
Dioxygen Activation with Molybdenum Complexes Bearing Amide-Functionalized Iminophenolate Ligands

 and
and
Abstract
:
1. Introduction
2. Results and Discussion
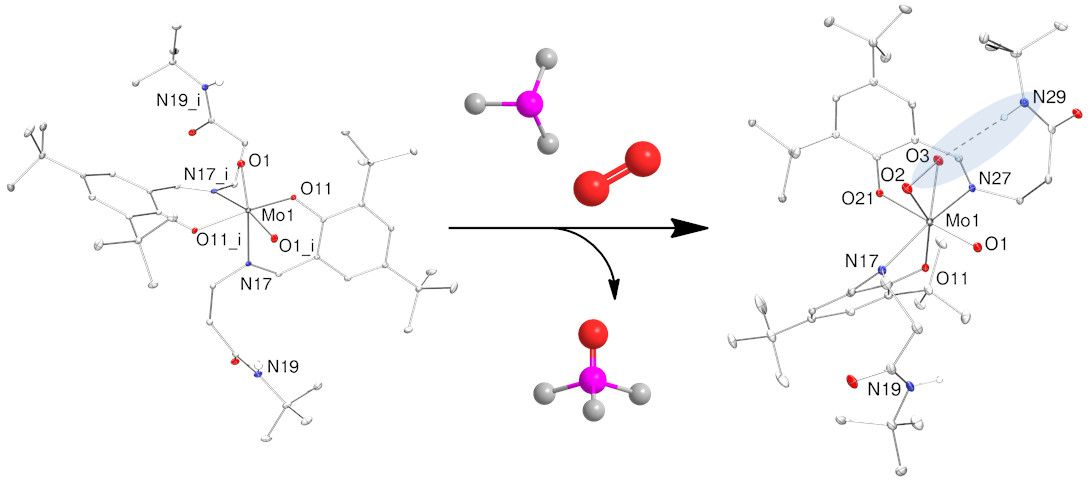
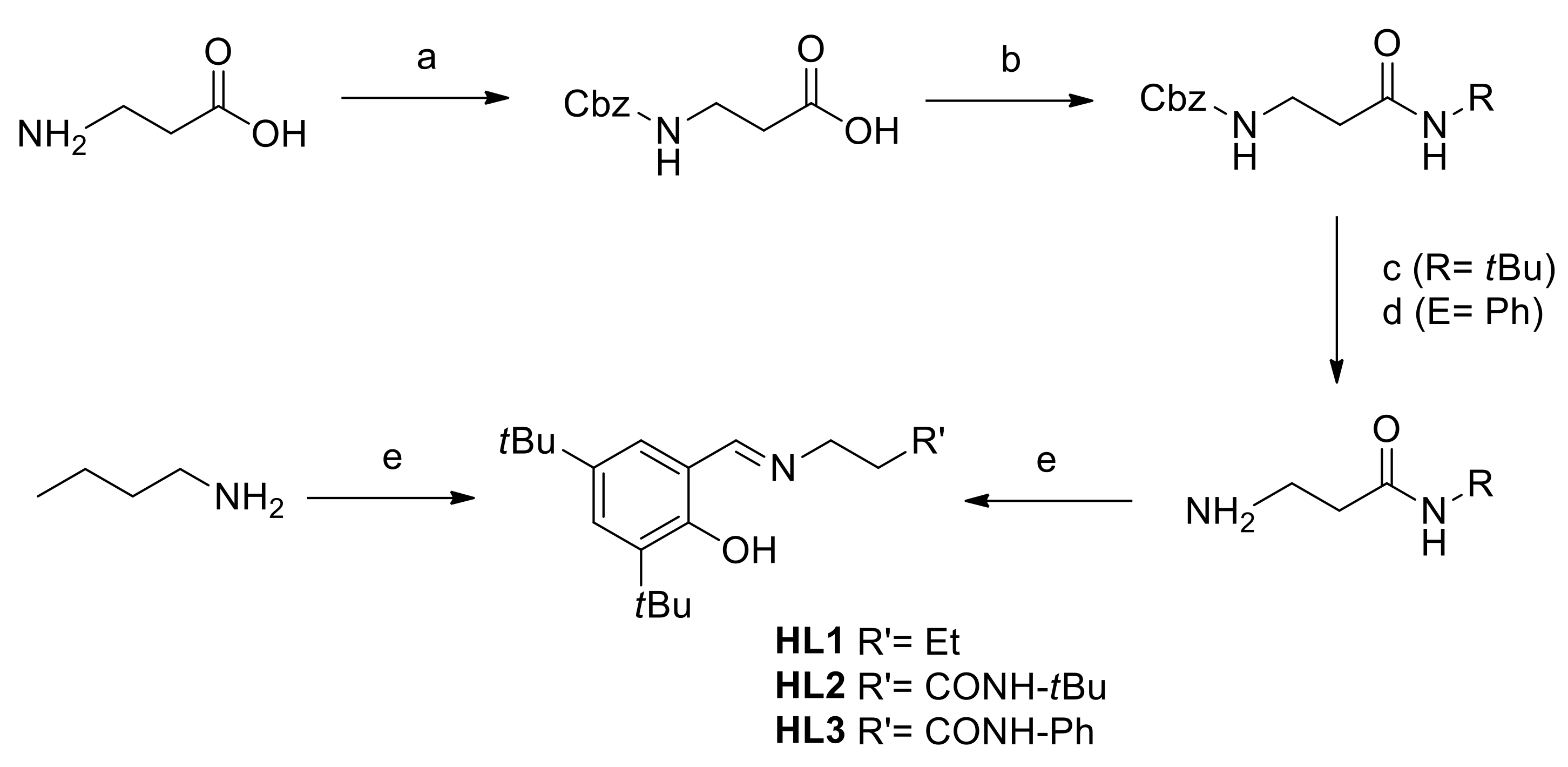
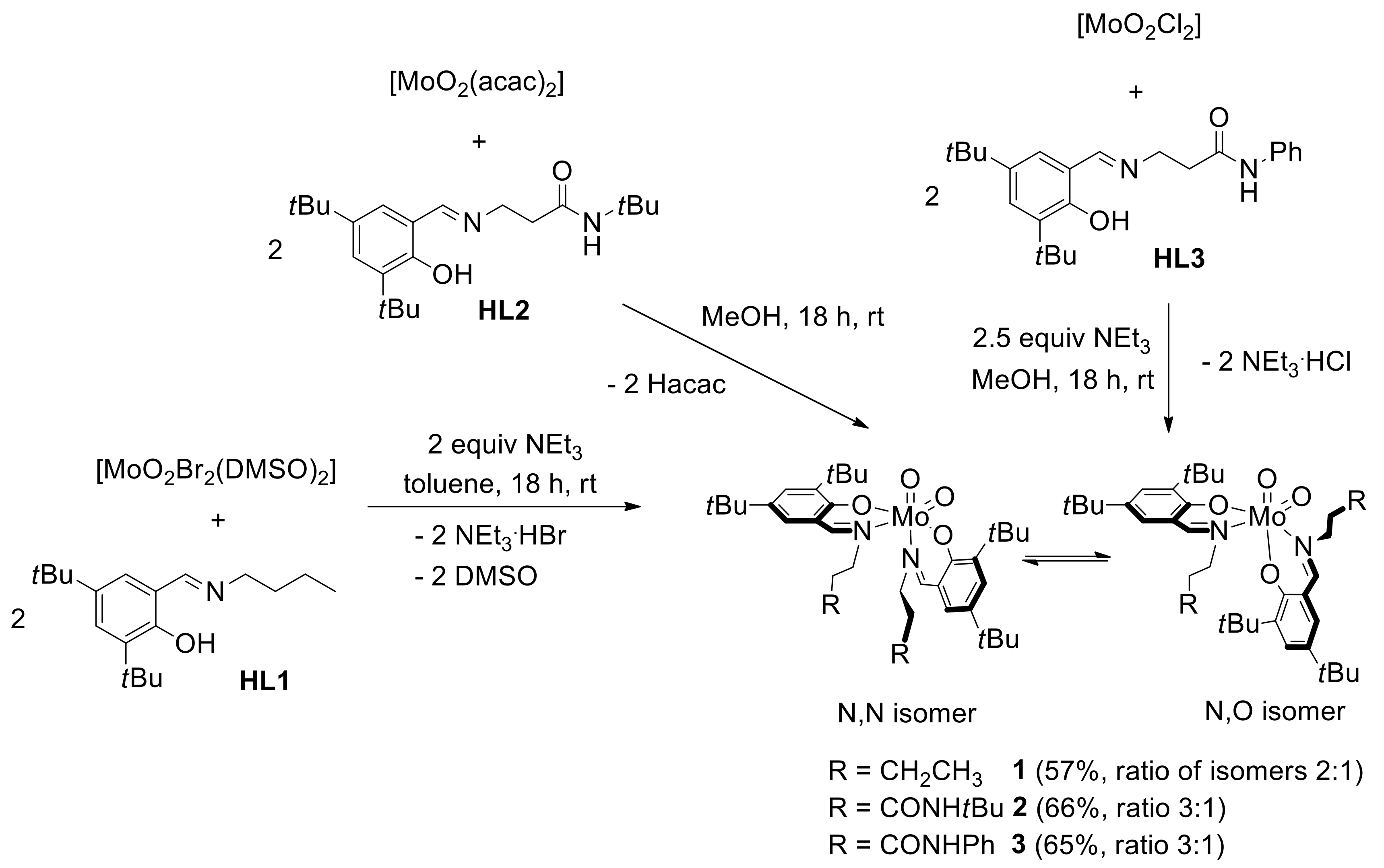
2.1. Ligand Synthesis
2.2. Synthesis of Dioxidomolybdenum(VI) Complexes
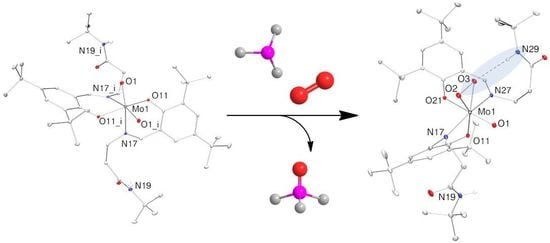
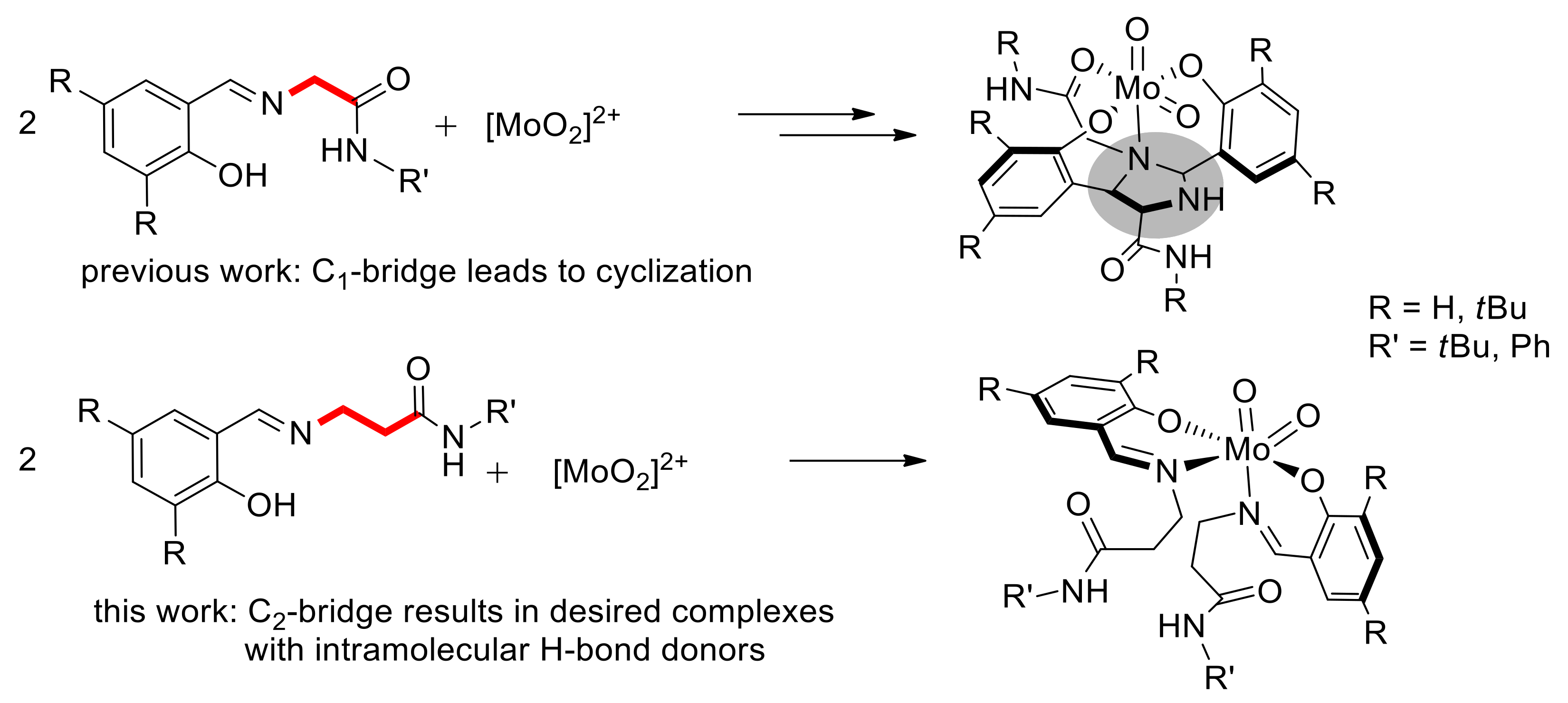
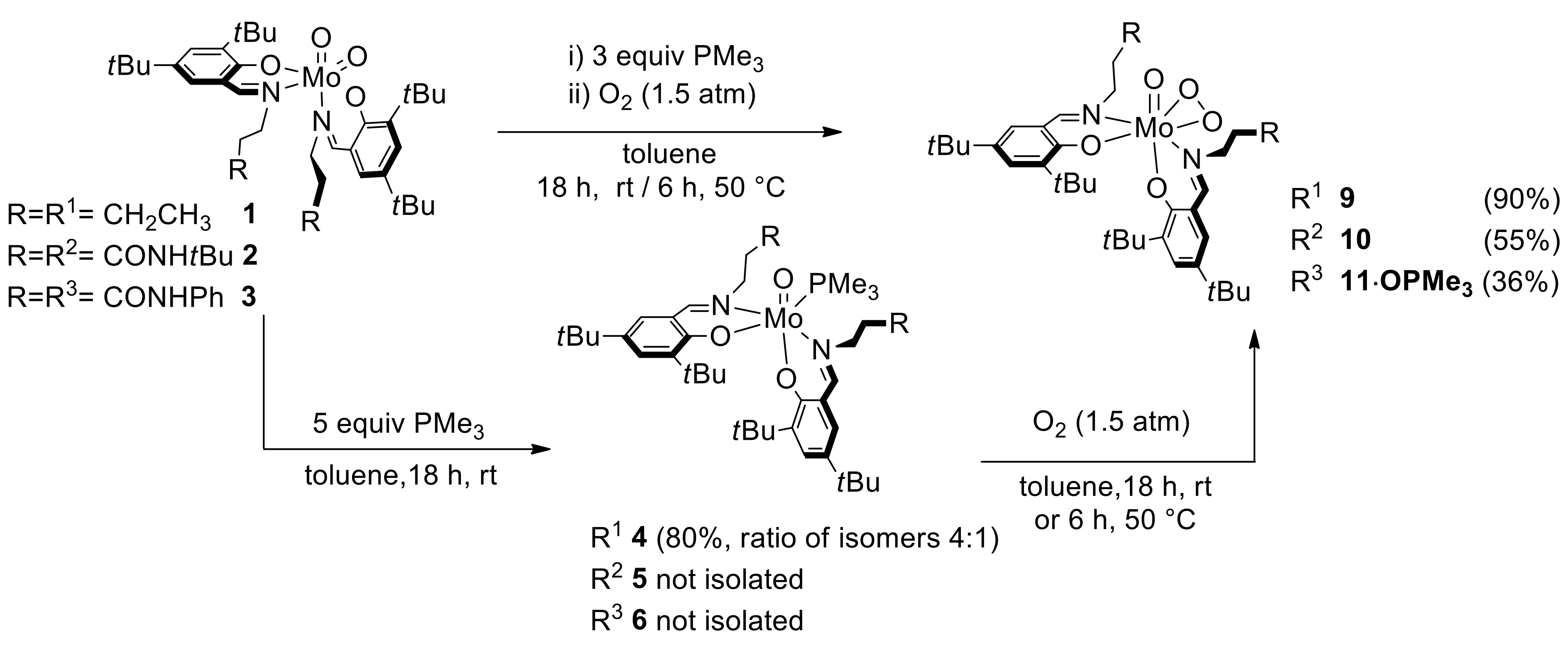
2.3. Reduction of Complexes 1–3 and Activation of Dioxygen
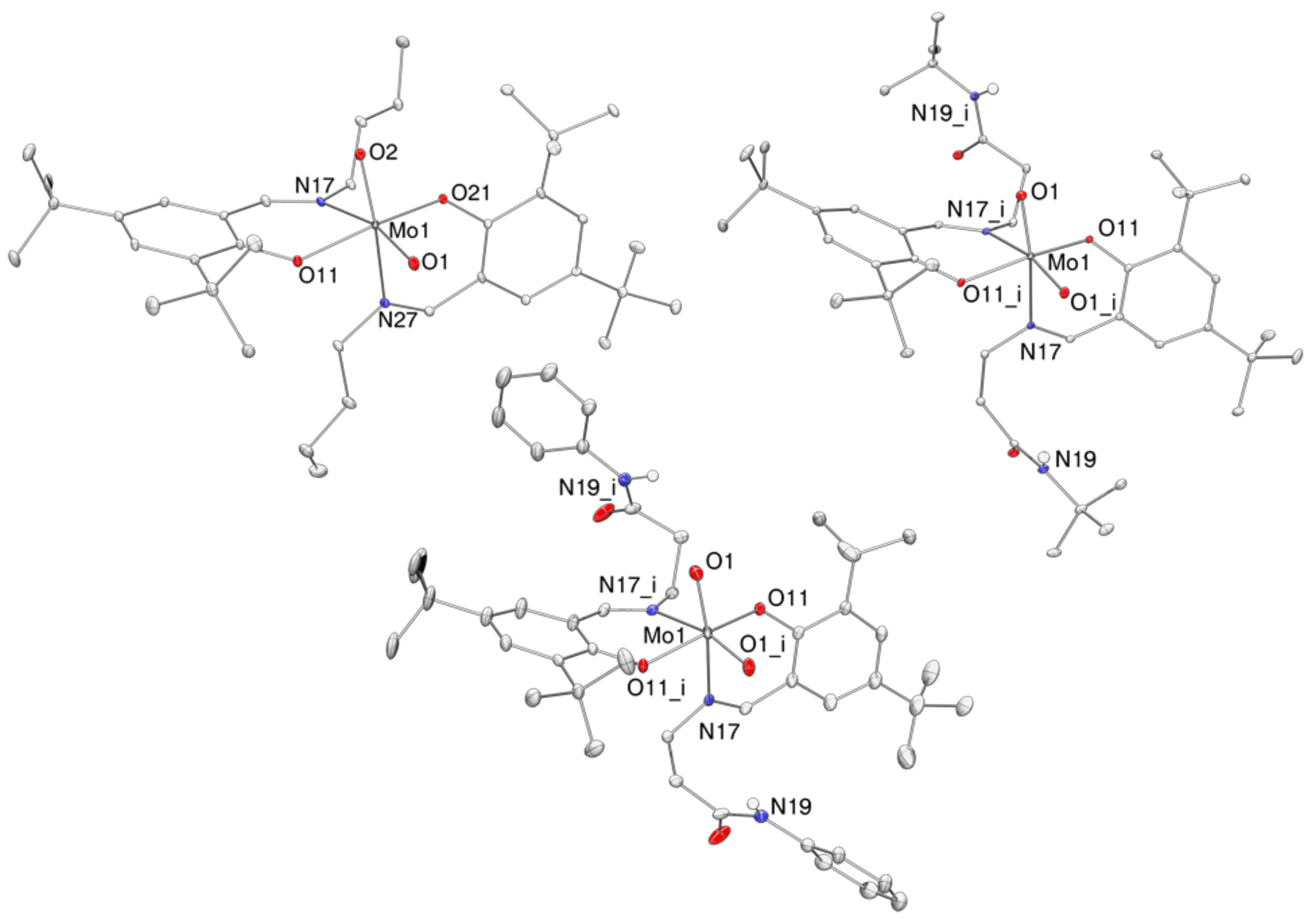
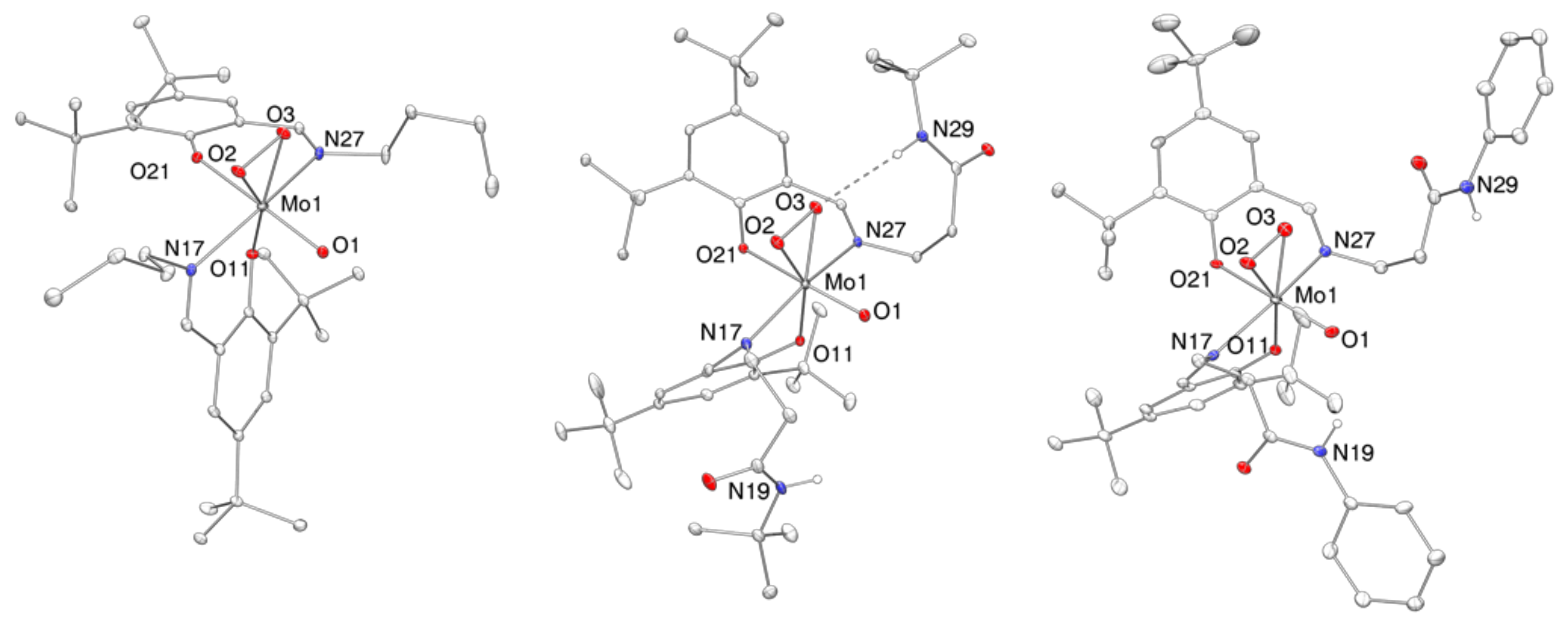
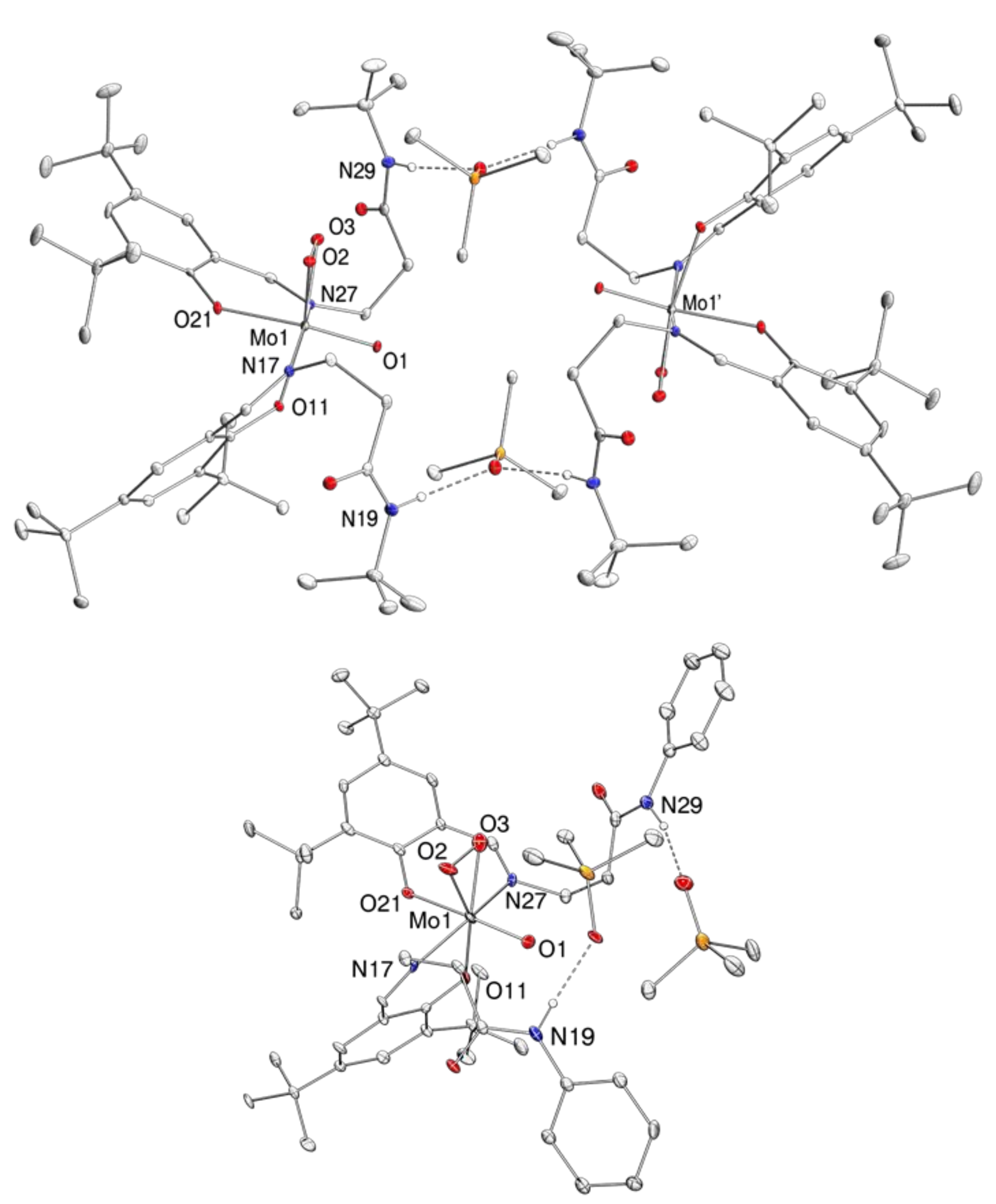
2.4. Molecular Structures
2.5. Catalysis

3. Experimental Section
3.1. General
3.2. X–ray Diffraction Analyses
3.3. Ligand Syntheses
3.3.1. Synthesis of 2,4-Di-tert-butyl-6-((butylimino)methyl)phenol (HL1)
3.3.2. Synthesis of N-(tert-Butyl)-3-((3,5-di-tert-butyl-2-hydroxybenzylidene)amino)-propanamide (HL2)
3.3.3. Synthesis of 3-((3,5-Di-tert-butyl-2-hydroxybenzylidene)amino)-N-phenylpropan-amide (HL3)
3.4. Complex Syntheses
3.4.1. Alternative Synthesis of [MoO2(L1)2] (1)
3.4.2. Synthesis of [MoO2(L2)2] (2)
3.4.3. Synthesis of [MoO2(L3)2] (3)
3.4.4. Synthesis of [MoO(PMe3)(L1)2] (4)
3.4.5. Characterization of [MoO(PMe3)(L2)2] (5)
3.4.6. Synthesis of [MoO(O2)(L1)2] (9)
3.4.7. Synthesis of [MoO(O2)(L2)2] (10)
3.4.8. Synthesis of [MoO(O2)(L3)2] (11)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hille, R.; Hall, J.; Basu, P. The Mononuclear Molybdenum Enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef]
- Heinze, K. Bioinspired functional analogs of the active site of molybdenum enzymes: Intermediates and mechanisms. Coord. Chem. Rev. 2015, 300, 121–141. [Google Scholar] [CrossRef]
- Hille, R.; Mendel, R. Molybdenum in living systems. Coord. Chem. Rev. 2011, 255, 991–992. [Google Scholar] [CrossRef]
- Hine, F.J.; Taylor, A.J.; Garner, C.D. Dithiolene complexes and the nature of molybdopterin. Coord. Chem. Rev. 2010, 254, 1570–1579. [Google Scholar] [CrossRef]
- Basu, P.; Burgmayer, S.J.N. Pterin chemistry and its relationship to the molybdenum cofactor. Coord. Chem. Rev. 2011, 255, 1016–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühn, F.E.; Santos, A.M.; Abrantes, M. Mononuclear organomolybdenum(VI) dioxo complexes: synthesis, reactivity, and catalytic applications. Chem. Rev. 2006, 106, 2455–2475. [Google Scholar] [CrossRef]
