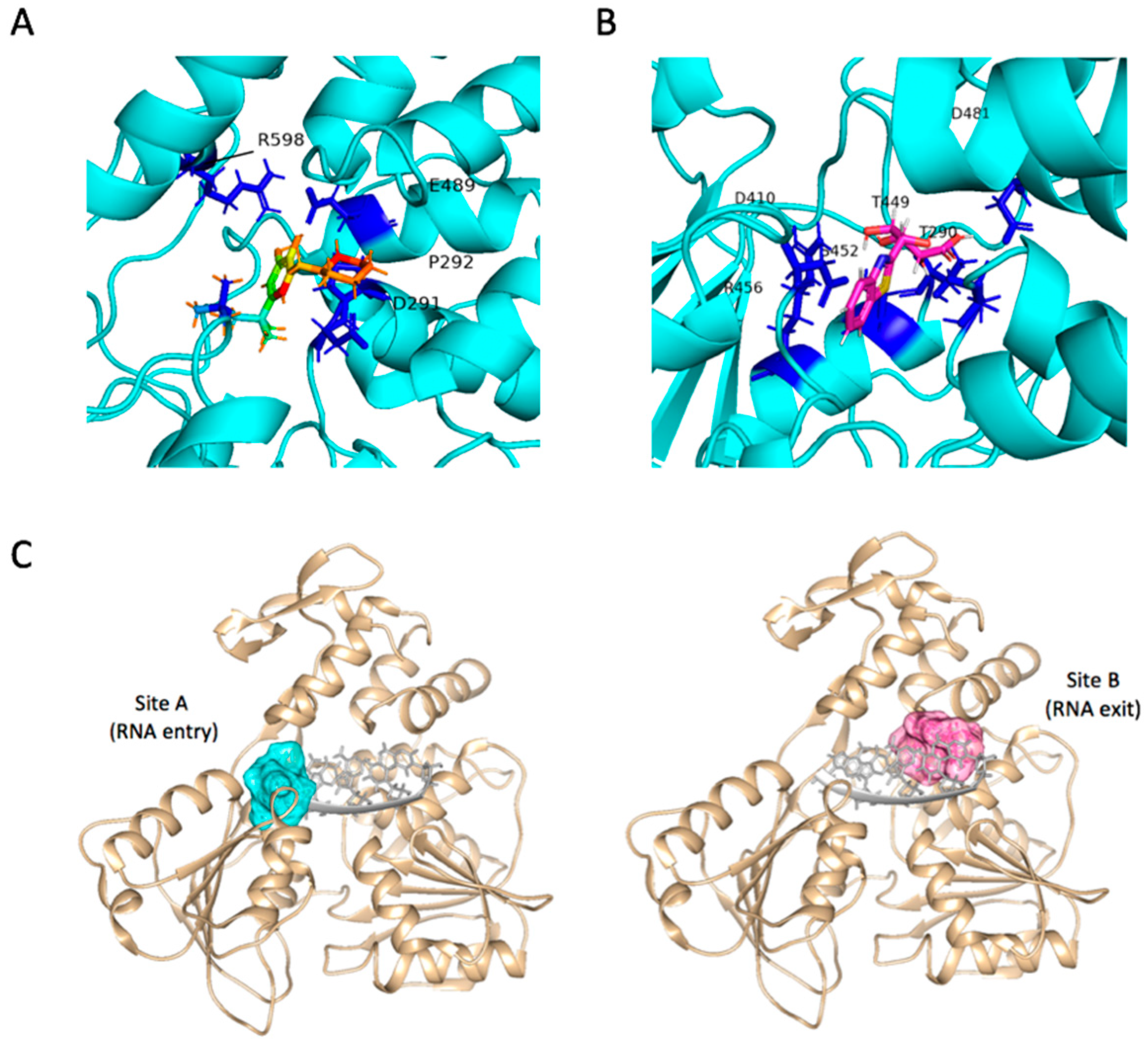
2.4. Binding Modes and Interactions with NS3 in Silico
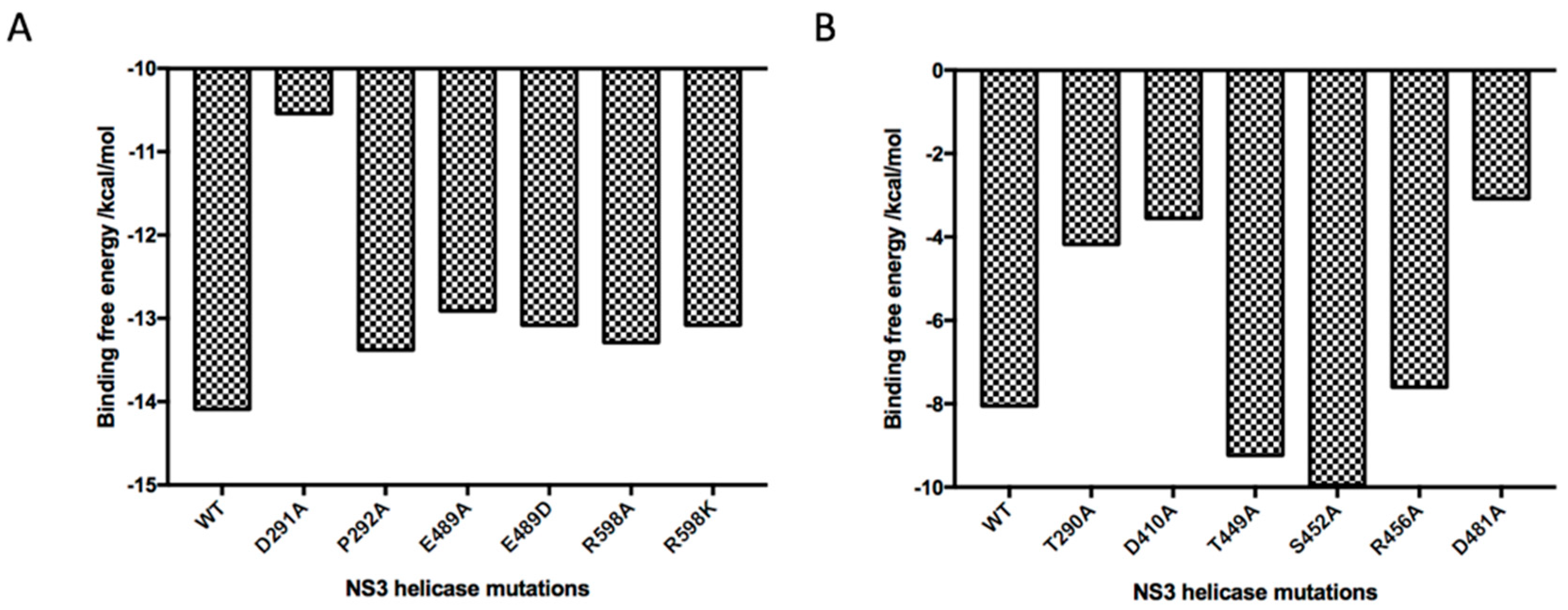
2.5. Absolute Binding Free Energy Calculation
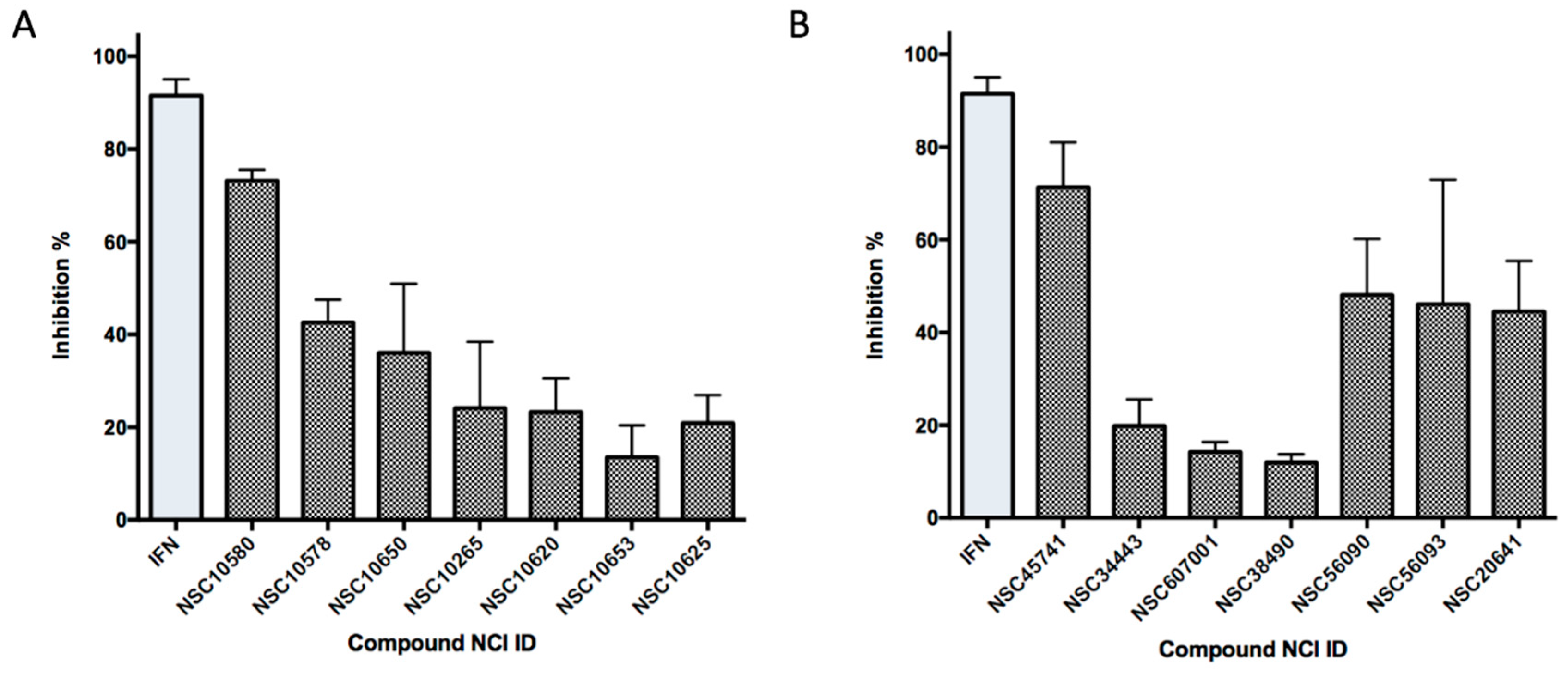
2.6. Ligand-Based Search and Structure-Activity Relationship Analysis of Derivatives
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Hardware and Software
5.2. Protein Refinement and Compound Database Preparation
5.3. Virtual Screening using Surflex-Dock
5.4. In silico Molecular Interaction Analysis
5.5. Molecular Dynamics Simulation
5.6. Ab Initio Binding Free Energy Calculation
5.7. Cell Culture and Compounds Preparation
5.8. Virus Preparation and in Vitro Infection Assay
5.9. Virus Plaque Reduction Assay
5.10. Cytopathic Effect Inhibition Assay
5.11. Time-of-Drug-Addition Assay
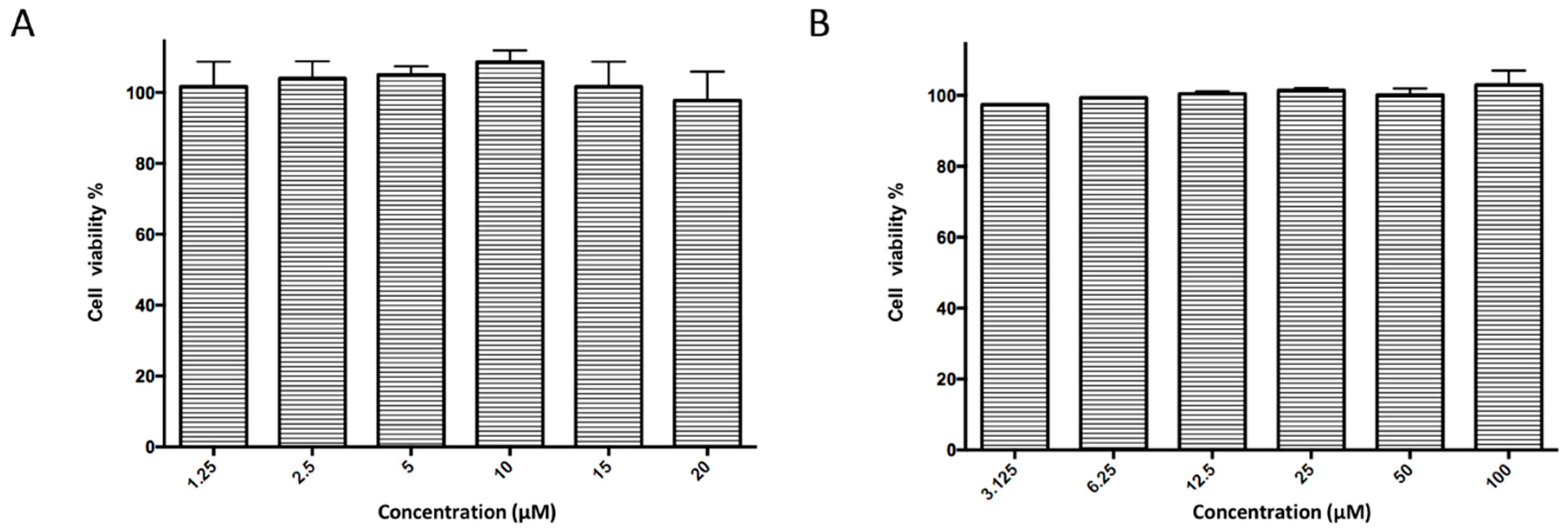
5.12. Cytotoxicity Assay
5.13. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ZIKV | Zika virus; |
| HCV | Hepatitis C Virus; |
| DENV | Dengue virus; |
| WNV | West Nile virus; |
| JEV | Japanese Encephalitis Virus; |
| DAA | direct-acting antiviral agents; |
| NSP | non-structural protein; |
| NS3 | non-structural protein 3; |
| NS3PRO | non-structural protein 3 protease; |
| NS3HEL | non-structural protein 3 helicase; |
| ssRNA | single-stranded RNA; |
| NTP | nucleoside triphosphate; |
| PDB | Protein Data Bank; |
| NCI | National Cancer Institute; |
| SBDD | Structure-based drug discovery; |
| SAR | Structure-activity relationship; |
| SFXC | Surflex-Dock algorithm; |
References
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Petersen, L.R.; Jamieson, D.J.; Powers, A.M.; Honein, M.A. Zika virus. N. Engl. J. Med. 2016, 374, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, M.; Fischer, M.; Staples, J.E. Zika Virus Spreads to New Areas—Region of the Americas, May 2015–January 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 55–58. [Google Scholar]
- Teixeira, M.G.; de Costa, M.N.C.; de Oliveira, W.K.; Nunes, M.L.; Rodrigues, L.C. The Epidemic of Zika Virus-Related Microcephaly in Brazil: Detection, Control, Etiology, and Future Scenarios. Am. J. Public Health 2016, 106, 601–605. [Google Scholar] [CrossRef]
