
A Comparison of Different Approaches to Quantify Nitric Oxide Release from NO-Releasing Materials in Relevant Biological Media




Abstract
:1. Introduction
2. Results and Discussion
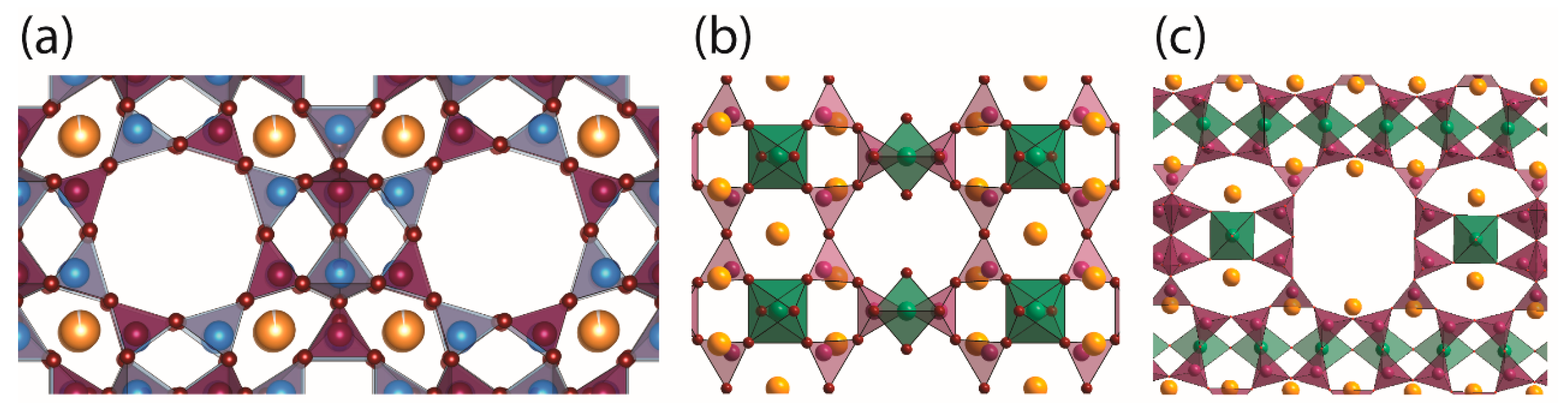
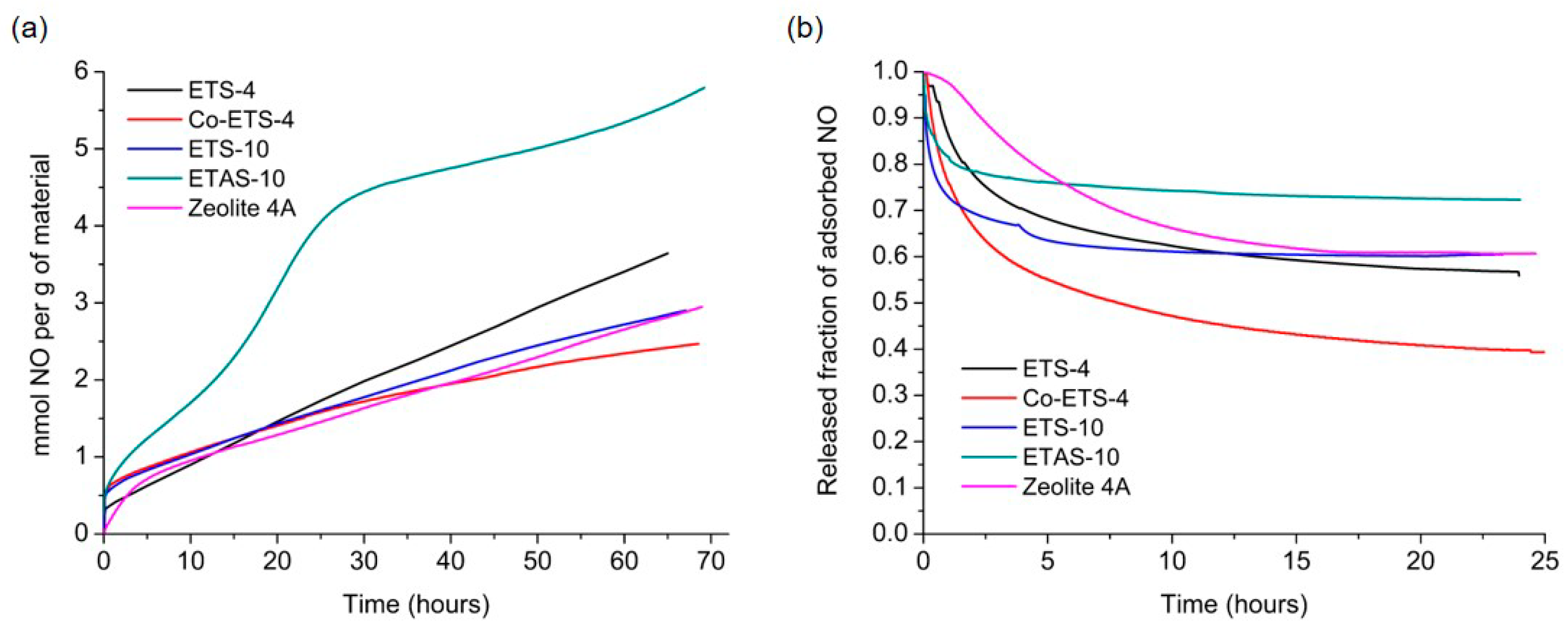
2.1. NO Adsorption Capacity of Different Porous Materials
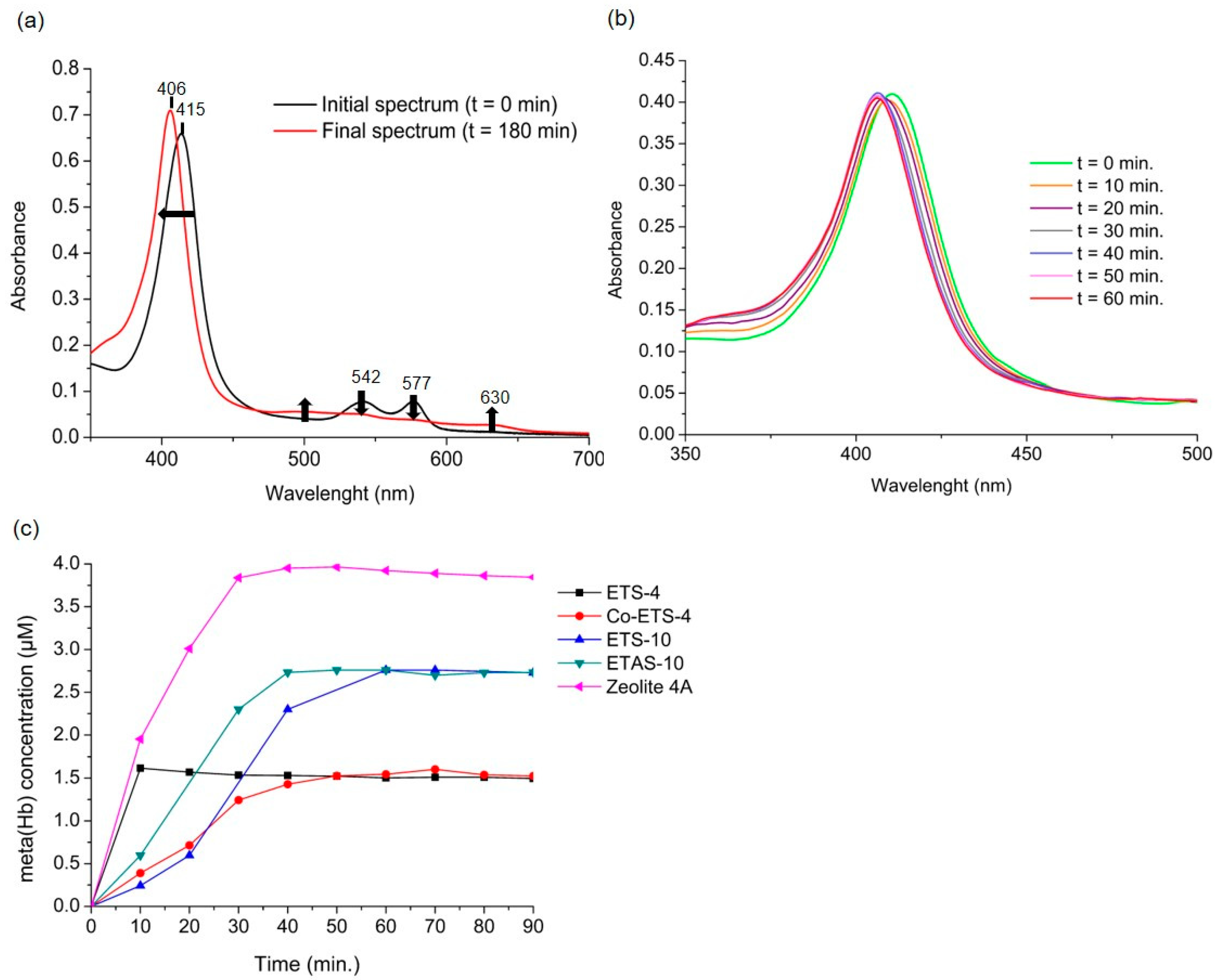
2.2. Oxyhaemoglobin Assay
2.3. Griess Assay
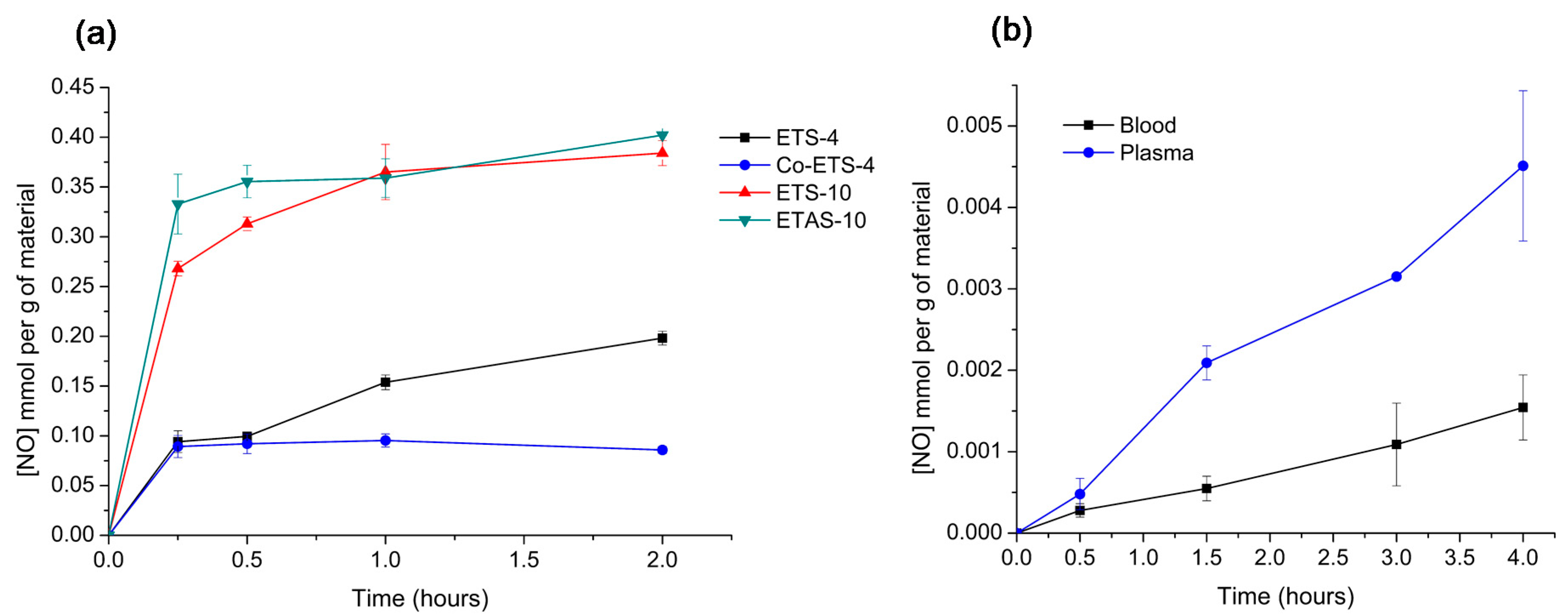
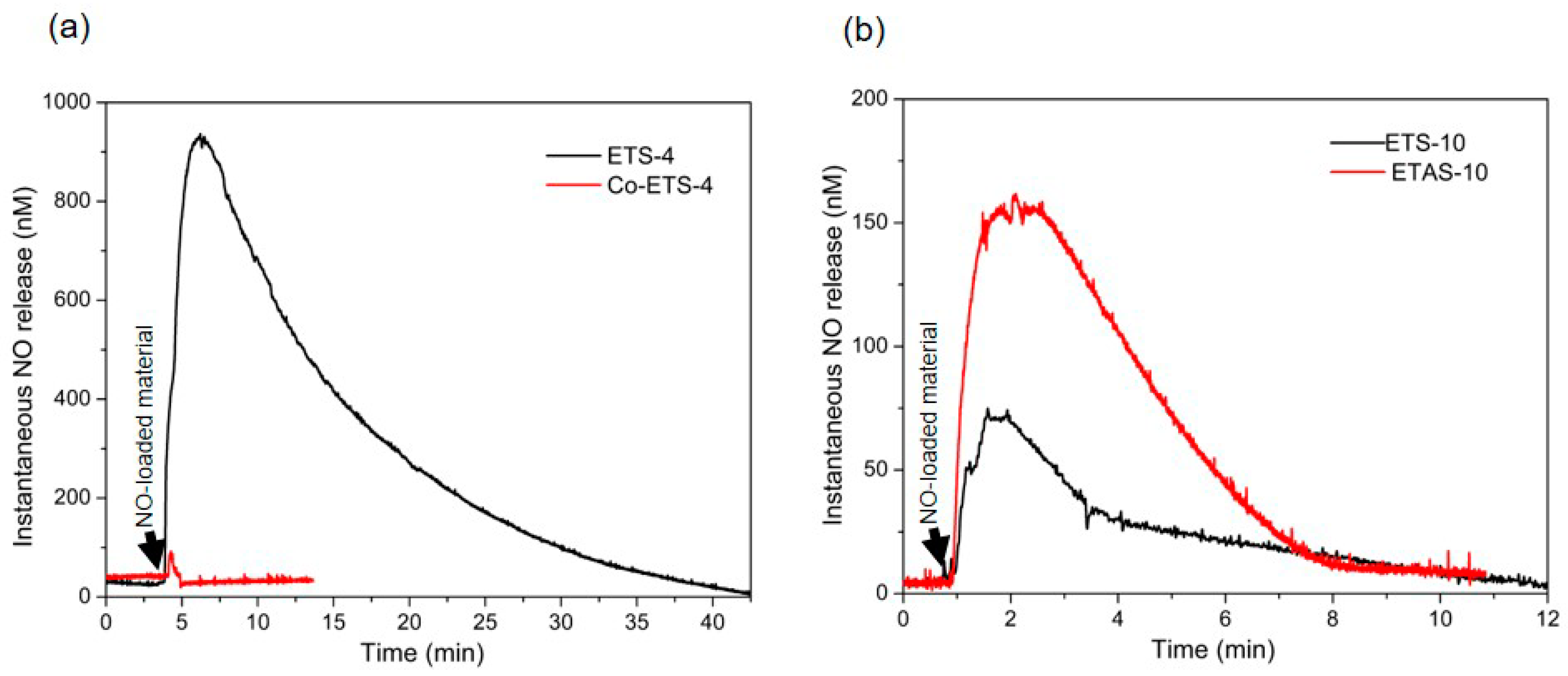
2.4. NO Electrochemical Sensor
3. Materials and Methods
3.1. NO-Releasing Porous Materials and Their Synthesis
3.2. Nitric Oxide Adsorption/Desorption Isotherms
3.3. NO Loading
3.4. Nitric Oxide Release Quantification in Liquid Phase
3.4.1. Oxyhaemoglobin Assay
3.4.2. Griess Assay
3.4.3. Electrochemical Sensor
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Carpenter, A.W.; Schoenfisch, M.H. Nitric oxide release: Part II. Therapeutic applications. Chem. Soc. Rev. 2012, 41, 3742–3752. [Google Scholar] [CrossRef] [PubMed]
- Riccio, D.A.; Schoenfisch, M.H. Nitric oxide release: Part I. Macromolecular scaffolds. Chem. Soc. Rev. 2012, 41, 3731–3741. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.V.; Wang, S.; Tavares, S.R.; Pires, J.; Antunes, F.; Vimont, A.; Clet, G.; Daturi, M.; Maurin, G.; Serre, C.; et al. Tuning Cellular Biological Functions Through the Controlled Release of NO from a Porous Ti-MOF. Angew. Chem. Int. Ed. 2020, 59, 5135–5143. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.E.; Wheatley, P.S. Gas storage in nanoporous materials. Angew. Chem. Int. Ed. 2008, 47, 4966–4981. [Google Scholar] [CrossRef]
- Pinto, R.V.; Fernandes, A.C.; Antunes, F.; Lin, Z.; Rocha, J.; Pires, J.; Pinto, M.L. New generation of nitric oxide-releasing porous materials: Assessment of their potential to regulate biological functions. Nitric Oxide Biol. Chem. 2019, 90, 29–36. [Google Scholar] [CrossRef]
