Synthesis, Molecular Recognition Study and Liquid Membrane-Based Applications of Highly Lipophilic Enantiopure Acridino-Crown Ethers



Abstract
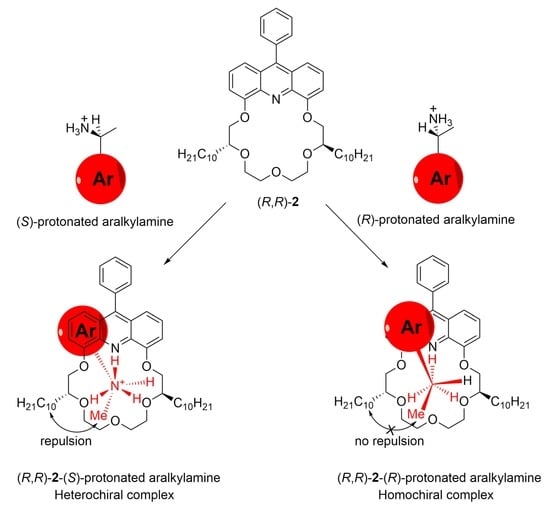
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Membrane Transport Studies
2.2.1. Investigation of the Influence of Structure on Molecular Recognition
2.2.2. Enantioselective Phase Transport Studies
2.3. Spectrophotometric Investigation of Enantiomeric Recognition
2.4. Development of Enantioselective Potentiometric Sensors
3. Conclusions
4. Experimental
4.1. General
4.2. Synthesis of New Compounds
4.2.1. (2R)-1-(Benzyloxy)dodecan-2-yl-4-Methylbenzene-1-Sulfonate [(R)-4]: See Scheme 1
4.2.2. (2S)-1-(Benzyloxy)dodecan-2-yl-4-Methylbenzene-1-Sulfonate [(S)-4]: See Scheme 1
4.2.3. (8R,16R)-8,16-Bis(decyl)-6,9,12,15,18-Pentaoxa-25-Azatetracyclo [2 1.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23-Hexaen-27-one [(R,R)-1]: See Scheme 2
4.2.4. (8S,16S)-8,16-Bis(decyl)-6,9,12,15,18-Pentaoxa-25-Azatetracyclo [21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23-Hexaen-27-one [(S,S)-1]: See Scheme 2
4.2.5. (8R,16R)-8,16-Bis(decyl)-27-Phenyl-6,9,12,15,18-Pentaoxa-25-Azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-Heptaene [(R,R)-2]: See Scheme 2
Procedure “A”
Procedure “B”
4.2.6. (8S,16S)-8,16-Bis(decyl)-27-Phenyl-6,9,12,15,18-Pentaoxa-25-Azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-Heptaene [(S,S)-2]: See Scheme 2
Procedure “A”
Procedure “B”
4.3. Transport Studies
4.4. Spectrophotometric Investigations
4.5. Electrochemical Measurements
Supplementary Materials
Author Contributions
Funding

Acknowledgments
Conflicts of Interest
References
- Stoddart, J.F.; Szarek, W.A. Medium heterocyclic rings from carbohydrate precursors. Can. J. Chem. 1968, 46, 3061–3069. [Google Scholar] [CrossRef]
- Kyba, E.P.; Siegel, M.G.; Sousa, L.R.; Sogah, G.D.; Cram, D.J. Chiral, hinged, and functionalized multiheteromacrocycles. J. Am. Chem. Soc. 1973, 95, 2691–2692. [Google Scholar] [CrossRef]
- Kyba, E.P.; Koga, K.; Sousa, L.R.; Siegel, M.G.; Cram, D.J. Chiral recognition in molecular complexing. J. Am. Chem. Soc. 1973, 95, 2692–2693. [Google Scholar] [CrossRef]
- Qing, G.Y.; He, Y.B.; Zhao, Y.; Hu, C.G.; Liu, S.Y.; Yang, X. Calix [4] arene-based chromogenic chemosensor for the α-phenylglycine anion: Synthesis and chiral recognition. Eur. J. Org. Chem. 2006, 1574–1580. [Google Scholar] [CrossRef]
- Schnopp, M.; Haberhauer, G. Highly Selective Recognition of α-Chiral Primary Organoammonium Ions by C3-Symmetric Peptide Receptors. Eur. J. Org. Chem. 2009, 2009, 4458–4467. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Pappalardo, A.; Tomaselli, G.A.; Toscano, R.M.; Sfrazzetto, G.T. Heteroditopic chiral uranyl–salen receptor for molecular recognition of amino acid ammonium salts. Eur. J. Org. Chem. 2010, 2010, 3806–3810. [Google Scholar] [CrossRef]
- Wang, C.; Wu, E.; Pu, L. Enantioselective Fluorescent Recognition by Using a 1,1′-Binaphthyl-2,2′-diamine Derivative. Eur. J. Org. Chem. 2017, 2017, 4736–4739. [Google Scholar] [CrossRef]