- Ventura, C.V.; Maia, M.; Bravo-Filho, V.; Gois, A.L.; Belfort, R., Jr. Zika virus in brazil and macular atrophy in a child with microcephaly. Lancet 2016, 387, 228. [Google Scholar] [CrossRef]
- Singh, R.K.; Dhama, K.; Malik, Y.S.; Ramakrishnan, M.A.; Karthik, K.; Tiwari, R.; Sachan, S.; Joshi, S.K. Zika virus—Emergence, evolution, pathology, diagnosis, and control: Current global scenario and future perspectives—A comprehensive review. Vet. Q. 2016, 36, 150–175. [Google Scholar] [CrossRef]
- Lazear, H.M.; Diamond, M.S. Zika virus: New clinical syndromes and its emergence in the western hemisphere. J. Virol. 2016, 90, 4864–4875. [Google Scholar] [CrossRef] [PubMed]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastere, S.; Valour, F.; Baudouin, L.; Mallet, H.; Musso, D.; Ghawche, F. Zika virus infection complicated by guillain-barre syndrome--case report, french polynesia, december 2013. Euro Surveill. 2014, 19, 20720. [Google Scholar] [CrossRef]
- Oliveira Melo, A.S.; Malinger, G.; Ximenes, R.; Szejnfeld, P.O.; Alves Sampaio, S.; Bispo de Filippis, A.M. Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: Tip of the iceberg? Ultrasound Obstet. Gynecol. 2016, 47, 6–7. [Google Scholar] [CrossRef]
- Chibueze, E.C.; Tirado, V.; Lopes, K.D.; Balogun, O.O.; Takemoto, Y.; Swa, T.; Dagvadorj, A.; Nagata, C.; Morisaki, N.; Menendez, C.; et al. Zika virus infection in pregnancy: A systematic review of disease course and complications. Reprod. Health 2017, 14, 28. [Google Scholar] [CrossRef]
- Song, H.; Qi, J.; Haywood, J.; Shi, Y.; Gao, G.F. Zika virus ns1 structure reveals diversity of electrostatic surfaces among flaviviruses. Nat. Struct. Mol. Biol. 2016, 23, 456–458. [Google Scholar] [CrossRef]
- Brown, W.C.; Akey, D.L.; Konwerski, J.R.; Tarrasch, J.T.; Skiniotis, G.; Kuhn, R.J.; Smith, J.L. Extended surface for membrane association in zika virus ns1 structure. Nat. Struct. Mol. Biol. 2016, 23, 865–867. [Google Scholar] [CrossRef]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R. Crystal structure of zika virus ns2b-ns3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Loh, Y.R.; Phoo, W.W.; Hung, A.W.; Kang, C.; Luo, D. Crystal structure of unlinked ns2b-ns3 protease from zika virus. Science 2016, 354, 1597–1600. [Google Scholar] [CrossRef]
