
Involvement of Integrin-Activating Peptides Derived from Tenascin-C in Cancer Aggression and New Anticancer Strategy Using the Fibronectin-Derived Integrin-Inactivating Peptide



Abstract
:1. Introduction
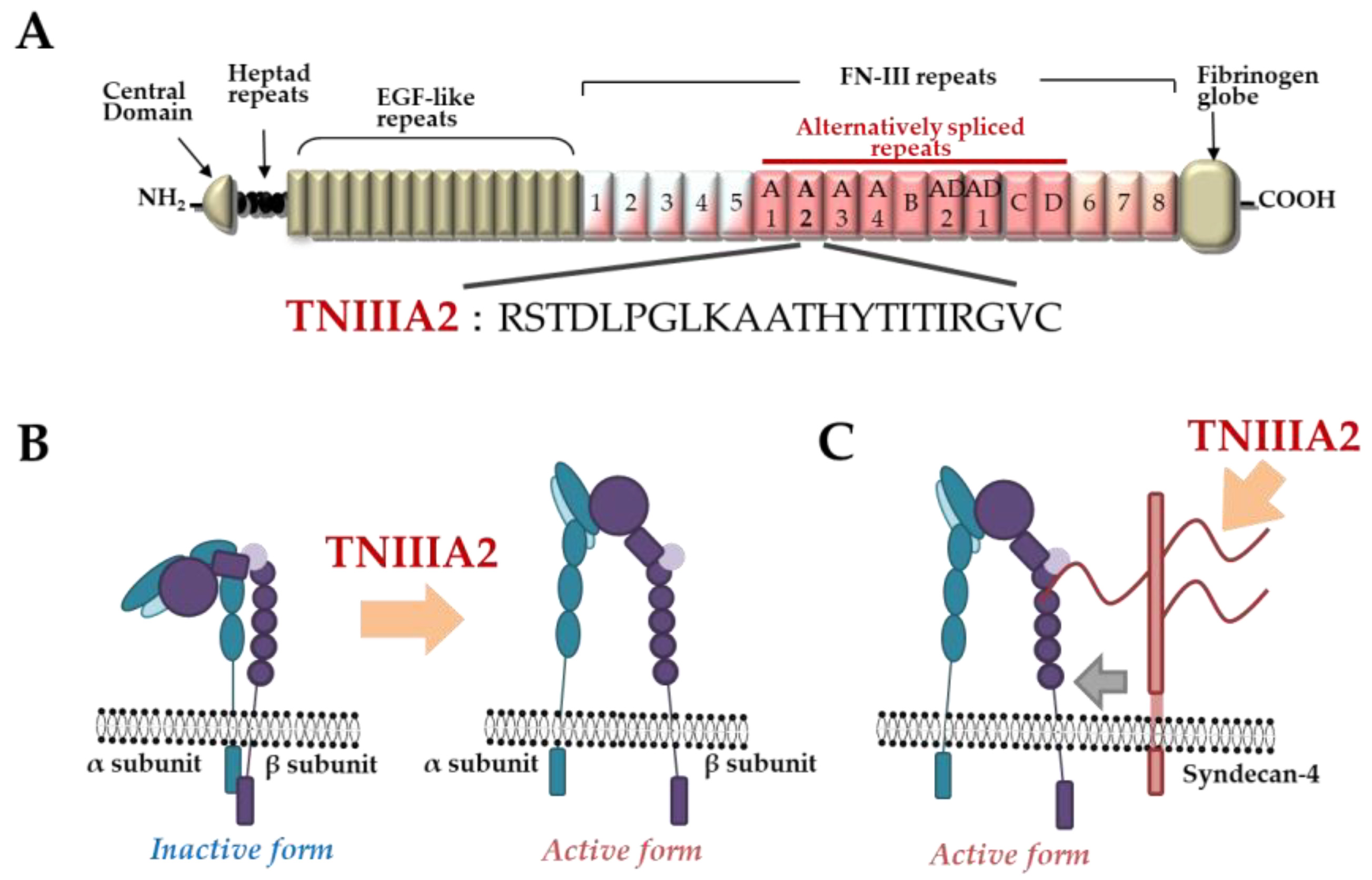
2. Tenascin-C-Derived Peptide, TNIIIA2
2.1. Glioma/Glioblastoma
2.2. Colitis-Associated Colorectal Cancer
3. Fibronectin-Derived Peptide, FNIII14
3.1. Glioma/Glioblastoma
3.2. Colitis-Associated Colorectal Cancer
4. Perspectives and Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAMTS | a disintegrin and metalloproteinase with thrombospondin motifs |
| ADME | absorption, distribution, metabolism and excretion |
| AML | acute myelogenous leukemia |
| AOM | azoxymethane |
| Ara C | cytosine arabinoside |
| CAC | colitis-associated cancer |
| CAFs | cancer-associated fibroblasts |
| CAM-DR | cell adhesion-mediated drug resistance |
| Dox | doxorubicin |
| DPPIV | dipeptidyl peptidase IV |
| DSS | dextran sulfate sodium |
| ECM | extracellular matrix |
| eEF | eukaryotic elongation factor |
| EGF | epidermal growth factor |
| ER | estrogen receptor |
| FN | fibronectin |
| GBM | glioblastoma multiforme |
| IBD | inflammatory bowel disease |
| LDV | Leu–Asp–Val |
| MAPK | mitogen-activated protein kinase |
| MCAM | melanoma cellular adhesion molecule |
| MGMT | O6-methylguanine–DNA methyltransferase |
| MIA | melanoma inhibitory activity |
| MMP | matrix metalloproteinase |
| MRD | minimal residual disease |
| OSCC | oral squamous cell carcinoma |
| PDGF | platelet-derived growth factor |
| PDGF-R | platelet-derived growth factor-receptor |
| pFN | plasma fibronectin |
| PD | pharmacodynamics |
| PK | pharmacokinetics |
| RGD | Arg–Gly–Asp |
| SPARC | secreted protein acidic and rich in cysteine |
| TLR | toll-like receptor |
| TME | tumor microenvironment |
| TMZ | temozolomide |
| TNC | tenascin-C |
| UC | ulcerative colitis |
| 5-FU | 5-Fluorouracil |
References
- Bornstein, P.; Sage, E.H. Matricellular proteins: Extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002, 14, 608–616. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Sage, E.H. Revisiting the matricellular concept. Matrix Biol. 2014, 37, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Kim, C.; Ginsberg, M.H. Reconstruction of integrin activation. Blood 2012, 119, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Sugimori, T.; Griffith, D.L.; Arnaout, M.A. Emerging paradigms of integrin ligand binding and activation. Kidney Int. 1997, 51, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef] [Green Version]
- De Castro Brás, L.E.; Frangogiannis, N.G. Extracellular matrix-derived peptides in tissue remodeling and fibrosis. Matrix Biol. 2020. [Google Scholar] [CrossRef]
- Davis, G.E.; Bayless, K.J.; Davis, M.J.; Meininger, G.A. Regulation of Tissue Injury Responses by the Exposure of Matricryptic Sites within Extracellular Matrix Molecules. Am. J. Pathol. 2000, 156, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.M.; Pritchard, J.M.; Macdonald, S.J.F.; Jamieson, C.; Watson, A.J.B. Emergence of Small-Molecule Non-RGD-Mimetic Inhibitors for RGD Integrins. J. Med. Chem. 2017, 60, 3241–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, K.T.; Lamb, P.; Deng, J.-S. Matrikines and matricryptins: Implications for cutaneous cancers and skin repair. J. Dermatol. Sci. 2005, 40, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S.; Vallet, S.D. Matricryptins Network with Matricellular Receptors at the Surface of Endothelial and Tumor Cells. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikkawa, Y.; Hozumi, K.; Katagiri, F.; Nomizu, M.; Kleinman, H.K.; Koblinski, J.E. Laminin-111-derived peptides and cancer. Cell Adhes. Migr. 2013, 7, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Kuratomi, Y.; Nomizu, M.; Tanaka, K.; Ponce, M.L.; Komiyama, S.; Kleinman, H.K.; Yamada, Y. Laminin γ1 chain peptide, C-16 (KAFDITYVRLKF), promotes migration, MMP-9 secretion, and pulmonary metastasis of B16–F10 mouse melanoma cells. Br. J. Cancer 2002, 86, 1169–1173. [Google Scholar] [CrossRef]
- Song, S.-Y.; Nomizu, M.; Yamada, Y.; Kleinman, H.K. Liver metastasis formation by laminin-1 peptide (LQVQLSIR)-adhesion selected B16-F10 melanoma cells. Int. J. Cancer 1997, 71, 436–441. [Google Scholar] [CrossRef]
- Paolillo, M.; Galiazzo, M.C.; Daga, A.; Ciusani, E.; Serra, M.; Colombo, L.; Schinelli, S. An RGD small-molecule integrin antagonist induces detachment-mediated anoikis in glioma cancer stem cells. Int. J. Oncol. 2018, 53, 2683–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alday-Parejo, B.; Stupp, R.; Rüegg, C. Are Integrins Still Practicable Targets for Anti-Cancer Therapy? Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itagaki, K.; Naito, T.; Iwakiri, R.; Haga, M.; Miura, S.; Saito, Y.; Owaki, T.; Kamiya, S.; Iyoda, T.; Yajima, H.; et al. Eukaryotic translation elongation factor 1A induces anoikis by triggering cell detachment. J. Biol. Chem. 2012, 287, 16037–16046. [Google Scholar] [CrossRef] [Green Version]
- Meiners, S.; Nur-e-Kamal, M.S.A.; Mercado, M.L.T. Identification of a Neurite Outgrowth-Promoting Motif within the Alternatively Spliced Region of Human Tenascin-C. J. Neurosci. 2001, 21, 7215–7225. [Google Scholar] [CrossRef]