- Gregg, S.T.; Yuan, Q.; Morris, R.E.; Xiao, B. Functionalised solids delivering bioactive nitric oxide gas for therapeutic applications. Mater. Today Commun. 2017, 12, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [Green Version]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R., Jr. The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA 2001, 98, 355–360. [Google Scholar] [CrossRef]
- Pullano, S.A.; Falcone, F.; Critello, D.C.; Bianco, M.G.; Menniti, M.; Fiorillo, A.S. An Affordable Fabrication of a Zeolite-Based Capacitor for Gas Sensing. Sensors 2020, 20, 2143. [Google Scholar] [CrossRef] [Green Version]
- Coneski, P.N.; Schoenfisch, M.H. Nitric oxide release: Part III. Measurement and reporting. Chem. Soc. Rev. 2012, 41, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Bryan, N.S.; Grisham, M.B. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic. Biol. Med. 2007, 43, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Kleinbongard, P.; Rassaf, T.; Dejam, A.; Kerber, S.; Kelm, M. Griess method for nitrite measurement of aqueous and protein-containing samples. In Nitric Oxide, Part D: Oxide Detection, Mitochondria and Cell Functions, and Peroxynitrite Reactions; Academic Press: Cambridge, MA, USA, 2002; Volume 359, pp. 158–168. ISBN 0076-6879. [Google Scholar]
- Sun, J.; Zhang, X.; Broderick, M.; Fein, H. Measurement of Nitric Oxide Production in Biological Systems by Using Griess Reaction Assay. Sensors 2003, 3, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Griveau, S.; Bedioui, F. Overview of significant examples of electrochemical sensor arrays designed for detection of nitric oxide and relevant species in a biological environment. Anal. Bioanal. Chem. 2013, 405, 3475–3488. [Google Scholar] [CrossRef]