- Phoo, W.W.; Li, Y.; Zhang, Z.; Lee, M.Y.; Loh, Y.R.; Tan, Y.B.; Ng, E.Y.; Lescar, J.; Kang, C.; Luo, D. Structure of the NS2b-NS3 protease from zika virus after self-cleavage. Nat. Commun. 2016, 7, 13410. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Coloma, J.; Garcia-Sastre, A.; Aggarwal, A.K. Structure of the ns3 helicase from zika virus. Nat. Struct. Mol. Biol. 2016, 23, 752–754. [Google Scholar] [CrossRef]
- Coloma, J.; Jain, R.; Rajashankar, K.R.; Garcia-Sastre, A.; Aggarwal, A.K. Structures of ns5 methyltransferase from zika virus. Cell Rep. 2016, 16, 3097–3102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Feng, T.; Cheng, J.; Li, Y.; Yin, X.; Zeng, W.; Jin, X.; Li, Y.; Guo, F.; Jin, T. Structure of the ns5 methyltransferase from zika virus and implications in inhibitor design. Biochem. Biophys. Res. Commun. 2017, 492, 624–630. [Google Scholar] [CrossRef]
- Coutard, B.; Barral, K.; Lichiere, J.; Selisko, B.; Martin, B.; Aouadi, W.; Lombardia, M.O.; Debart, F.; Vasseur, J.J.; Guillemot, J.C.; et al. Zika virus methyltransferase: Structure and functions for drug design perspectives. J. Virol. 2017, 91, e02202-16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Yi, G.; Du, F.; Chuang, Y.C.; Vaughan, R.C.; Sankaran, B.; Kao, C.C.; Li, P. Structure and function of the zika virus full-length ns5 protein. Nat. Commun. 2017, 8, 14762. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.L.; Jones, C.T.; Rice, C.M. Architects of assembly: Roles of flaviviridae non-structural proteins in virion morphogenesis. Nat. Rev. Microbiol. 2008, 6, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Z.; Phoo, W.W.; Loh, Y.R.; Wang, W.; Liu, S.; Chen, M.W.; Hung, A.W.; Keller, T.H.; Luo, D.; et al. Structural dynamics of zika virus ns2b-ns3 protease binding to dipeptide inhibitors. Structure 2017, 25, 1242–1250. [Google Scholar] [CrossRef]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef]
- Garcia, L.L.; Padilla, L.; Castano, J.C. Inhibitors compounds of the flavivirus replication process. Virol. J. 2017, 14, 95. [Google Scholar] [CrossRef]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein. J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, D.; Dhanda, S.K.; Chauhan, J.S.; Bhardwaj, A.; Brahmachari, S.K.; Consortium, Open Source Drug Discovery; Raghava, G.P. Open source software and web services for designing therapeutic molecules. Curr. Top. Med. Chem. 2013, 13, 1172–1191. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I.M. Computer-aided drug discovery and development (caddd): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef]