- Zhao, F.; Du, Y.; Tian, J.; Shi, D.; Wang, Y.; Hu, L.; Yu, S.; Yu, X.; Pu, L. Enantioselective Fluorescent Recognition of Amino Acids in Aqueous Solution by Using a Chiral Aldehyde Probe. Eur. J. Org. Chem. 2018, 2018, 1891–1895. [Google Scholar] [CrossRef]
- Scaramuzzo, F.A.; Badetti, E.; Licini, G.; Zonta, C. Second-generation tris (2-pyridylmethyl) amine–zinc complexes as probes for enantiomeric excess determination of amino acids. Eur. J. Org. Chem. 2017, 2017, 1438–1442. [Google Scholar] [CrossRef]
- Xiao, M.; Yu, S.; Chen, L.; Huang, Z.; Wen, K.; Xu, Y.; Zhao, F.; Yu, X.; Pu, L. Fluorous-Phase-Based Chiral Assay with Circular Dichroism Spectroscopy. Eur. J. Org. Chem. 2017, 2017, 1413–1417. [Google Scholar] [CrossRef]
- Iuliano, A.; Attolino, E.; Salvadori, P. (S)-Leucine and [(S)-1-(1-Naphthyl) ethyl]amine as Chiral Building Blocks for a Bifunctional System − Synthesis of a New Chiral Stationary Phase and Evaluation of Its Biselector Properties in the HPLC Resolution of Racemic Compounds. Eur. J. Org. Chem. 2001, 2001, 3523–3529. [Google Scholar] [CrossRef]
- Verkuijl, B.J.; Schoonen, A.K.; Minnaard, A.J.; de Vries, J.G.; Feringa, B.L. The Use of N-Type Ligands in the Enantioselective Liquid–Liquid Extraction of Underivatized Amino Acids. Eur. J. Org. Chem. 2010, 2010, 5197–5202. [Google Scholar] [CrossRef]
- Adhikari, B.B.; Fujii, A.; Schramm, M.P. Calixarene-mediated liquid-membrane transport of choline conjugates. Eur. J. Org. Chem. 2014, 2014, 2972–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.X.; Bradshaw, J.S.; Izatt, R.M. Enantiomeric recognition of amine compounds by chiral macrocyclic receptors. Chem. Rev. 1997, 97, 3313–3362. [Google Scholar] [CrossRef]
- Redd, J.T.; Bradshaw, J.S.; Huszthy, P.; Izatt, R.M. New pyrimidino-crown ether ligands. J. Heterocycl. Chem. 1994, 31, 1047–1052. [Google Scholar] [CrossRef]
- Redd, J.T.; Bradshaw, J.S.; Huszthy, P.; Izatt, R.M. Pyrimidino-and proton-ionizable pyrimidono-crown ether ligands: Synthesis and preliminary complexation studies. J. Inclusion Phenom. Mol. Recognit. Chem. 1997, 29, 301–308. [Google Scholar] [CrossRef]
- Wang, T.; Bradshaw, J.S.; Huszthy, P.; Kou, X.; Dalley, N.K.; Izatt, R.M. Recognition by a new chiral dimethyl-substituted phenanthrolino-18-crown-6 diester ligand of the enantiomers of various organic ammonium perchlorates. J. Heterocycl. Chem. 1994, 31, 1–10. [Google Scholar] [CrossRef]
- Bradshaw, J.S.; Chamberlin, D.A.; Harrison, P.E.; Wilson, B.E.; Arena, G.; Dalley, N.K.; Lamb, J.D.; Izatt, R.M.; Morin, F.G.; Grant, D.M. Proton-ionizable crown compounds. 1. Synthesis, complexation properties, and structural studies of macrocyclic polyether-diester ligands containing a triazole subcyclic unit. J. Org. Chem. 1985, 50, 3065–3069. [Google Scholar] [CrossRef]
- Jones, B.A.; Bradshaw, J.S.; Izatt, R.M. The synthesis of chiral dimethyl substituted macrocyclic polyether-diester ligands. J. Heterocycl. Chem. 1982, 19, 551–556. [Google Scholar] [CrossRef]
- Izatt, R.M.; Wang, T.; Hathaway, J.K.; Zhang, X.X.; Curtis, J.C.; Bradshaw, J.S.; Zhu, C.Y.; Huszthy, P. Factors influencing enantiomeric recognition of primary alkylammonium salts by pyridino-18-crown-6 type ligands. J. Inclusion Phenom. Mol. Recognit. Chem. 1994, 17, 157–175. [Google Scholar] [CrossRef]