- Mösch-Zanetti, N.C.; Wurm, D.; Volpe, M.; Lyashenko, G.; Harum, B.N.; Belaj, F.; Baumgartner, J. Replacement of an Oxo by an Imido Group in Oxotransferase Model Compounds: Influence on the Oxygen Atom Transfer. Inorg. Chem. 2010, 49, 8914–8921. [Google Scholar] [CrossRef] [PubMed]
- Volpe, M.; Mösch-Zanetti, N.C. Molybdenum(VI) Dioxo and Oxo-Imido Complexes of Fluorinated β-Ketiminato Ligands and Their Use in OAT Reactions. Inorg. Chem. 2012, 51, 1440–1449. [Google Scholar] [CrossRef]
- Majumdar, A.; Sarkar, S. Bioinorganic chemistry of molybdenum and tungsten enzymes: A structural–functional modeling approach. Coord. Chem. Rev. 2011, 255, 1039–1054. [Google Scholar] [CrossRef]
- Young, C.G. Biomimetic chemistry of molybdenum. In Biomimetic Oxidations Catalyzed by Transition Metal Complexes; Meunier, B., Ed.; World Scientific: Singapore, 2000; pp. 415–459. [Google Scholar]
- Groysman, S.; Holm, R.H. Biomimetic Chemistry of Iron, Nickel, Molybdenum, and Tungsten in Sulfur-Ligated Protein Sites. Biochemistry 2009, 48, 2310–2320. [Google Scholar] [CrossRef]
- Holm, R.H.; Solomon, E.I.; Majumdar, A.; Tenderholt, A. Comparative molecular chemistry of molybdenum and tungsten and its relation to hydroxylase and oxotransferase enzymes. Coord. Chem. Rev. 2011, 255, 993–1015. [Google Scholar] [CrossRef]
- Hauser, S.A.; Cokoja, M.; Kühn, F.E. Epoxidation of olefins with homogeneous catalysts—Guo vadis? Catal. Sci. Technol. 2013, 3, 552–561. [Google Scholar] [CrossRef]
- Lyashenko, G.; Saischek, G.; Judmaier, M.E.; Volpe, M.; Baumgartner, J.; Belaj, F.; Jancik, V.; Herbst-Irmer, R.; Mösch-Zanetti, N.C. Oxo-molybdenum and oxo-tungsten complexes of Schiff bases relevant to molybdoenzymes. Dalton Trans. 2009, 5655–5665. [Google Scholar] [CrossRef] [PubMed]
- Judmaier, M.E.; Sala, C.H.; Belaj, F.; Volpe, M.; Mösch-Zanetti, N.C. Dimeric μ-oxo bridged molybdenum(VI) dioxo complexes as catalysts in the epoxidation of internal and terminal alkenes. New J. Chem. 2013, 37, 2139. [Google Scholar] [CrossRef]