- Huang, Z. Drug Discovery Research: New Frontiers in the Post-Genomic Era; Wiley-Interscience: Hoboken, NJ, USA, 2007; Volume 521, p. 516. [Google Scholar]
- Lim, S.P.; Wang, Q.Y.; Noble, C.G.; Chen, Y.L.; Dong, H.; Zou, B.; Yokokawa, F.; Nilar, S.; Smith, P.; Beer, D.; et al. Ten years of dengue drug discovery: Progress and prospects. Antivir. Res. 2013, 100, 500–519. [Google Scholar] [CrossRef]
- Gotte, M.; Feld, J.J. Direct-acting antiviral agents for hepatitis c: Structural and mechanistic insights. Nat. Rev Gastroenterol. Hepatol. 2016, 13, 338–351. [Google Scholar] [CrossRef]
- Cao, X.; Li, Y.; Jin, X.; Li, Y.; Guo, F.; Jin, T. Molecular mechanism of divalent-metal-induced activation of ns3 helicase and insights into zika virus inhibitor design. Nucleic Acids Res. 2016, 44, 10505–10514. [Google Scholar] [CrossRef]
- Tian, H.; Ji, X.; Yang, X.; Zhang, Z.; Lu, Z.; Yang, K.; Chen, C.; Zhao, Q.; Chi, H.; Mu, Z.; et al. Structural basis of zika virus helicase in recognizing its substrates. Protein Cell 2016, 7, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Ji, X.; Yang, X.; Xie, W.; Yang, K.; Chen, C.; Wu, C.; Chi, H.; Mu, Z.; Wang, Z.; et al. The crystal structure of zika virus helicase: Basis for antiviral drug design. Protein Cell 2016, 7, 450–454. [Google Scholar] [CrossRef]
- Mottin, M.; Braga, R.C.; da Silva, R.A.; Silva, J.H.; Perryman, A.L.; Ekins, S.; Andrade, C.H. Molecular dynamics simulations of zika virus ns3 helicase: Insights into rna binding site activity. Biochem. Biophys. Res. Commun. 2016, 22, 1101–1107. [Google Scholar] [CrossRef]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef]