- Kormos, A.; Móczár, I.; Pál, D.; Baranyai, P.; Kupai, J.; Tóth, K.; Huszthy, P. Synthesis and enantiomeric recognition studies of a novel 5,5-dioxophenothiazine-1,9 bis (thiourea) containing glucopyranosyl groups. Tetrahedron Asymmetry 2013, 24, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Huszthy, P.; Samu, E.; Vermes, B.; Mezey-Vándor, G.; Nógrádi, M.; Bradshaw, J.S.; Izatt, R.M. Synthesis of novel acridino-and phenazino-18-crown-6 ligands and their optically pure dimethyl-substituted analogues for molecular recognition studies. Tetrahedron 1999, 55, 1491–1504. [Google Scholar] [CrossRef]
- Székely, G.; Csordás, B.; Farkas, V.; Kupai, J.; Pogány, P.; Sánta, Z.; Szakács, Z.; Tóth, T.; Hollósi, M.; Nyitrai, J.; et al. Synthesis and Preliminary Structural and Binding Characterization of New Enantiopure Crown Ethers Containing an Alkyl Diarylphosphinate or a Proton-Ionizable Diarylphosphinic Acid Unit. Eur. J. Org. Chem. 2012, 2012, 3396–3407. [Google Scholar] [CrossRef]
- Prodi, L.; Bolletta, F.; Montalti, M.; Zaccheroni, N.; Huszthy, P.; Samu, E.; Vermes, B. Luminescence signalled enantiomeric recognition of chiral organic ammonium ions by an enantiomerically pure dimethylacridino-18-crown-6 ligand. New J. Chem. 2000, 24, 781–785. [Google Scholar] [CrossRef]
- Kertész, J.; Móczár, I.; Kormos, A.; Baranyai, P.; Kubinyi, M.; Tóth, K.; Huszthy, P. Synthesis and enantiomeric recognition studies of dialkyl-substituted 18-crown-6 ethers containing an acridine fluorophore unit. Tetrahedron: Asymmetry 2011, 22, 684–689. [Google Scholar] [CrossRef]
- Pu, L. Supramolecular optical chemosensors for organic analytes. Chem. Rev. 2004, 104, 1687–1716. [Google Scholar] [CrossRef]
- Kertész, J.; Huszthy, P.; Kormos, A.; Bertha, F.; Horváth, V.; Horvai, G. Synthesis of new optically active acridino-18-crown-6 ligands and studies of their potentiometric selectivity toward the enantiomers of protonated 1-phenylethylamine and metal ions. Tetrahedron: Asymmetry 2009, 20, 2795–2801. [Google Scholar] [CrossRef]
- Lakatos, S.; Fetter, J.; Bertha, F.; Huszthy, P.; Tóth, T.; Farkas, V.; Orosz, G.; Hollósi, M. Preparation of a new chiral acridino-18-crown-6 ether-based stationary phase for enantioseparation of racemic protonated primary aralkyl amines. Tetrahedron 2008, 64, 1012–1022. [Google Scholar] [CrossRef]
- Németh, T.; Lévai, S.; Kormos, A.; Kupai, J.; Tóth, T.; Balogh, G.T.; Huszthy, P. Preparation and studies of chiral stationary phases containing enantiopure acridino-18-crown-6 ether selectors. Chirality 2014, 26, 651–654. [Google Scholar] [CrossRef]
- Lévai, S.; Németh, T.; Fődi, T.; Kupai, J.; Tóth, T.; Huszthy, P.; Balogh, G.T. Studies of a pyridino-crown ether-based chiral stationary phase on the enantioseparation of biogenic chiral aralkylamines and α-amino acid esters by high-performance liquid chromatography. J. Pharm. Biomed. Anal. 2015, 115, 192–195. [Google Scholar] [CrossRef]