- Judmaier, M.E.; Holzer, C.; Volpe, M.; Mösch-Zanetti, N.C. Molybdenum(VI) Dioxo Complexes Employing Schiff Base Ligands with an Intramolecular Donor for Highly Selective Olefin Epoxidation. Inorg. Chem. 2012, 51, 9956–9966. [Google Scholar] [CrossRef] [PubMed]
- Schachner, J.A.; Traar, P.; Sala, C.H.; Melcher, M.; Harum, B.N.; Sax, A.F.; Volpe, M.; Belaj, F.; Mösch-Zanetti, N.C. Dioxomolybdenum(VI) Complexes with Pyrazole Based Aryloxide Ligands: Synthesis, Characterization and Application in Epoxidation of Olefins. Inorg. Chem. 2012, 51, 7642–7649. [Google Scholar] [CrossRef] [PubMed]
- Zwettler, N.; Schachner, J.A.; Belaj, F.; Mösch-Zanetti, N.C. Hydrogen bond donor functionalized dioxido-molybdenum(VI) complexes as robust and highly efficient precatalysts for alkene epoxidation. Mol. Catal. 2017, 443, 209–219. [Google Scholar] [CrossRef]
- Dupé, A.; Hossain, M.K.; Schachner, J.A.; Belaj, F.; Lehtonen, A.; Nordlander, E.; Mösch-Zanetti, N.C. Dioxomolybdenum(VI) and -Tungsten(VI) Complexes with Multidentate Aminobisphenol Ligands as Catalysts for Olefin Epoxidation. Eur. J. Inorg. Chem. 2015, 3572–3579. [Google Scholar] [CrossRef]
- Gómez, M.; Jansat, S.; Muller, G.; Noguera, G.; Teruel, H.; Moliner, V.; Cerrada, E.; Hursthouse, M.B. First Dioxomolybdenum(VI) Complexes Containing Chiral Oxazoline Ligands: Synthesis, Characterization and Catalytic Activity. Eur. J. Inorg. Chem. 2001, 1071–1076. [Google Scholar] [CrossRef]
- Bagherzadeh, M.; Tahsini, L.; Latifi, R.; Woo, L.K. cis-Dioxo-molybdenum(VI)-oxazoline complex catalyzed epoxidation of olefins by tert-butyl hydrogen peroxide. Inorg. Chim. Acta 2009, 362, 3698–3702. [Google Scholar] [CrossRef]
- Bäckvall, J.-E. Modern Oxidation Methods; Wiley-VCH Verlag GmbH & Co KGaA: Weinheim, Germany, 2010. [Google Scholar]
- MacBeth, C.E.; Golombek, A.P.; Young, V.G., Jr.; Yang, C.; Kuczera, K.; Hendrich, K.; Borovik, A.S. O2 Activation by Nonheme Iron Complexes: A Monomeric Fe(III)-Oxo Complex Derived From O2. Science 2000, 289, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Shook, R.L.; Peterson, S.M.; Greaves, J.; Moore, C.; Rheingold, A.L.; Borovik, A.S. Catalytic reduction of dioxygen to water with a monomeric manganese complex at room temperature. J. Am. Chem. Soc. 2011, 133, 5810–5817. [Google Scholar] [CrossRef] [PubMed]