- Jarocki, M.; Sallouh, O.; Weberskirch, R.; Faissner, A. The Tenascin-C-Derived Peptide VSWRAPTA Promotes Neuronal Branching Via Transcellular Activation of the Focal Adhesion Kinase (FAK) and the ERK1/2 Signaling Pathway In Vitro. Mol. Neurobiol. 2019, 56, 632–647. [Google Scholar] [CrossRef]
- Schneider, H.; Harbottle, R.P.; Yokosaki, Y.; Kunde, J.; Sheppard, D.; Coutelle, C. A novel peptide, PLAEIDGIELTY, for the targeting of α9β1-integrins. FEBS Lett. 1998, 429, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Sheets, A.R.; Demidova-Rice, T.N.; Shi, L.; Ronfard, V.; Grover, K.V.; Herman, I.M. Identification and Characterization of Novel Matrix-Derived Bioactive Peptides: A Role for Collagenase from Santyl® Ointment in Post-Debridement Wound Healing? PLoS ONE 2016, 11, e0159598. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Erickson, H.P.; Ikeda, Y.; Takada, Y. Identification of Amino Acid Sequences in Fibrinogen γ-Chain and Tenascin C C-terminal Domains Critical for Binding to Integrin αvβ3. J. Biol. Chem. 2000, 275, 16891–16898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Imazeki, H.; Miura, S.; Yoshimura, T.; Okutsu, H.; Harada, Y.; Ohwaki, T.; Nagao, O.; Kamiya, S.; Hayashi, R.; et al. A peptide derived from tenascin-C induces beta1 integrin activation through syndecan-4. J. Biol. Chem. 2007, 282, 34929–34937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Sasada, M.; Kamiya, S.; Ito, Y.; Watanabe, H.; Okada, Y.; Ishibashi, K.; Iyoda, T.; Yanaka, A.; Fukai, F. The Promoting Effect of the Extracellular Matrix Peptide TNIIIA2 Derived from Tenascin-C in Colon Cancer Cell Infiltration. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.-J.; Schwaiger, M.; Weinmüller, M.; Räder, A.; Steiger, K.; Kessler, H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9. [Google Scholar] [CrossRef]
- Komoriya, A.; Green, L.J.; Mervic, M.; Yamada, S.S.; Yamada, K.M.; Humphries, M.J. The minimal essential sequence for a major cell type-specific adhesion site (CS1) within the alternatively spliced type III connecting segment domain of fibronectin is leucine-aspartic acid-valine. J. Biol. Chem. 1991, 266, 15075–15079. [Google Scholar]
- Aota, S.; Nomizu, M.; Yamada, K.M. The short amino acid sequence Pro-His-Ser-Arg-Asn in human fibronectin enhances cell-adhesive function. J. Biol. Chem. 1994, 269, 24756–24761. [Google Scholar]
- Doñate, F.; Parry, G.C.; Shaked, Y.; Hensley, H.; Guan, X.; Beck, I.; Tel-Tsur, Z.; Plunkett, M.L.; Manuia, M.; Shaw, D.E.; et al. Pharmacology of the novel antiangiogenic peptide ATN-161 (Ac-PHSCN-NH2): Observation of a U-shaped dose-response curve in several preclinical models of angiogenesis and tumor growth. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 2137–2144. [Google Scholar] [CrossRef] [Green Version]
- Massia, S.P.; Hubbell, J.A. Vascular endothelial cell adhesion and spreading promoted by the peptide REDV of the IIICS region of plasma fibronectin is mediated by integrin alpha 4 beta 1. J. Biol. Chem. 1992, 267, 14019–14026. [Google Scholar]
- Gee, E.P.S.; Yüksel, D.; Stultz, C.M.; Ingber, D.E. SLLISWD sequence in the 10FNIII domain initiates fibronectin fibrillogenesis. J. Biol. Chem. 2013, 288, 21329–21340. [Google Scholar] [CrossRef] [Green Version]
- Moyano, J.V.; Carnemolla, B.; Domínguez-Jiménez, C.; García-Gila, M.; Albar, J.P.; Sánchez-Aparicio, P.; Leprini, A.; Querzé, G.; Zardi, L.; Garcia-Pardo, A. Fibronectin type III5 repeat contains a novel cell adhesion sequence, KLDAPT, which binds activated alpha4beta1 and alpha4beta7 integrins. J. Biol. Chem. 1997, 272, 24832–24836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.-F.; Gotwals, P.J.; Koteliansky, V.E.; Sheppard, D.; Van De Water, L. The EIIIA segment of fibronectin is a ligand for integrins alpha 9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J. Biol. Chem. 2002, 277, 14467–14474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mould, A.P.; Humphries, M.J. Identification of a novel recognition sequence for the integrin alpha 4 beta 1 in the COOH-terminal heparin-binding domain of fibronectin. EMBO J. 1991, 10, 4089–4095. [Google Scholar] [CrossRef] [PubMed]
- Okochi, M.; Nomura, S.; Kaga, C.; Honda, H. Peptide array-based screening of human mesenchymal stem cell-adhesive peptides derived from fibronectin type III domain. Biochem. Biophys. Res. Commun. 2008, 371, 85–89. [Google Scholar] [CrossRef]
- Woods, A.; McCarthy, J.B.; Furcht, L.T.; Couchman, J.R. A synthetic peptide from the COOH-terminal heparin-binding domain of fibronectin promotes focal adhesion formation. Mol. Biol. Cell 1993, 4, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Stack, M.S.; Pizzo, S.V. Modulation of tissue plasminogen activator-catalyzed plasminogen activation by synthetic peptides derived from the amino-terminal heparin binding domain of fibronectin. J. Biol. Chem. 1993, 268, 18924–18928. [Google Scholar]
- Wong, J.Y.; Weng, Z.; Moll, S.; Kim, S.; Brown, C.T. Identification and validation of a novel cell-recognition site (KNEED) on the 8th type III domain of fibronectin. Biomaterials 2002, 23, 3865–3870. [Google Scholar] [CrossRef]