- Barrows, N.J.; Campos, R.K.; Powell, S.T.; Prasanth, K.R.; Schott-Lerner, G.; Soto-Acosta, R.; Galarza-Munoz, G.; McGrath, E.L.; Urrabaz-Garza, R.; Gao, J.; et al. A screen of fda-approved drugs for inhibitors of zika virus infection. Cell Host Microbe 2016, 20, 259–270. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Farhy, C.; Pinto, A.; Huang, C.T.; Simonetti, N.; Ngono, A.E.; Dewing, A.; Shresta, S.; Pinkerton, A.B.; Cieplak, P.; et al. Characterization of the zika virus two-component ns2b-ns3 protease and structure-assisted identification of allosteric small-molecule antagonists. Antivir. Res. 2017, 143, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.T.; Hassounah, S.A.; Colby-Germinario, S.P.; Oliveira, M.; Fogarty, C.; Quan, Y.; Han, Y.; Golubkov, O.; Ibanescu, I.; Brenner, B.; et al. Purification of zika virus rna-dependent rna polymerase and its use to identify small-molecule zika inhibitors. J. Antimicrob. Chemother. 2017, 72, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ren, J.; Nocadello, S.; Rice, A.J.; Ojeda, I.; Light, S.; Minasov, G.; Vargas, J.; Nagarathnam, D.; Anderson, W.F.; et al. Identification of novel small molecule inhibitors against ns2b/ns3 serine protease from zika virus. Antivir. Res. 2017, 139, 49–58. [Google Scholar] [CrossRef]
- Brecher, M.; Li, Z.; Liu, B.; Zhang, J.; Koetzner, C.A.; Alifarag, A.; Jones, S.A.; Lin, Q.; Kramer, L.D.; Li, H. A conformational switch high-throughput screening assay and allosteric inhibition of the flavivirus ns2b-ns3 protease. PLoS Pathog. 2017, 13, e1006411. [Google Scholar] [CrossRef]
- Li, K.; Frankowski, K.J.; Hanson, A.M.; Ndjomou, J.; Shanahan, M.A.; Mukherjee, S.; Kolli, R.; Shadrick, W.R.; Sweeney, N.L.; Belon, C.A.; et al. Hepatitis C Virus NS3 Helicase Inhibitor Discovery. In Probe Reports from the NIH Molecular Libraries Program; Bethesda: Rockville, MD, USA, 2010. [Google Scholar]
- Mastrangelo, E.; Pezzullo, M.; De Burghgraeve, T.; Kaptein, S.; Pastorino, B.; Dallmeier, K.; de Lamballerie, X.; Neyts, J.; Hanson, A.M.; Frick, D.N.; et al. Ivermectin is a potent inhibitor of flavivirus replication specifically targeting ns3 helicase activity: New prospects for an old drug. J. Antimicrob. Chemother. 2012, 67, 1884–1894. [Google Scholar] [CrossRef] [PubMed]
- Byrd, C.M.; Grosenbach, D.W.; Berhanu, A.; Dai, D.; Jones, K.F.; Cardwell, K.B.; Schneider, C.; Yang, G.; Tyavanagimatt, S.; Harver, C.; et al. Novel benzoxazole inhibitor of dengue virus replication that targets the ns3 helicase. Antimicrob. Agents Chemother. 2013, 57, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Byler, K.G.; Ogungbe, I.V.; Setzer, W.N. In-silico screening for anti-zika virus phytochemicals. J. Mol. Graph. Model. 2016, 69, 78–91. [Google Scholar] [CrossRef]