- Pinto, M.L.; Rocha, J.; Gomes, J.R.B.; Pires, J. Slow release of NO by microporous titanosilicate ETS-4. J. Am. Chem. Soc. 2011, 133, 6396–6402. [Google Scholar] [CrossRef]
- Pinto, M.L.; Fernandes, A.C.; Rocha, J.; Ferreira, A.; Antunes, F.; Pires, J. Microporous titanosilicates Cu2+– and Co2+–ETS-4 for storage and slow release of therapeutic nitric oxide. J. Mater. Chem. B 2014, 2, 224–230. [Google Scholar] [CrossRef]
- Pinto, M.L.; Fernandes, A.C.; Antunes, F.; Pires, J.; Rocha, J. Storage and delivery of nitric oxide by microporous titanosilicate ETS-10 and Al and Ga substituted analogues. Microporous Mesoporous Mater. 2016, 229, 83–89. [Google Scholar] [CrossRef]
- Wheatley, P.S.; Butler, A.R.; Crane, M.S.; Fox, S.; Xiao, B.; Rossi, A.G.; Megson, I.L.; Morris, R.E. NO-releasing zeolites and their antithrombotic properties. J. Am. Chem. Soc. 2006, 128, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Henao, J.D.; Córdoba, L.F.; de Correa, C.M. Theoretical and experimental study of NO/NO2 adsorption over Co-exchanged type-A zeolite. J. Mol. Catal. A Chem. 2004, 207, 195–204. [Google Scholar] [CrossRef]
- Xiao, B.; Wheatley, P.S.; Zhao, X.; Fletcher, A.J.; Fox, S.; Rossi, A.G.; Megson, I.L.; Bordiga, S.; Regli, L.; Thomas, K.M.; et al. High-capacity hydrogen and nitric oxide adsorption and storage in a metal-organic framework. J. Am. Chem. Soc. 2007, 129, 1203–1209. [Google Scholar] [CrossRef]
- Gow, A.J.; Luchsinger, B.P.; Pawloski, J.R.; Singel, D.J.; Stamler, J.S. The oxyhemoglobin reaction of nitric oxide. Proc. Natl. Acad. Sci. USA 1999, 96, 9027–9032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feelisch, M.; Kubitzek, D.; Werringloer, J. The Oxyhemoglobin Assay. In Methods in Nitric Oxide Research; Feelisch, M., Stamler, J.S., Eds.; John Wiley & Sons, Ltd.: New York, NY, USA, 1996; pp. 455–478. [Google Scholar]