- Gui, L.; Wojciechowski, K.; Gildner, C.D.; Nedelkovska, H.; Hocking, D.C. Identification of the heparin-binding determinants within fibronectin repeat III1: Role in cell spreading and growth. J. Biol. Chem. 2006, 281, 34816–34825. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.; Zhu, J.; Tonnesen, M.G.; Taira, B.R.; McClain, S.A.; Singer, A.J.; Clark, R.A.F. Fibronectin peptides that bind PDGF-BB enhance survival of cells and tissue under stress. J. Invest. Dermatol. 2014, 134, 1119–1127. [Google Scholar] [CrossRef] [Green Version]
- Drake, S.L.; Klein, D.J.; Mickelson, D.J.; Oegema, T.R.; Furcht, L.T.; McCarthy, J.B. Cell surface phosphatidylinositol-anchored heparan sulfate proteoglycan initiates mouse melanoma cell adhesion to a fibronectin-derived, heparin-binding synthetic peptide. J. Cell Biol. 1992, 117, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Zoppi, N.; De Petro, G.; Marchina, E.; Gardella, R.; Tavian, D.; Ferraboli, S.; Barlati, S. Matrix assembly induction and cell migration and invasion inhibition by a 13-amino acid fibronectin peptide. J. Biol. Chem. 2003, 278, 14346–14355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoll, R.; Renner, C.; Zweckstetter, M.; Brüggert, M.; Ambrosius, D.; Palme, S.; Engh, R.A.; Golob, M.; Breibach, I.; Buettner, R.; et al. The extracellular human melanoma inhibitory activity (MIA) protein adopts an SH3 domain-like fold. EMBO J. 2001, 20, 340–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosserhoff, A.-K.; Stoll, R.; Sleeman, J.P.; Bataille, F.; Buettner, R.; Holak, T.A. Active Detachment Involves Inhibition of Cell-Matrix Contacts of Malignant Melanoma Cells by Secretion of Melanoma Inhibitory Activity. Lab. Invest. 2003, 83, 1583–1594. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.R.; Spellman, S.; Kim, J.; Duan, W.-M.; McCarthy, J.B.; Low, W.C. Synthetic fibronectin peptide exerts neuroprotective effects on transient focal brain ischemia in rats. Brain Res. 2005, 1054, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ingham, K.C.; Brew, S.A.; Migliorini, M. An unusual heparin-binding peptide from the carboxy-terminal hep-2 region of fibronectin. Arch. Biochem. Biophys. 1994, 314, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-C.; Abdel-Ghany, M.; Pauli, B.U. A novel consensus motif in fibronectin mediates dipeptidyl peptidase IV adhesion and metastasis. J. Biol. Chem. 2003, 278, 24600–24607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, F.; Hasebe, S.; Ueki, M.; Mutoh, M.; Ohgi, C.; Takahashi, H.; Takeda, K.; Katayama, T. Identification of the anti-adhesive site buried within the heparin-binding domain of fibronectin. J. Biochem. (Tokyo) 1997, 121, 189–192. [Google Scholar] [PubMed]
- Giblin, S.P.; Midwood, K.S. Tenascin-C: Form versus function. Cell Adhes. Migr. 2015, 9, 48–82. [Google Scholar] [CrossRef] [Green Version]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef]
- Suzuki, H.; Fujimoto, M.; Kawakita, F.; Liu, L.; Nakano, F.; Nishikawa, H.; Okada, T.; Imanaka-Yoshida, K.; Yoshida, T.; Shiba, M. Toll-Like Receptor 4 and Tenascin-C Signaling in Cerebral Vasospasm and Brain Injuries After Subarachnoid Hemorrhage. Acta Neurochir. Suppl. 2020, 127, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Tajiri, K.; Sato, A.; Sakai, S.; Wang, Z.; Yoshida, T.; Uede, T.; Hiroe, M.; Aonuma, K.; Ieda, M.; et al. Tenascin-C accelerates adverse ventricular remodelling after myocardial infarction by modulating macrophage polarization. Cardiovasc. Res. 2019, 115, 614–624. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Wang, W.; Morales-Nebreda, L.; Feng, G.; Wu, M.; Zhou, X.; Lafyatis, R.; Lee, J.; Hinchcliff, M.; Feghali-Bostwick, C.; et al. Tenascin-C drives persistence of organ fibrosis. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.S.; Hussenet, T.; Langlois, B.; Orend, G. Advances in tenascin-C biology. Cell. Mol. Life Sci. CMLS 2011, 68, 3175–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppänen, J.; Lindholm, V.; Isohookana, J.; Haapasaari, K.-M.; Karihtala, P.; Lehenkari, P.P.; Saarnio, J.; Kauppila, J.H.; Karttunen, T.J.; Helminen, O.; et al. Tenascin C, Fibronectin, and Tumor-Stroma Ratio in Pancreatic Ductal Adenocarcinoma. Pancreas 2019, 48, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, W.; Yang, Z.; Li, H.; Cui, Y.; Xuan, Y. The role of Tenascin-C and Twist1 in gastric cancer: Cancer progression and prognosis. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2019, 127, 64–71. [Google Scholar] [CrossRef]
- Murakami, T.; Kikuchi, H.; Ishimatsu, H.; Iino, I.; Hirotsu, A.; Matsumoto, T.; Ozaki, Y.; Kawabata, T.; Hiramatsu, Y.; Ohta, M.; et al. Tenascin C in colorectal cancer stroma is a predictive marker for liver metastasis and is a potent target of miR-198 as identified by microRNA analysis. Br. J. Cancer 2017, 117, 1360–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.-T.; Yeo, S.-Y.; Yin, Y.-X.; Lin, Z.-H.; Lee, H.-M.; Xuan, Y.-H.; Cui, Y.; Kim, S.-H. Tenascin-C, a Prognostic Determinant of Esophageal Squamous Cell Carcinoma. PloS One 2016, 11, e0145807. [Google Scholar] [CrossRef]