- Nam, W. Synthetic Mononuclear Nonheme Iron–Oxygen Intermediates. Acc. Chem. Res. 2015, 48, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Pfaff, F.F.; Wang, B.; Nam, W. Status of Reactive Non-Heme Metal–Oxygen Intermediates in Chemical and Enzymatic Reactions. J. Am. Chem. Soc. 2014, 136, 13942–13958. [Google Scholar] [CrossRef]
- Arzoumanian, H.; Petrignani, J.F.; Pierrot, M.; Ridouane, F.; Sanchez, J. Preparation of an oxoperoxocyanomolybdate(VI) complex by dioxygen oxidation of an oxocyanomolybdate(IV) anion. Structure and reactivity toward phosphines and olefins. Inorg. Chem. 1988, 27, 3377–3381. [Google Scholar] [CrossRef]
- Tachibana, J.; Imamura, T.; Sasaki, Y. Synthesis and characterization of a novel dioxygen complex of molybdenum porphyrin. J. Chem. Soc. Chem. Commun. 1993, 1436–1438. [Google Scholar] [CrossRef]
- Minato, M.; Zhou, D.-Y.; Sumiura, K.-I.; Oshima, Y.; Mine, S.; Ito, T.; Kakeya, M.; Hoshino, K.; Asaeda, T.; Nakada, T.; et al. Reactivity Patterns of O2, CO2, Carboxylic Acids, and Triflic Acid with Molybdenum Silyl Hydrido Complexes Bearing Polydentate Phosphinoalkyl–Silyl Ligands: Pronounced Effects of Silyl Ligands on Reactions. Organometallics 2012, 31, 4941–4949. [Google Scholar] [CrossRef]
- Suzuki, M. Ligand effects on dioxygen activation by copper and nickel complexes: Reactivity and intermediates. Acc. Chem. Res. 2007, 40, 609–617. [Google Scholar] [CrossRef]
- Duan, P.-C.; Manz, D.-H.; Dechert, S.; Demeshko, S.; Meyer, F. Reductive O2 Binding at a Dihydride Complex Leading to Redox Interconvertible μ-1,2-Peroxo and μ-1,2-Superoxo Dinickel(II) Intermediates. J. Am. Chem. Soc. 2018. [Google Scholar] [CrossRef]
- DeRosha, D.E.; Mercado, B.Q.; Lukat-Rodgers, G.; Rodgers, K.R.; Holland, P.L. Enhancement of C-H Oxidizing Ability in Co-O2 Complexes through an Isolated Heterobimetallic Oxo Intermediate. Angew. Chem. Int. Ed. 2017, 56, 3211–3215. [Google Scholar] [CrossRef]
- Lyashenko, G.; Saischek, G.; Pal, A.; Herbst-Irmer, R.; Mösch-Zanetti, N.C. Molecular oxygen activation by a molybdenum(IV) monooxo bis(b-ketiminato) complex. Chem. Commun. 2007, 701–703. [Google Scholar] [CrossRef]