- Németh, T.; Tóth, T.; Balogh, G.T.; Huszthy, P. Synthesis and Fluorescence Spectroscopic Studies of Novel 9-phenylacridino-18-crown-6 Ether Type Sensor Molecules. Period. Polytech. Chem. Eng. 2017, 61, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Szabó, T.; Hirsch, E.; Tóth, T.; Huszthy, P. Synthesis and transport studies of new enantiopure lipophilic crown ethers containing a diarylphosphinic acid unit. Tetrahedron: Asymmetry 2014, 25, 1443–1449. [Google Scholar] [CrossRef]
- Huszthy, P.; Köntös, Z.; Vermes, B.; Pintér, Á. Synthesis of novel fluorescent acridono-and thioacridono-18-crown-6 ligands. Tetrahedron 2001, 57, 4967–4975. [Google Scholar] [CrossRef]
- Samu, E.; Huszthy, P.; Somogyi, L.; Hollósi, M. Enantiomerically pure chiral phenazino-crown ethers: Synthesis, preliminary circular dichroism spectroscopic studies and complexes with the enantiomers of 1-arethyl ammonium salts. Tetrahedron: Asymmetry 1999, 10, 2775–2795. [Google Scholar] [CrossRef]
- Szalay, L.; Farkas, V.; Vass, E.; Hollósi, M.; Móczár, I.; Pintér, Á.; Huszthy, P. Synthesis and selective lead (II) binding of achiral and enantiomerically pure chiral acridono-18-crown-6 ether type ligands. Tetrahedron: Asymmetry 2004, 15, 1487–1493. [Google Scholar] [CrossRef]
- Huszthy, P.; Farkas, V.; Tóth, T.; Székely, G.; Hollósi, M. Synthesis and preliminary studies on novel enantiopure crown ethers containing an alkyl diarylphosphinate or a proton-ionizable diarylphosphinic acid unit. Tetrahedron 2008, 64, 10107–10115. [Google Scholar] [CrossRef]
- Weissberger, A.; Taylor, E.C. An Acridines the Chemistry of Heterocyclic Compounds; Acheson, R.M., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1973; Volume 2, pp. 9–378. ISBN 0-471-37753-8. [Google Scholar]
- Bartsch, R.A.; Way, J.D. Chemical Separations with Liquid Membranes: An Overview; Chapter 1 of ACS Symposium Series; Chemical Separations with Liquid Membranes; American Chemical Society: Washington, DC, USA, 1996; Volume 642, pp. 1–10. ISBN 9780841234475. [Google Scholar] [CrossRef] [Green Version]
- Chrisstoffels, L.A.J.; de Jong, F.; Reinhoudt, D.N. Mechanistic Studies of Carrier-Mediated Transport Through Supported Liquid Membranes; Chapter 3 of ACS Symposium Series; Chemical Separations with Liquid Membranes; American Chemical Society: Washington, DC, USA, 1996; Volume 642, pp. 18–56. ISBN 9780841234475. [Google Scholar] [CrossRef] [Green Version]
- Peterson, R.T.; Lamb, J.D. Rational Design of Liquid Membrane Separation Systems; Chapter 4 of ACS Symposium Series; Chemical Separations with Liquid Membranes; American Chemical Society: Washington, DC, USA, 1996; Volume 642, pp. 57–74. ISBN 9780841234475. [Google Scholar] [CrossRef]
- Steed, J.E.; Atwood, J.L. (Eds.) Supramolecular Chemistry; John Wiley & Sons Ltd.: West Sussex, UK, 2013; Volume 2, pp. 180–194. ISBN 978-0-470-51233-3. [Google Scholar]
- Fogassy, E.; Lopata, A.; Faigl, F.; Darvas, F.; Ács, M.; Tőke, L. A quantitative approach to optical resolution. Tetrahedron Lett. 1980, 21, 647–650. [Google Scholar] [CrossRef]