- Gocheva, V.; Naba, A.; Bhutkar, A.; Guardia, T.; Miller, K.M.; Li, C.M.-C.; Dayton, T.L.; Sanchez-Rivera, F.J.; Kim-Kiselak, C.; Jailkhani, N.; et al. Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival. Proc. Natl. Acad. Sci. USA 2017, 114, E5625–E5634. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, A.; Yoshida, T.; Tamaki, H.; Sakakura, T. Tenascin expression in cancer cells and stroma of human breast cancer and its prognostic significance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1995, 1, 1035–1041. [Google Scholar]
- Yang, Z.; Ni, W.; Cui, C.; Fang, L.; Xuan, Y. Tenascin C is a prognostic determinant and potential cancer-associated fibroblasts marker for breast ductal carcinoma. Exp. Mol. Pathol. 2017, 102, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Nong, Y.; Wu, D.; Lin, Y.; Zhang, Y.; Bai, L.; Tang, H. Tenascin-C expression is associated with poor prognosis in hepatocellular carcinoma (HCC) patients and the inflammatory cytokine TNF-α-induced TNC expression promotes migration in HCC cells. Am. J. Cancer Res. 2015, 5, 782–791. [Google Scholar] [PubMed]
- Aishima, S.; Taguchi, K.; Terashi, T.; Matsuura, S.; Shimada, M.; Tsuneyoshi, M. Tenascin Expression at the Invasive Front Is Associated with Poor Prognosis in Intrahepatic Cholangiocarcinoma. Mod. Pathol. 2003, 16, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, W.-D.; Yang, Z.-T.; Cui, C.-A.; Cui, Y.; Fang, L.-Y.; Xuan, Y.-H. Tenascin-C is a potential cancer-associated fibroblasts marker and predicts poor prognosis in prostate cancer. Biochem. Biophys. Res. Commun. 2017, 486, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, E.; Kauppila, J.H.; Veijola, J.; Mroueh, R.; Lehenkari, P.; Laitinen, S.; Risteli, J.; Soini, Y.; Kosma, V.-M.; Sawazaki-Calone, I.; et al. Tenascin-C and fibronectin expression divide early stage tongue cancer into low- and high-risk groups. Br. J. Cancer 2017, 116, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Esfahani, D.R.; Huang, T.; Ozark, P.; Bartom, E.; Hashizume, R.; Bonner, E.R.; An, S.; Horbinski, C.M.; James, C.D.; et al. Tenascin-C expression contributes to pediatric brainstem glioma tumor phenotype and represents a novel biomarker of disease. Acta Neuropathol. Commun. 2019, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Steitz, A.M.; Steffes, A.; Finkernagel, F.; Unger, A.; Sommerfeld, L.; Jansen, J.M.; Wagner, U.; Graumann, J.; Müller, R.; Reinartz, S. Tumor-associated macrophages promote ovarian cancer cell migration by secreting transforming growth factor beta induced (TGFBI) and tenascin C. Cell Death Dis. 2020, 11, 249. [Google Scholar] [CrossRef]
- Ribeiro Franco, P.I.; Rodrigues, A.P.; de Menezes, L.B.; Pacheco Miguel, M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Mizushima, H.; Chinen, I.; Moribe, H.; Yagi, S.; Hoffman, R.M.; Kimura, T.; Yoshino, K.; Ueda, Y.; Enomoto, T.; et al. HB-EGF and PDGF mediate reciprocal interactions of carcinoma cells with cancer-associated fibroblasts to support progression of uterine cervical cancers. Cancer Res. 2011, 71, 6633–6642. [Google Scholar] [CrossRef] [Green Version]
- Ishii, G.; Ochiai, A.; Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 2016, 99, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.E.; Cittelly, D.M.; Tan, A.-C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin. Cancer Res. 2017, 23, 1710–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brechbuhl, H.M.; Barrett, A.S.; Kopin, E.; Hagen, J.C.; Han, A.L.; Gillen, A.E.; Finlay-Schultz, J.; Cittelly, D.M.; Owens, P.; Horwitz, K.B.; et al. Fibroblast subtypes define a metastatic matrisome in breast cancer. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [Green Version]
- Dueck, M.; Riedl, S.; Hinz, U.; Tandara, A.; Möller, P.; Herfarth, C.; Faissner, A. Detection of tenascin-C isoforms in colorectal mucosa, ulcerative colitis, carcinomas and liver metastases. Int. J. Cancer 1999, 82, 477–483. [Google Scholar] [CrossRef]
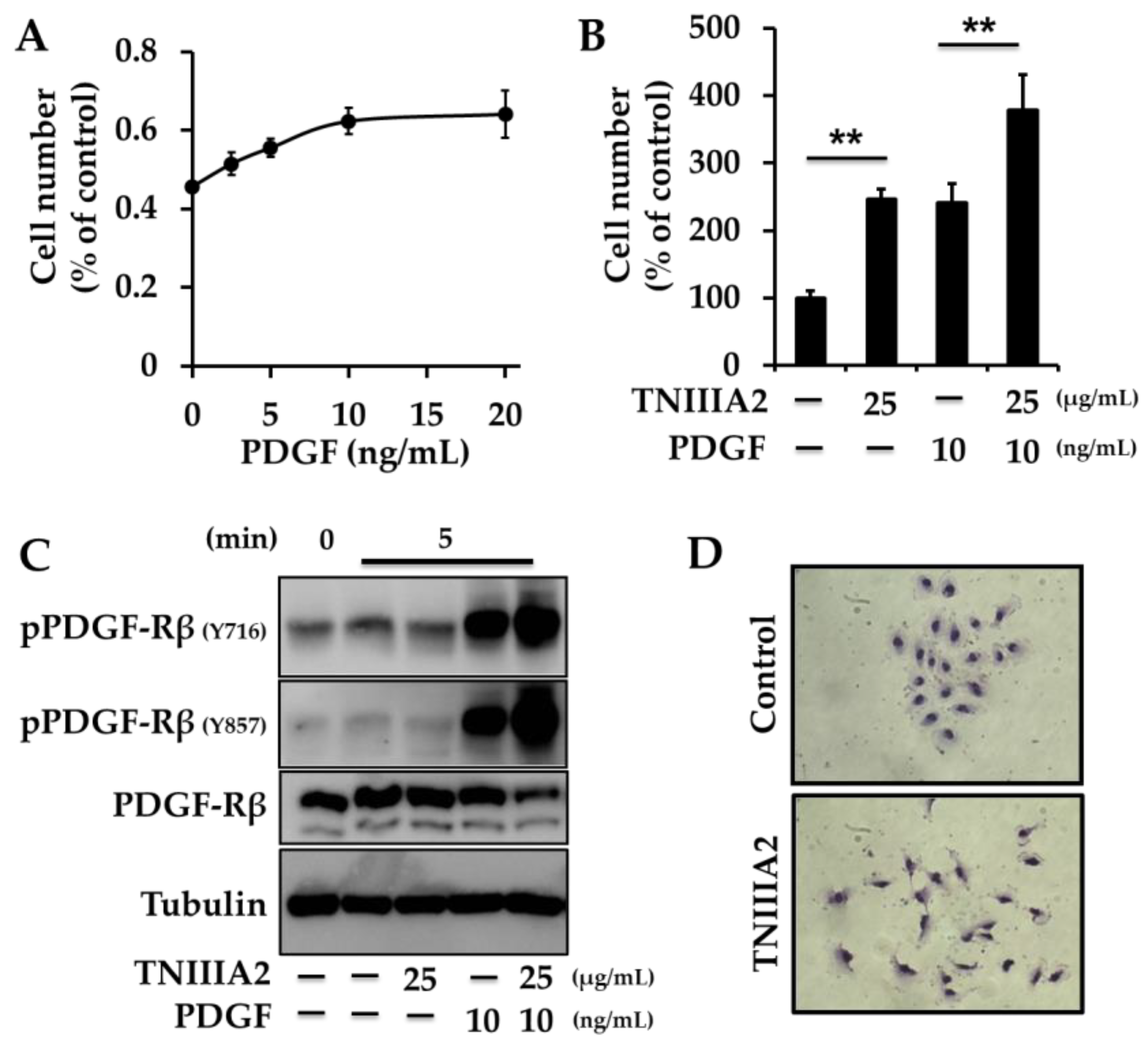
- Tanaka, R.; Seki, Y.; Saito, Y.; Kamiya, S.; Fujita, M.; Okutsu, H.; Iyoda, T.; Takai, T.; Owaki, T.; Yajima, H.; et al. Tenascin-C-derived peptide TNIIIA2 highly enhances cell survival and platelet-derived growth factor (PDGF)-dependent cell proliferation through potentiated and sustained activation of integrin α5β1. J. Biol. Chem. 2014, 289, 17699–17708. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Mirzaei, R.; Sarkar, S.; Dzikowski, L.; Rawji, K.S.; Khan, L.; Faissner, A.; Bose, P.; Yong, V.W. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oncoimmunology 2018, 7, e1478647. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Mirzaei, R.; Zemp, F.J.; Wei, W.; Senger, D.L.; Robbins, S.M.; Yong, V.W. Activation of NOTCH Signaling by Tenascin-C Promotes Growth of Human Brain Tumor-Initiating Cells. Cancer Res. 2017, 77, 3231–3243. [Google Scholar] [CrossRef] [Green Version]