- Dupé, A.; Judmaier, M.E.; Belaj, F.; Zangger, K.; Mösch-Zanetti, N.C. Activation of molecular oxygen by a molybdenum complex for catalytic oxidation. Dalton Trans. 2015, 44, 20514–20522. [Google Scholar] [CrossRef] [Green Version]
- Zwettler, N.; Grover, N.; Belaj, F.; Kirchner, K.; Mösch-Zanetti, N.C. Activation of Molecular Oxygen by a Molybdenum(IV) Imido Compound. Inorg. Chem. 2017, 56, 10147–10150. [Google Scholar] [CrossRef]
- Zwettler, N.; Judmaier, M.E.; Strohmeier, L.; Belaj, F.; Mösch-Zanetti, N.C. Oxygen activation and catalytic aerobic oxidation by Mo(IV)/(VI) complexes with functionalized iminophenolate ligands. Dalton Trans. 2016, 45, 14549–14560. [Google Scholar] [CrossRef]
- Zwettler, N.; Walg, S.; Belaj, F.; Mösch-Zanetti, N.C. Heterolytic Si-H bond cleavage at a molybdenum oxido based Lewis pair. Chem. Eur. J. 2018, 24, 7149–7160. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Gupta, R.; Lassalle-Kaiser, B.; Boyce, D.W.; Yachandra, V.K.; Tolman, W.B.; Yano, J.; Hendrich, M.P.; Borovik, A.S. Preparation and properties of a monomeric high-spin Mn(V)-oxo complex. J. Am. Chem. Soc. 2012, 134, 1996–1999. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.A.; Borovik, A.S. Molecular Designs for Controlling the Local Environments around Metal Ions. Acc. Chem. Res. 2015, 48, 2407–2414. [Google Scholar] [CrossRef] [Green Version]
- Cook, S.A.; Hill, E.A.; Borovik, A.S. Lessons from Nature: A Bio-Inspired Approach to Molecular Design. Biochemistry 2015, 54, 4167–4180. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, P.; Prokopchuk, D.E.; Mock, M.T. Exploring the role of pendant amines in transition metal complexes for the reduction of N2 to hydrazine and ammonia. Coord. Chem. Rev. 2017, 334, 67–83. [Google Scholar] [CrossRef]
- Baba, K.; Okamura, T.-A.; Suzuki, C.; Yamamoto, H.; Yamamoto, T.; Ohama, M.; Ueyama, N. O-Atom-Transfer Oxidation of [Molybdenum(IV) Oxo{3,6-(acylamino)2-1,2-benzenedithiolato}2]2− Promoted by Intramolecular NH···S Hydrogen Bonds. Inorg. Chem. 2006, 45, 894–901. [Google Scholar] [CrossRef]
- Ueyama, N.; Okamura, T.; Nakamura, A. Structure and properties of molybdenum(IV,V) arenethiolates with a neighboring amide group. Significant contribution of NH.cntdot.cntdot.cntdot.S hydrogen bond to the positive shift of redox potential of Mo(V)/Mo(IV). J. Am. Chem. Soc. 1992, 114, 8129–8137. [Google Scholar] [CrossRef]