- Sahoo, M.; Jena, L.; Daf, S.; Kumar, S. Virtual screening for potential inhibitors of ns3 protein of zika virus. Genomics Inform. 2016, 14, 104–111. [Google Scholar] [CrossRef]
- Ramharack, P.; Soliman, M.E.S. Zika virus ns5 protein potential inhibitors: An enhanced in silico approach in drug discovery. J. Biomol. Struct. Dyn. 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Stephen, P.; Baz, M.; Boivin, G.; Lin, S.X. Structural insight into ns5 of zika virus leading to the discovery of mtase inhibitors. J. Am. Chem. Soc. 2016, 138, 16212–16215. [Google Scholar] [CrossRef]
- Allen, J.E.; Krigsfeld, G.; Patel, L.; Mayes, P.A.; Dicker, D.T.; Wu, G.S.; El-Deiry, W.S. Identification of trail-inducing compounds highlights small molecule onc201/tic10 as a unique anti-cancer agent that activates the trail pathway. Mol. Cancer 2015, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Ishitani, R.; Sato, T.; Suga, T.; Kitagawa, H. Studies on the ultrastructural distribution of h 3 -dimetacrine in rat cerebral cortex. Jpn. J. Pharmacol. 1972, 22, 313–323. [Google Scholar] [CrossRef]
- Joshi, S.K.; Lamb, K.; Bielefeldt, K.; Gebhart, G.F. Arylacetamide kappa-opioid receptor agonists produce a tonic- and use-dependent block of tetrodotoxin-sensitive and -resistant sodium currents in colon sensory neurons. J. Pharmacol. Exp. Ther. 2003, 307, 367–372. [Google Scholar] [CrossRef]
- Castellani, G.; Zagaria, F. Antiphosphodiesterase activity of papaveroline/sulfonic acid. In vitro spectrophotometric determination. Farmacol. Prat. 1978, 33, 187–193. [Google Scholar]
- Spitzer, R.; Jain, A.N. Surflex-dock: Docking benchmarks and real-world application. J. Comput. Aided Mol. Des. 2012, 26, 687–699. [Google Scholar] [CrossRef]
- Heang, V.; Yasuda, C.Y.; Sovann, L.; Haddow, A.D.; Travassos da Rosa, A.P.; Tesh, R.B.; Kasper, M.R. Zika virus infection, cambodia, 2010. Emerg. Infect. Dis. 2012, 18, 349–351. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the Compounds 1 and 2 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No | NCI ID, ZINC ID and Molecular Name | Structure | Molecular Weight (g/mol) | Log-P Value | H-Bond Acceptor Count | H-Bond Donor Count | Inhibitory Activities % |