- El-Rahman, M.K.A.; Salem, M.Y. Ion selective electrode (in-line analyzer) versus UV-spectroscopy (at-line analyzer); which strategy offers more opportunities for real time monitoring of the degradation kinetics of pyridostigmine bromide. Sens. Actuators B 2015, 220, 255–262. [Google Scholar] [CrossRef]
- Morf, W.E. The Principles of Ion-Selective Electrodes and of Membrane Transport; Morf, W.E., Ed.; Elsevier: New York, NY, USA, 1981; Volume 2, pp. 264–296. ISBN 0-444-99749-0. [Google Scholar]
- Riddick, J.A.; Bunger, W.B.; Sakano, T.K. Organic Solvents: Physical Properties and Methods of Purification; Techniques of Chemistry; Weissberger, A., Ed.; Wiley-Interscience: New York, NY, USA, 1986; Volume 4, pp. 1344–1400. ISBN 0471084670. [Google Scholar]
- Van de Weert, M.; Stella, L. Fluorescence quenching and ligand binding: A critical discussion of a popular methodology. J. Mol. Struct. 2011, 998, 144–150. [Google Scholar] [CrossRef]
- Bakker, E.; Bühlmann, P.; Pretsch, E. Carrier-based ion-selective electrodes and bulk optodes. 1. General characteristics. Chem. Rev. 1997, 97, 3083–3132. [Google Scholar] [CrossRef]
Sample Availability: Samples of the new crown compounds are available from the authors. |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Counter Ions | Transported Amount (%) | Optical Purity (%) |
|---|---|---|
| acetate | 45 | 5 |
| t-butyl sulfonate | 42 | 7 |
| chloride | 42 | 7 |
| p-toluenesulfonate | 38 | 5 |
| Temperature (°C) | Transported Amount (%) | Optical Purity (%) |
|---|---|---|
| 10 | 10 | 68 |
| 15 | 12 | 62 |
| 20 | 16 | 53 |
| Macrocycle | Ammonium Salts | ΔEMF 1 (mV) | Potentiometric Selectivity 2 | Enantioselectivity 3 | Estimated ΔlogK(R/S) 4 |
|---|---|---|---|---|---|
| 1 | 21 | 2.5 | 0.89 ± 0.02 | 1.12 ± 0.03 | 0.05 ± 0.01 |
| 1 | 22 | ≈0 | ≈1 | ≈1 | ≈0 |
| 1 | 23 | ≈0 | ≈1 | ≈1 | ≈0 |
| 1 | 24 | ≈0 | ≈1 | ≈1 | ≈0 |
| 2 | 21 | 13.9 | 0.55 ± 0.02 | 1.83 ± 0.07 | 0.26 ± 0.02 |
| 2 | 22 | 11.3 | 0.60 ± 0.03 | 1.66 ± 0.09 | 0.22 ± 0.02 |
| 2 | 23 | −13.4 | 1.79 ± 0.10 | 0.56 ± 0.03 | −0.25 ± 0.02 |
| 2 | 24 | 3.9 | 0.85 ± 0.03 | 1.18 ± 0.05 | 0.07 ± 0.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golcs, Á.; Ádám, B.Á.; Horváth, V.; Tóth, T.; Huszthy, P. Synthesis, Molecular Recognition Study and Liquid Membrane-Based Applications of Highly Lipophilic Enantiopure Acridino-Crown Ethers. Molecules 2020, 25, 2571. https://doi.org/10.3390/molecules25112571
Golcs Á, Ádám BÁ, Horváth V, Tóth T, Huszthy P. Synthesis, Molecular Recognition Study and Liquid Membrane-Based Applications of Highly Lipophilic Enantiopure Acridino-Crown Ethers. Molecules. 2020; 25(11):2571. https://doi.org/10.3390/molecules25112571
Chicago/Turabian StyleGolcs, Ádám, Bálint Árpád Ádám, Viola Horváth, Tünde Tóth, and Péter Huszthy. 2020. "Synthesis, Molecular Recognition Study and Liquid Membrane-Based Applications of Highly Lipophilic Enantiopure Acridino-Crown Ethers" Molecules 25, no. 11: 2571. https://doi.org/10.3390/molecules25112571
APA StyleGolcs, Á., Ádám, B. Á., Horváth, V., Tóth, T., & Huszthy, P. (2020). Synthesis, Molecular Recognition Study and Liquid Membrane-Based Applications of Highly Lipophilic Enantiopure Acridino-Crown Ethers. Molecules, 25(11), 2571. https://doi.org/10.3390/molecules25112571