- Hirata, E.; Arakawa, Y.; Shirahata, M.; Yamaguchi, M.; Kishi, Y.; Okada, T.; Takahashi, J.A.; Matsuda, M.; Hashimoto, N. Endogenous tenascin-C enhances glioblastoma invasion with reactive change of surrounding brain tissue. Cancer Sci. 2009, 100, 1451–1459. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Zemp, F.J.; Senger, D.; Robbins, S.M.; Yong, V.W. ADAM-9 is a novel mediator of tenascin-C-stimulated invasiveness of brain tumor-initiating cells. Neuro-Oncol. 2015, 17, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Rupp, T.; Langlois, B.; Koczorowska, M.M.; Radwanska, A.; Sun, Z.; Hussenet, T.; Lefebvre, O.; Murdamoothoo, D.; Arnold, C.; Klein, A.; et al. Tenascin-C Orchestrates Glioblastoma Angiogenesis by Modulation of Pro- and Anti-angiogenic Signaling. Cell Rep. 2016, 17, 2607–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.-Y.; Cheng, Y.-J.; Lin, Y.-P.; Lin, H.-C.; Su, C.-C.; Juliano, R.; Yang, B.-C. Extracellular matrix of glioblastoma inhibits polarization and transmigration of T cells: The role of tenascin-C in immune suppression. J. Immunol. Baltim. Md 1950 2010, 185, 1450–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarenko, I.; Hede, S.-M.; He, X.; Hedrén, A.; Thompson, J.; Lindström, M.S.; Nistér, M. PDGF and PDGF receptors in glioma. Ups. J. Med. Sci. 2012, 117, 99–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermark, B. Platelet-derived growth factor in glioblastoma-driver or biomarker? Ups. J. Med. Sci. 2014, 119, 298–305. [Google Scholar] [CrossRef]
- Guha, A.; Dashner, K.; Black, P.M.; Wagner, J.A.; Stiles, C.D. Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int. J. Cancer 1995, 60, 168–173. [Google Scholar] [CrossRef]
- Jiang, Y.; Boije, M.; Westermark, B.; Uhrbom, L. PDGF-B Can sustain self-renewal and tumorigenicity of experimental glioma-derived cancer-initiating cells by preventing oligodendrocyte differentiation. Neoplasia 2011, 13, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, T.; Mao, Q.; Lin, J.; Jia, J.; Li, S.; Xiong, W.; Lin, Y.; Liu, Z.; Liu, X.; et al. PDGFR-β-activated ACK1-AKT signaling promotes glioma tumorigenesis. Int. J. Cancer 2015, 136, 1769–1780. [Google Scholar] [CrossRef]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef]
- DeLay, M.; Jahangiri, A.; Carbonell, W.S.; Hu, Y.-L.; Tsao, S.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Aghi, M.K. Microarray analysis verifies two distinct phenotypes of glioblastomas resistant to antiangiogenic therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2930–2942. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Yamamoto, T.; Iyoda, T.; Fujisawa, T.; Sasada, M.; Nagai, R.; Kudo, C.; Otsuka, K.; Kamiya, S.; Kodama, H.; et al. Aggressive Progression in Glioblastoma Cells through Potentiated Activation of Integrin α5β1 by the Tenascin-C-Derived Peptide TNIIIA2. Mol. Cancer Ther. 2019, 18, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Yamamoto, T.; Iyoda, T.; Fujisawa, T.; Nagai, R.; Kudo, C.; Sasada, M.; Kodama, H.; Fukai, F. Autocrine Production of PDGF Stimulated by the Tenascin-C-Derived Peptide TNIIIA2 Induces Hyper-Proliferation in Glioblastoma Cells. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jess, T.; Loftus, E.V.; Velayos, F.S.; Harmsen, W.S.; Zinsmeister, A.R.; Smyrk, T.C.; Schleck, C.D.; Tremaine, W.J.; Melton, L.J.; Munkholm, P.; et al. Risk of intestinal cancer in inflammatory bowel disease: A population-based study from olmsted county, Minnesota. Gastroenterology 2006, 130, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Zisman, T.L.; Rubin, D.T. Colorectal cancer and dysplasia in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 2662–2669. [Google Scholar] [CrossRef]
- Yaeger, R.; Paroder, V.; Bates, D.; Capanu, M.; Chou, J.; Tang, L.; Chatila, W.; Schultz, N.; Hersch, J.; Kelsen, D. Systemic Chemotherapy for Metastatic Colitis-Associated Cancer has a Worse Outcome than for Sporadic Colorectal Cancer: A Matched Case Cohort Analysis. Clin. Colorectal Cancer 2020. [Google Scholar] [CrossRef]
- Geboes, K.; El-Zine, M.Y.; Dalle, I.; El-Haddad, S.; Rutgeerts, P.; Van Eyken, P. Tenascin and strictures in inflammatory bowel disease: An immunohistochemical study. Int. J. Surg. Pathol. 2001, 9, 281–286. [Google Scholar] [CrossRef]
- Kawamura, T.; Yamamoto, M.; Suzuki, K.; Suzuki, Y.; Kamishima, M.; Sakata, M.; Kurachi, K.; Setoh, M.; Konno, H.; Takeuchi, H. Tenascin-C Produced by Intestinal Myofibroblasts Promotes Colitis-associated Cancer Development Through Angiogenesis. Inflamm. Bowel Dis. 2019, 25, 732–741. [Google Scholar] [CrossRef]
- Fujita, M.; Ito-Fujita, Y.; Iyoda, T.; Sasada, M.; Okada, Y.; Ishibashi, K.; Osawa, T.; Kodama, H.; Fukai, F.; Suzuki, H. Peptide TNIIIA2 Derived from Tenascin-C Contributes to Malignant Progression in Colitis-Associated Colorectal Cancer via β1-Integrin Activation in Fibroblasts. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [Green Version]
- Farrar, C.S.; Rouin, G.T.; Miller, B.L.; Raeman, C.H.; Mooney, N.A.; Hocking, D.C. A Matricryptic Conformation of the Integrin-Binding Domain of Fibronectin Regulates Platelet-Derived Growth Factor-Induced Intracellular Calcium Release. Cells 2019, 8. [Google Scholar] [CrossRef]