|---|---|---|---|---|---|---|---|
| 1 | NSC10580 ZINC01706300 3-(1-piperidinylmethyl)-5-(1,1,3,3-tetramethylbutyl)-1,2-benzenediol |  | 319.489 | 4.798 | 3 | 3 | 73.241 ± 3.336 |
| 2 | NSC45741 ZINC263598830 5-(1,3-benzothiazol-2-yl)-2,3,4,5-tetrahydroxypentanoate |  | 321.279 | −0.503 | 8 | 4 | 54.475 ± 14.460 |
| 3 | NSC99676 ZINC100132692 1,3-dimethyl-8-(3-phenylpropylsulfanyl)-6-sulfanylidene-7H-purin-2-one |  | 346.467 | 3.054 | 4 | 1 | 39.024 ± 4.243 |
| 4 | NSC95910 ZINC1621537 8-((benzylthio)methyl)-1,3-dimethyl-3,9-dihydro-1H-purine-2,6-dione |  | 316.386 | 1.394 | 6 | 1 | 34.920 ± 14.255 |
| 5 | NSC20172 ZINC2046417 N-acetyl-3-(1-naphthyl) alanine |  | 257.288 | 1.972 | 3 | 2 | 30.965 ± 4.292 |
| 6 | NSC100297 ZINC00001260 3-(9,9-dimethylacridin-10-yl)-N,N-dimethylpropan-1-amine |  | 294.210 | 4.260 | 2 | 0 | 25.455 ± 2.185 |
| 7 | NSC299209 ZINC1871679 4-((3-quinolinylmethyl) amino) benzenesulfonamide |  | 313.373 | 2.494 | 5 | 2 | 21.595 ± 0.573 |
| 8 | NSC99799 ZINC22910125 1-(3,4-dihydroxybenzyl)-6,7-isoquinolinediol |  | 283.283 | 2.648 | 4 | 5 | 20.080 ± 0.113 |
| Compound No | NCI ID, ZINC ID and Molecular Name | Structure | Molecular Weight (g/mol) | Log-P Value | H-bond Acceptor Count | H-bond Donor Count | Inhibitory Activities % |
|---|---|---|---|---|---|---|---|
| 1 | NSC10580 ZINC01706300 3-(1-piperidinylmethyl)-5-(1,1,3,3-tetramethylbutyl)-1,2-benzenediol | | 319.489 | 4.798 | 3 | 3 | 73.161 ± 2.363 |
| 1A | NSC10578 ZINC31613795 3-(morpholin-4-ylmethyl)-5-(2,4,4-trimethylpentan-2-yl)benzene-1,2-diol |  | 321.461 | 3.644 | 4 | 2 | 42.602 ± 4.906 |
| 1B | NSC10650 ZINC1706378 2-(piperidin-1-ylmethyl)-4-(2,4,4-trimethylpentan-2-yl)phenol |  | 303.490 | 5.092 | 2 | 1 | 36.027 ± 14.915 |
| 1C | NSC100265 ZINC1662241 2,4-bis(2-methylbutan-2-yl)-6-(piperidin-1-ylmethyl)phenol |  | 331.544 | 5.753 | 2 | 1 | 24.118 ± 14.307 |
| 1D | NSC10620 ZINC1706337 2-[[bis(2-hydroxyethyl)amino]methyl]-4-tert-butylphenol |  | 267.369 | 1.476 | 4 | 3 | 23.266 ± 7.250 |
| 1E | NSC10653 ZINC1706381 2,6-bis[(dimethylamino)methyl]-4-(2,4,4-trimethylpentan-2-yl)phenol |  | 320.521 | 4.229 | 3 | 1 | 20.885 ± 6.033 |