- Zwettler, N.; Dupé, A.; Schachner, J.A.; Belaj, F.; Mösch-Zanetti, N.C. Templated C–C and C–N Bond Formation Facilitated by a Molybdenum(VI) Metal Center. Inorg. Chem. 2015, 54, 11969–11976. [Google Scholar] [CrossRef]
- Traar, P.; Schachner, J.A.; Steiner, L.; Sachse, A.; Volpe, M.; Mösch-Zanetti, N.C. Oxorhenium(V) complexes with pyrazole based aryloxide ligands and application in olefin epoxidation. Inorg. Chem. 2011, 50, 1983–1990. [Google Scholar] [CrossRef] [PubMed]
- Safaei, E.; Kabir, M.M.; Wojtczak, A.; Jagličić, Z.; Kozakiewicz, A.; Lee, Y.-I. Synthesis, crystal structure, magnetic and redox properties of copper(II) complexes of N-alkyl(aryl) tBu-salicylaldimines. Inorg. Chim. Acta 2011, 366, 275–282. [Google Scholar] [CrossRef]
- Nahrwold, M.; Stoncius, A.; Penner, A.; Neumann, B.; Stammler, H.-G.; Sewald, N. 2-Phenyl-tetrahydropyrimidine-4(1H)-ones--cyclic benzaldehyde aminals as precursors for functionalised beta-amino acids. Beilstein J. Org. Chem. 2009, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, H.; Veal, J. Acetylacetonate complexes of molybdenum(V) and molybdenum(VI). I. Inorg. Chim. Acta 1969, 3, 623–627. [Google Scholar] [CrossRef]
- Robin, T.; Montilla, F.; Galindo, A.; Ruiz, C.; Hartmann, J. Synthesis and characterization of dioxocomplexes of molybdenum with (η-C5H5)Co{P(O)(OEt)2}3, C5H4(SiMe3) and 1,3-C5H3(SiMe3)2 ligands. X-ray crystal structure of [(η-C5H5)Co{P(O)(OEt)2}3]MoO2Cl. Polyhedron 1999, 18, 1485–1490. [Google Scholar] [CrossRef]
- Sengar, R.S.; Miller, J.J.; Basu, P. Design, syntheses, and characterization of dioxo-molybdenum(VI) complexes with thiolate ligands: effects of intraligand NH…S hydrogen bonding. Dalton Trans. 2008, 2569–2577. [Google Scholar] [CrossRef]
- Kato, M.; Okamura, T.-A.; Yamamoto, H.; Ueyama, N. Effects of the intramolecular NH…S hydrogen bond in mononuclear platinum(II) and palladium(II) complexes with 2,2′-bipyridine and benzenethiol derivatives. Inorg. Chem. 2005, 44, 1966–1972. [Google Scholar] [CrossRef]
- Kato, M.; Kojima, K.; Okamura, T.-A.; Yamamoto, H.; Yamamura, T.; Ueyama, N. Relation between intramolecular NH…S hydrogen bonds and coordination number in mercury(II) complexes with carbamoylbenzenethiol derivatives. Inorg. Chem. 2005, 44, 4037–4044. [Google Scholar] [CrossRef] [PubMed]