- Tanaka, R.; Owaki, T.; Kamiya, S.; Matsunaga, T.; Shimoda, K.; Kodama, H.; Hayashi, R.; Abe, T.; Harada, Y.P.; Shimonaka, M.; et al. VLA-5-mediated adhesion to fibronectin accelerates hemin-stimulated erythroid differentiation of K562 cells through induction of VLA-4 expression. J. Biol. Chem. 2009, 284, 19817–19825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ejiri, S. Moonlighting functions of polypeptide elongation factor 1: From actin bundling to zinc finger protein R1-associated nuclear localization. Biosci. Biotechnol. Biochem. 2002, 66, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Sasada, M.; Iyoda, T.; Asayama, T.; Suenaga, Y.; Sakai, S.; Kase, N.; Kodama, H.; Yokoi, S.; Isohama, Y.; Fukai, F. Inactivation of beta1 integrin induces proteasomal degradation of Myc oncoproteins. Oncotarget 2019, 10, 4960–4972. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Fukai, F.; Miura, S.; Nakane, Y.; Owaki, T.; Kodama, H.; Tanaka, M.; Nagaya, T.; Takimoto, R.; Takayama, T.; et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia 2008, 22, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, R.; Ishikawa, T.; Kamiya, S.; Oguma, F.; Ueki, M.; Goto, S.; Nakamura, H.; Katayama, T.; Fukai, F. A new type of antimetastatic peptide derived from fibronectin. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 2455–2462. [Google Scholar]
- Iyoda, T.; Nagamine, Y.; Nakane, Y.; Tokita, Y.; Akari, S.; Otsuka, K.; Fujita, M.; Itagaki, K.; Takizawa, Y.-I.; Orita, H.; et al. Coadministration of the FNIII14 Peptide Synergistically Augments the Anti-Cancer Activity of Chemotherapeutic Drugs by Activating Pro-Apoptotic Bim. PLoS ONE 2016, 11, e0162525. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Nakayama, H.; Nagata, M.; Yoshida, R.; Kawahara, K.; Hirosue, A.; Tanaka, T.; Yuno, A.; Matsuoka, Y.; Kojima, T.; et al. Overexpression of fibronectin confers cell adhesion-mediated drug resistance (CAM-DR) against 5-FU in oral squamous cell carcinoma cells. Int. J. Oncol. 2014, 44, 1376–1384. [Google Scholar] [CrossRef] [Green Version]
- Janouskova, H.; Maglott, A.; Leger, D.Y.; Bossert, C.; Noulet, F.; Guerin, E.; Guenot, D.; Pinel, S.; Chastagner, P.; Plenat, F.; et al. Integrin α5β1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Cancer Res. 2012, 72, 3463–3470. [Google Scholar] [CrossRef] [Green Version]
- Ellert-Miklaszewska, A.; Poleszak, K.; Pasierbinska, M.; Kaminska, B. Integrin Signaling in Glioma Pathogenesis: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [Green Version]
- Malric, L.; Monferran, S.; Gilhodes, J.; Boyrie, S.; Dahan, P.; Skuli, N.; Sesen, J.; Filleron, T.; Kowalski-Chauvel, A.; Cohen-Jonathan Moyal, E.; et al. Interest of integrins targeting in glioblastoma according to tumor heterogeneity and cancer stem cell paradigm: An update. Oncotarget 2017, 8, 86947–86968. [Google Scholar] [CrossRef] [Green Version]
- Arun, A.S.; Tepper, C.G.; Lam, K.S. Identification of integrin drug targets for 17 solid tumor types. Oncotarget 2018, 9, 30146–30162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandin, A.-F.; Noulet, F.; Renner, G.; Mercier, M.-C.; Choulier, L.; Vauchelles, R.; Ronde, P.; Carreiras, F.; Etienne-Selloum, N.; Vereb, G.; et al. Glioma cell dispersion is driven by α5 integrin-mediated cell-matrix and cell-cell interactions. Cancer Lett. 2016, 376, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinkova, E.; Maglott, A.; Leger, D.Y.; Bonnet, D.; Stiborova, M.; Takeda, K.; Martin, S.; Dontenwill, M. alpha5beta1 integrin antagonists reduce chemotherapy-induced premature senescence and facilitate apoptosis in human glioblastoma cells. Int. J. Cancer 2010, 127, 1240–1248. [Google Scholar] [CrossRef]
- Carbonell, W.S.; DeLay, M.; Jahangiri, A.; Park, C.C.; Aghi, M.K. β1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013, 73, 3145–3154. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Rodansky, E.S.; Sauder, K.L.; Horowitz, J.C.; Mih, J.D.; Tschumperlin, D.J.; Higgins, P.D. Matrix stiffness corresponding to strictured bowel induces a fibrogenic response in human colonic fibroblasts. Inflamm. Bowel Dis. 2013, 19, 891–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terasaki, M.; Ikuta, M.; Kojima, H.; Tanaka, T.; Maeda, H.; Miyashita, K.; Mutoh, M. Dietary Fucoxanthin Induces Anoikis in Colorectal Adenocarcinoma by Suppressing Integrin Signaling in a Murine Colorectal Cancer Model. J. Clin. Med. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, K.; Harimoto, N.; Yokobori, T.; Muranushi, R.; Hoshino, K.; Gantumur, D.; Yamanaka, T.; Ishii, N.; Tsukagoshi, M.; Igarashi, T.; et al. High Co-expression of Large Tenascin C Splice Variants in Stromal Tissue and Annexin A2 in Cancer Cell Membranes is Associated with Poor Prognosis in Pancreatic Cancer. Ann. Surg. Oncol. 2020, 27, 924–930. [Google Scholar] [CrossRef]
- Mai, J.; Sameni, M.; Mikkelsen, T.; Sloane, B.F. Degradation of extracellular matrix protein tenascin-C by cathepsin B: An interaction involved in the progression of gliomas. Biol. Chem. 2002, 383, 1407–1413. [Google Scholar] [CrossRef]