- Murphy, M.E.; Noack, E. Nitric oxide assay using hemoglobin method. In Oxygen Radicals in Biological Systems Part C. Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1994; Volume 233, pp. 240–250. [Google Scholar]
- Griess, P. Bemerkungen zu der Abhandlung der HH. Weselsky und Benedikt „Ueber einige Azoverbindungen”. Ber. Dtsch. Chem. Ges. 1879, 12, 426–428. [Google Scholar] [CrossRef] [Green Version]
- Grisham, M.B.; Johnson, G.G.; Lancaster, J.R., Jr. Quantitation of nitrate and nitrite in extracellular fluids. In Nitric Oxide Part A: Sources and Detection of NO.; NO Synthase; Academic Press: Cambridge, MA, USA, 1996; Volume 268, pp. 237–246. ISBN 0076-6879. [Google Scholar]
- Bellavia, L.; Kim-Shapiro, D.B.; King, S.B. Detecting and monitoring NO, SNO and nitrite in vivo. Futur. Sci. OA 2015, 1, FSO36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Zhang, Q.; Qian, X.; Yang, Y. Practical Assay for Nitrite and Nitrosothiol as an Alternative to the Griess Assay or the 2,3-Diaminonaphthalene Assay. Anal. Chem. 2015, 87, 1274–1280. [Google Scholar] [CrossRef]
- Csonka, C.; Páli, T.; Bencsik, P.; Görbe, A.; Ferdinandy, P.; Csont, T. Measurement of NO in biological samples. Br. J. Pharmacol. 2015, 172, 1620–1632. [Google Scholar] [CrossRef] [Green Version]
- Napoli, C.; Paolisso, G.; Casamassimi, A.; Al-Omran, M.; Barbieri, M.; Sommese, L.; Infante, T.; Ignarro, L.J. Effects of Nitric Oxide on Cell Proliferation: Novel Insights. J. Am. Coll. Cardiol. 2013, 62, 89–95. [Google Scholar] [CrossRef]
- Fox, S.; Wilkinson, T.S.; Wheatley, P.S.; Xiao, B.; Morris, R.E.; Sutherland, A.; Simpson, A.J.; Barlow, P.G.; Butler, A.R.; Megson, I.L.; et al. NO-loaded Zn2+-exchanged zeolite materials: A potential bifunctional anti-bacterial strategy. Acta Biomater. 2010, 6, 1515–1521. [Google Scholar] [CrossRef]