| 1F | NSC10625 ZINC610623 4-tert-butyl-2,6-bis[(dimethylamino)methyl]phenol |  | 264.413 | 2.813 | 3 | 1 | 13.538 ± 6.838 |
| 2 | NSC45741 ZINC263598830 5-(1,3-benzothiazol-2-yl)-2,3,4,5-tetrahydroxypentanoate | | 321.279 | −0.503 | 8 | 4 | 71.316 ± 9.711 |
| 2A | NSC34443 ZINC4777682 1-(1,3-benzothiazol-2-yl)pentane-1,2,3,4,5-pentol |  | 285.314 | −0.595 | 7 | 5 | 19.868 ± 5.688 |
| 2B | NSC607001 ZINC1609998 1-(benzo[d]thiazol-2-yl)-5-hydroxypentan-1-one |  | 235.301 | 2.642 | 4 | 1 | 14.238 ± 2.179 |
| 2C | NSC38490 ZINC1670696 Ethyl 3-(1,3-benzothiazol-2-yl)-2-oxopropanoate |  | 249.284 | 1.971 | 5 | 0 | 11.938 ± 1.782 |
| 2D | NSC56090 ZINC286970 (1S)-1-(1H-benzimidazol-2-yl)pentane-1,2,3,4,5-pentol |  | 268.269 | −1.329 | 6 | 6 | 48.107 ± 12.078 |
| 2E | NSC56093 ZINC4533419 (1R)-1-(1H-benzimidazol-2-yl)hexane-1,2,3,4,5,6-hexol |  | 298.295 | −1.968 | 7 | 7 | 46.108 ± 26.869 |
| 2F | NSC20641 ZINC4692139 N,N-dicyclohexyl-2,3,4,5,6-pentahydroxyhexanamide |  | 359.463 | −0.084 | 6 | 5 | 44.509 ± 10.951 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, S.; Zhang, C.; Huang, L.S.; Zhang, X.-Q.; Xu, Y.; Fang, X.; Zhou, J.; Wu, M.; Schooley, R.T.; Huang, Z.; et al. Discovery and Computational Analyses of Novel Small Molecule Zika Virus Inhibitors. Molecules 2019, 24, 1465. https://doi.org/10.3390/molecules24081465
Zhu S, Zhang C, Huang LS, Zhang X-Q, Xu Y, Fang X, Zhou J, Wu M, Schooley RT, Huang Z, et al. Discovery and Computational Analyses of Novel Small Molecule Zika Virus Inhibitors. Molecules. 2019; 24(8):1465. https://doi.org/10.3390/molecules24081465
Chicago/Turabian StyleZhu, Siyu, Chaozai Zhang, Lina S. Huang, Xing-Quan Zhang, Yan Xu, Xiong Fang, Jiao Zhou, Meixian Wu, Robert T. Schooley, Ziwei Huang, and et al. 2019. "Discovery and Computational Analyses of Novel Small Molecule Zika Virus Inhibitors" Molecules 24, no. 8: 1465. https://doi.org/10.3390/molecules24081465
APA StyleZhu, S., Zhang, C., Huang, L. S., Zhang, X. -Q., Xu, Y., Fang, X., Zhou, J., Wu, M., Schooley, R. T., Huang, Z., & An, J. (2019). Discovery and Computational Analyses of Novel Small Molecule Zika Virus Inhibitors. Molecules, 24(8), 1465. https://doi.org/10.3390/molecules24081465