- Cai, M.; Onoda, K.; Takao, M.; Kyoko, I.-Y.; Shimpo, H.; Yoshida, T.; Yada, I. Degradation of tenascin-C and activity of matrix metalloproteinase-2 are associated with tumor recurrence in early stage non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 1152–1156. [Google Scholar]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell. Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.M.; Kaushik, S.; Bainer, R.O.; Sa, J.K.; Woods, E.C.; Kai, F.; Przybyla, L.; Lee, M.; Lee, H.W.; Tung, J.C.; et al. A tension-mediated glycocalyx-integrin feedback loop promotes mesenchymal-like glioblastoma. Nat. Cell Biol. 2018, 20, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Nabors, L.B.; Fink, K.L.; Mikkelsen, T.; Grujicic, D.; Tarnawski, R.; Nam, D.H.; Mazurkiewicz, M.; Salacz, M.; Ashby, L.; Zagonel, V.; et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: Results of the open-label, controlled, randomized phase II CORE study. Neuro-Oncol. 2015, 17, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Neyns, B.; Goldbrunner, R.; Schlegel, U.; Clement, P.M.J.; Grabenbauer, G.G.; Ochsenbein, A.F.; Simon, M.; Dietrich, P.-Y.; et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2712–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Fink, K.L.; Mikkelsen, T.; Cloughesy, T.F.; O’Neill, A.; Plotkin, S.; Glantz, M.; Ravin, P.; Raizer, J.J.; Rich, K.M.; et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5610–5617. [Google Scholar] [CrossRef]
- Nabors, L.B.; Mikkelsen, T.; Hegi, M.E.; Ye, X.; Batchelor, T.; Lesser, G.; Peereboom, D.; Rosenfeld, M.R.; Olsen, J.; Brem, S.; et al. A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306). Cancer 2012, 118, 5601–5607. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Guigay, J.; Mesia, R.; Trigo, J.M.; Keilholz, U.; Kerber, A.; Bethe, U.; Picard, M.; Brummendorf, T.H. Phase I/II trial of cilengitide with cetuximab, cisplatin and 5-fluorouracil in recurrent and/or metastatic squamous cell cancer of the head and neck: Findings of the phase I part. Br. J. Cancer 2011, 104, 1691–1696. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Peyrade, F.; Krauss, J.; Mesía, R.; Remenar, E.; Gauler, T.C.; Keilholz, U.; Delord, J.P.; Schafhausen, P.; Erfán, J.; et al. Cisplatin, 5-fluorouracil, and cetuximab (PFE) with or without cilengitide in recurrent/metastatic squamous cell carcinoma of the head and neck: Results of the randomized phase I/II ADVANTAGE trial (phase II part). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014, 25, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.; Barlesi, F.; Waller, C.F.; Bennouna, J.; Gridelli, C.; Goekkurt, E.; Verhoeven, D.; Szczesna, A.; Feurer, M.; Milanowski, J.; et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (NSCLC) patients: Results of an open-label, randomized, controlled phase II study (CERTO). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Alva, A.; Slovin, S.; Daignault, S.; Carducci, M.; Dipaola, R.; Pienta, K.; Agus, D.; Cooney, K.; Chen, A.; Smith, D.C.; et al. Phase II study of cilengitide (EMD 121974, NSC 707544) in patients with non-metastatic castration resistant prostate cancer, NCI-6735. A study by the DOD/PCF prostate cancer clinical trials consortium. Invest. New Drugs 2012, 30, 749–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, D.A.; Daignault, S.; Ryan, C.J.; Dipaola, R.S.; Cooney, K.A.; Smith, D.C.; Small, E.; Mathew, P.; Gross, M.E.; Stein, M.N.; et al. Cilengitide (EMD 121974, NSC 707544) in asymptomatic metastatic castration resistant prostate cancer patients: A randomized phase II trial by the prostate cancer clinical trials consortium. Invest. New Drugs 2011, 29, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Khalili, P.; Arakelian, A.; Chen, G.; Plunkett, M.L.; Beck, I.; Parry, G.C.; Doñate, F.; Shaw, D.E.; Mazar, A.P.; Rabbani, S.A. A non-RGD-based integrin binding peptide (ATN-161) blocks breast cancer growth and metastasis in vivo. Mol. Cancer Ther. 2006, 5, 2271–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoeltzing, O.; Liu, W.; Reinmuth, N.; Fan, F.; Parry, G.C.; Parikh, A.A.; McCarty, M.F.; Bucana, C.D.; Mazar, A.P.; Ellis, L.M. Inhibition of integrin alpha5beta1 function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int. J. Cancer 2003, 104, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, M.E.; Kimmel, K.A.; Gallo, J.; Cardoso, T.; Brown, M.M.; Hudes, G.; Lewis, N.; Weiner, L.; Lam, G.N.; Brown, S.C.; et al. Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH(2)), a beta integrin antagonist, in patients with solid tumours. Br. J. Cancer 2006, 94, 1621–1626. [Google Scholar] [CrossRef] [Green Version]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta BBA - Mol. Cell Res. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parental Molecule | Sequence | Function (In Vitro and In Vivo Settings) | Ref. |
|---|---|---|---|
| Tenascin-C | VFDNFVLK | Neurite outgrowth | [18] |
| VSWRAPTA | Glioma cell migration, neuronal branching | [19] | |
| PLAEIDGIELTY | Cell adhesion, binding to integrin α9β1 | [20] | |
| VSGNTVEYALPTLE | Fibroblast proliferation | [21] | |
| LDSPTAPTVQSTALTWRP | Fibroblast and endothelial cell proliferation | [21] | |
| WYRNCHRVNLMGRYGDNNHSQGVNWFHWKG | Cell adhesion, binding to integrin αvβ3 | [22] | |
| RSTDLPGLKAATHYTITIRGVC (TNIIIA2) | Cell adhesion, integrin activation Enhancement of lung metastatic nodule formation in a mouse model of metastasis of colon cancer cells | [23] [24] | |
| Fibronectin | RGD | Cell adhesion Antiangiogenic effect in vivo | [25] [26] |
| LDV | Cell adhesion, binding to integrin α4β1 | [27] | |