- Zhao, Y.-N.; Xu, X.; Wen, N.; Song, R.; Meng, Q.; Guan, Y.; Cheng, S.; Cao, D.; Dong, Y.; Qie, J.; et al. A Drug Carrier for Sustained Zero-Order Release of Peptide Therapeutics. Sci. Rep. 2017, 7, 5524. [Google Scholar] [CrossRef] [Green Version]
- Indika, P.N.; Bayachou, M. Eliminating absorbing interference using the H-point standard addition method: Case of Griess assay in the presence of interferent heme enzymes such as NOS. Anal. Bioanal. Chem. 2004, 379, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Hunter, R.A.; Storm, W.L.; Coneski, P.N.; Schoenfisch, M.H. Inaccuracies of Nitric Oxide Measurement Methods in Biological Media. Anal. Chem. 2013, 85, 1957–1963. [Google Scholar] [CrossRef] [Green Version]
- Rios-Ibarra, C.P.; Torres-De La Cruz, V.; Ochoa-Ruiz, A.G.; Rivas-Estilla, A.M. Quantification of nitric oxide by high-performance liquid chromatography-fluorometric method in subgenomic hepatitis C virus-replicon expressing Huh7 cells upon treatment with acetylsalicylic acid. Exp. Ther. Med. 2018, 16, 2621–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim-Shapiro, D.B.; Schechter, A.N.; Gladwin, M.T. Unraveling the Reactions of Nitric Oxide, Nitrite, and Hemoglobin in Physiology and Therapeutics. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.H.; Privett, B.J.; Kita, J.M.; Wightman, R.M.; Schoenfisch, M.H. Fluorinated xerogel-derived microelectrodes for amperometric nitric oxide sensing. Anal. Chem. 2008, 80, 6850–6859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMaster, E.G.; Quast, B.J.; Redfern, B.; Nagasawa, H.T. Reaction of Nitric Oxide with the Free Sulfhydryl Group of Human Serum Albumin Yields a Sulfenic Acid and Nitrous Oxide. Biochemistry 1995, 34, 11494–11499. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Simon, D.I.; Osborne, J.A.; Mullins, M.E.; Jaraki, O.; Michel, T.; Singel, D.J.; Loscalzo, J. S-nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA 1992, 89, 444–448. [Google Scholar] [CrossRef] [Green Version]
- Vishwakarma, A.; Wany, A.; Pandey, S.; Bulle, M.; Kumari, A.; Kishorekumar, R.; Igamberdiev, A.U.; Mur, L.A.J.; Gupta, K.J. Current approaches to measure nitric oxide in plants. J. Exp. Bot. 2019, 70, 4333–4343. [Google Scholar] [CrossRef] [Green Version]
- Pluth, J.J.; Smith, J.V. Accurate redetermination of crystal structure of dehydrated zeolite A. Absence of near zero coordination of sodium. Refinement of silicon, aluminum-ordered superstructure. J. Am. Chem. Soc. 1980, 102, 4704–4708. [Google Scholar] [CrossRef]
- Lopes, C.B.; Lito, P.F.; Otero, M.; Lin, Z.; Rocha, J.; Silva, C.M.; Pereira, E.; Duarte, A.C. Mercury removal with titanosilicate ETS-4: Batch experiments and modelling. Microporous Mesoporous Mater. 2008, 115, 98–105. [Google Scholar] [CrossRef]
- Anderson, M.W.; Terasaki, O.; Ohsuna, T.; Philippou, A.; MacKay, S.P.; Ferreira, A.; Rocha, J.; Lidin, S. Structure of the microporous titanosilicate ETS-10. Nature 1994, 367, 347–351. [Google Scholar] [CrossRef]
- Lin, Z.; Rocha, J.; Ferreira, A.; Anderson, M.W. Synthesis of microporous titano-alumino-silicate ETAS-10 with different framework aluminum contents. Colloids Surf. A Physicochem. Eng. Asp. 2001, 179, 133–138. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Sensitivity | NO Quantification Periods | Preferred Range for NO Quantification | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Oxyhaemoglobin assay | 1.3–2.8 nM [40] | Several hours 1 | up to 4 µM | Inexpensive; does not require specialized equipment; | Indirect; restricted to oxyhaemoglobin solutions; local measurements are not possible |