- Okamura, T.-A.; Iwamura, T.; Yamamoto, H.; Ueyama, N. Synthesis and molecular structures of S-2-FcNHCOC6H4SH and [MIII(OEP)(S-2-FcNHCOC6H4)] (Fc=ferrocenyl, M=Fe, Ga): Electrochemical contributions of intramolecular SH⋯OC and NH⋯S hydrogen bonds. J. Organomet. Chem. 2007, 692, 248–256. [Google Scholar] [CrossRef]
- Choujaa, H.; Johnson, A.L.; Kociok-Köhn, G.; Molloy, K.C. The synthesis of W-O-W μ-oxo clusters by hydrolysis of tungsten aminoalkoxides and their structural characterisation. Dalton Trans. 2012, 41, 11393–11401. [Google Scholar] [CrossRef]
- Ziegler, J.E.; Du, G.; Fanwick, P.E.; Abu-Omar, M.M. An efficient method for the preparation of oxo molybdenum salalen complexes and their unusual use as hydrosilylation catalysts. Inorg. Chem. 2009, 48, 11290–11296. [Google Scholar] [CrossRef]
- Leppin, J.; Förster, C.; Heinze, K. Molybdenum complex with bulky chelates as a functional model for molybdenum oxidases. Inorg. Chem. 2014, 53, 12416–12427. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ide, R.; Kosaka, K.; Hasegawa, S.; Mikurube, K.; Taira, M.; Naruke, H.; Koguchi, S. Polyoxomolybdate–Surfactant Layered Crystals Derived from Long-tailed Alkylamine and Ionic Liquid. Chem. Lett. 2013, 42, 1400–1402. [Google Scholar] [CrossRef]
- Mayer, J.M. Metal-oxygen multiple bond lengths: A statistical study. Inorg. Chem. 1988, 27, 3899–3903. [Google Scholar] [CrossRef]
- Bingham, A.L.; Drake, J.E.; Hursthouse, M.B.; Light, M.E.; Kumar, R.; Ratnani, R. Synthesis, spectroscopic characterization and structural studies of bromodioxodimethylsulfoxide (N,N′-dialkyldithiocarbamates and O-alkyl dithiocarbonate)molybdenum(VI) complexes: Crystal structures of MoO2Br2(OSMe2)2 and MoO2Br2(C12H8N2)•CH2Cl2. Polyhedron 2006, 25, 3238–3244. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Göblyös, A.; Lázár, L.; Fülöp, F. Ring-chain tautomerism of 2-aryl-substituted-hexahydropyrimidines and tetrahydroquinazolines. Tetrahedron 2002, 58, 1011–1016. [Google Scholar] [CrossRef]
Sample Availability: Not available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | |
|---|---|---|---|
| Mo1–O1 | 1.705(3) | 1.7078(10) | 1.708(3) |
| Mo1–O2 | 1.712(3) | - | - |