| Griess assay | 0.5 µM [14] | Days | 0.5 to 100 µM | Rapid and inexpensive; measures NO in a variety of biological fluids; available in ready-to-use kits | indirect; poor sensitivity; located measurements are not possible |
| Electrochemical sensor | 0.3–10 nM [15] | Minutes 2 | 30 nM to 1 µM 3 | Direct; real-time quantification; Measures NO in a variety of biological fluids; portable; located measurements are possible | Requires constant calibration; membrane has short life span; sensitive to tip position |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
V. Pinto, R.; Antunes, F.; Pires, J.; Silva-Herdade, A.; Pinto, M.L. A Comparison of Different Approaches to Quantify Nitric Oxide Release from NO-Releasing Materials in Relevant Biological Media. Molecules 2020, 25, 2580. https://doi.org/10.3390/molecules25112580
V. Pinto R, Antunes F, Pires J, Silva-Herdade A, Pinto ML. A Comparison of Different Approaches to Quantify Nitric Oxide Release from NO-Releasing Materials in Relevant Biological Media. Molecules. 2020; 25(11):2580. https://doi.org/10.3390/molecules25112580
Chicago/Turabian StyleV. Pinto, Rosana, Fernando Antunes, João Pires, Ana Silva-Herdade, and Moisés L. Pinto. 2020. "A Comparison of Different Approaches to Quantify Nitric Oxide Release from NO-Releasing Materials in Relevant Biological Media" Molecules 25, no. 11: 2580. https://doi.org/10.3390/molecules25112580
APA StyleV. Pinto, R., Antunes, F., Pires, J., Silva-Herdade, A., & Pinto, M. L. (2020). A Comparison of Different Approaches to Quantify Nitric Oxide Release from NO-Releasing Materials in Relevant Biological Media. Molecules, 25(11), 2580. https://doi.org/10.3390/molecules25112580