| Mo1–O11 | 1.952(3) | 1.9571(10) | 1.950(2) |
| Mo1–O21 | 1.955(3) | - | - |
| Mo1–N17 | 2.351(2) | 2.3548(12) | 2.333(3) |
| Mo1–N27 | 2.341(4) | - | - |
| O1–Mo1–O2 | 106.80(16) | 107.62(7) | 108.0(2) |
| O11-Mo1-O21 | 160.48(11) | 157.63(6) | 159.49(16) |
| N17-Mo1-N27 | 73.30(12) | 73.42(6) | 74.78(16) |
| 9 | 10 | 11·2OPMe3 | |
|---|---|---|---|
| Mo1–O1 | 1.6817(18) | 1.6959(13) | 1.698(3) |
| Mo1–O2 | 1.9573(19) | 1.9398(13) | 1.961(4) |
| Mo1-O3 | 1.9469(16) | 1.9651(14) | 1.945(3) |
| Mo1–O11 | 2.0353(10) | 2.0256(13) | 2.014(3) |
| Mo1–O21 | 2.0113(10) | 2.0283(12) | 2.036(3) |
| Mo1–N17 | 2.1670(14) | 2.1542(15) | 2.167(4) |
| Mo1–N27 | 2.2053(14) | 2.1818(15) | 2.189(4) |
| O2–O3 | 1.431(3) | 1.4426(18) | 1.406(4) |
| O1–Mo1–O2 | 101.97(9) | 100.50(6) | 99.89(15) |
| O1–Mo1–O3 | 99.60(8) | 98.18(6) | 98.13(14) |
| O2–Mo1–O3 | 43.01(7) | 43.35(5) | 42.21(13) |
| O11-Mo1-O21 | 80.51(4) | 81.56(5) | 82.21(12) |
| N17-Mo1-N27 | 159.62(5) | 161.89(6) | 161.32(13) |
| Product | - | 1 | 2 | 3 |
|---|---|---|---|---|
| OPMe3 | <5% | 46% | 53% | 23% |
| OP(OMe)Me2 | - | - | 4% | 2% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zwettler, N.; Ehweiner, M.A.; Schachner, J.A.; Dupé, A.; Belaj, F.; Mösch-Zanetti, N.C. Dioxygen Activation with Molybdenum Complexes Bearing Amide-Functionalized Iminophenolate Ligands. Molecules 2019, 24, 1814. https://doi.org/10.3390/molecules24091814
Zwettler N, Ehweiner MA, Schachner JA, Dupé A, Belaj F, Mösch-Zanetti NC. Dioxygen Activation with Molybdenum Complexes Bearing Amide-Functionalized Iminophenolate Ligands. Molecules. 2019; 24(9):1814. https://doi.org/10.3390/molecules24091814
Chicago/Turabian StyleZwettler, Niklas, Madeleine A. Ehweiner, Jörg A. Schachner, Antoine Dupé, Ferdinand Belaj, and Nadia C. Mösch-Zanetti. 2019. "Dioxygen Activation with Molybdenum Complexes Bearing Amide-Functionalized Iminophenolate Ligands" Molecules 24, no. 9: 1814. https://doi.org/10.3390/molecules24091814
APA StyleZwettler, N., Ehweiner, M. A., Schachner, J. A., Dupé, A., Belaj, F., & Mösch-Zanetti, N. C. (2019). Dioxygen Activation with Molybdenum Complexes Bearing Amide-Functionalized Iminophenolate Ligands. Molecules, 24(9), 1814. https://doi.org/10.3390/molecules24091814