| PHSRN | Synergistic interactions between integrin α5β1 and RGD Antiangiogenic effect in vivo | [28] [29] | |
| REDV | Binding to integrin α4β1 | [30] | |
| SLLISWD | Fibronectin fibril assembly | [31] | |
| KLDAPT | Binding to integrin α4β1 and α4β7 | [32] | |
| EDGIHEL | Binding to integrin α4β1 and α9β1 | [33] | |
| IDAPS | Binding to integrin α4β1 | [34] | |
| ALNGR | Cell adhesion, binding to β1-integrn | [35] | |
| WQPPRARI | Cell adhesion, binding to heparin | [36] | |
| SRNRCNDQ | Plasminogen activation | [37] | |
| KNEED | Cell adhesion, cell-recognition site | [38] | |
| RWRPKNSVGR | Cell spreading, cell growth, vasodilation | [39] | |
| PSHISKYILRWRPK | Binding to PDGF-BB, cell survival | [40] | |
| YEKPGSPPREVVPRPRPGV | Cell adhesion, heparin-binding region | [41] | |
| KNNQKSEPLIGRKKT | Heparin-binding region, neurite outgrowth | [41] | |
| YRVRVTPKEKTGPMKE | Cell adhesion, heparin-binding region | [41] | |
| AHEEICTTNEGVM | Matrix assembly, cell migration | [42] | |
| ETTIVITWTPAPR | Cell adhesion, binding to MIA protein Reduction of the size of lung nodules in a mouse model of melanoma metastasis | [43] [44] | |
| TSLLISWDAPAVT | Cell adhesion, binding to MIA protein | [43] | |
| NSLLVSWQPPRAR | Cell adhesion, binding to MIA protein | [43] | |
| GTQSTAIPAPTD | Cell adhesion, binding to MIA protein Reduction of the size of lung nodules in a mouse model of melanoma metastasis | [44] | |
| PRARIY | Cell adhesion, neuroprotective effect | [45] | |
| NVSPPRRARVTDATETTITISW | Binding to heparin | [46] | |
| VTEATITGLEPGTEYTIY | Binding to DPPIV Reduction of lung colonization in a mouse model of metastasis | [47] | |
| TEATITGLEPGTEYTIYVIAL (FNIII14) | Cell adhesion, integrin inactivation Antitumor effects in vivo (Table 2) | [48] |
| Cancer Type | Cell Type/Animal Model | Phenotypic Effects | Ref. |
|---|---|---|---|
| Glioma | T98G, 9L cells | Suppression of cell survival/proliferation | [91,92] |
| /Glioblastoma | T98G | Suppression of disseminative migration | [91] |
| T98G, 9L cells | Potentiation of temozolomide (TMZ) cytotoxicity | [91] | |
| T98G cells | Downregulation of O6–methylguanine–DNA methyltransferase (MGMT) levels | [91] | |
| Mouse subcutaneous xenograft (9L cells) | Suppression of tumor growth as monotherapy | [91] | |
| Mouse subcutaneous xenograft (9L cells) | Potentiation of TMZ action | [91] | |
| Neuroblastoma | IMR-32, NB-1, KELLY cells | Downregulation of N-myc levels by proteasomal degradation | [103] |
| IMR-32 cells | Suppression of cell survival/proliferation | [103] | |
| Mouse subcutaneous xenograft (IMR-32 cells) | Suppression of tumor growth as monotherapy | [103] | |
| Colitis-associated colorectal cancer (CAC) | Azoxymethane–dextran sodium sulfate (AOM-DSS) mouse model | Suppression of polyp development as monotherapy | [98] |
| Acute myelogenous leukemia (AML) | U937, HL-60, Fresh leukemic cells from AML patients | Disruption of cell adhesion-mediated drug resistance (CAM-DR) to cytosine arabinoside (Ara C) | [104] |
| Mouse model of minimal residual disease (MRD) in AML (U937 cells) | Eradication of bone marrow MRD in mice transplanted with U937 cells and improvement of survival mouse treated with Ara C | [104] | |
| Lymphoma | L5178Y-ML25 cells | Inhibition of cell migration | [105] |
| Mouse model of experimental tumor metastasis (L5178Y-ML25 cells) | Inhibition of the liver and spleen metastases as monotherapy | [105] | |
| Mammary tumor | 4T1 cells | Potentiation of doxorubicin (Dox) cytotoxicity | [106] |
| Mouse model of experimental tumor metastasis (4T1 cells) | Inhibition of the liver metastases when coadministered with Dox | [106] | |
| Melanoma | B16BL6 cells | Increasing chemosensitivity of antitumor drugs (e.g., Aclarubicin, Vinblastine, 5-Fluorouracil (5-FU)) | [106] |
| Oral squamous cell carcinoma (OSCC) | Ca9-22/FR2 cells | Potentiation of 5-FU cytotoxicity | [107] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujita, M.; Sasada, M.; Iyoda, T.; Fukai, F. Involvement of Integrin-Activating Peptides Derived from Tenascin-C in Cancer Aggression and New Anticancer Strategy Using the Fibronectin-Derived Integrin-Inactivating Peptide. Molecules 2020, 25, 3239. https://doi.org/10.3390/molecules25143239
Fujita M, Sasada M, Iyoda T, Fukai F. Involvement of Integrin-Activating Peptides Derived from Tenascin-C in Cancer Aggression and New Anticancer Strategy Using the Fibronectin-Derived Integrin-Inactivating Peptide. Molecules. 2020; 25(14):3239. https://doi.org/10.3390/molecules25143239
Chicago/Turabian StyleFujita, Motomichi, Manabu Sasada, Takuya Iyoda, and Fumio Fukai. 2020. "Involvement of Integrin-Activating Peptides Derived from Tenascin-C in Cancer Aggression and New Anticancer Strategy Using the Fibronectin-Derived Integrin-Inactivating Peptide" Molecules 25, no. 14: 3239. https://doi.org/10.3390/molecules25143239
APA StyleFujita, M., Sasada, M., Iyoda, T., & Fukai, F. (2020). Involvement of Integrin-Activating Peptides Derived from Tenascin-C in Cancer Aggression and New Anticancer Strategy Using the Fibronectin-Derived Integrin-Inactivating Peptide. Molecules, 25(14), 3239. https://doi.org/10.3390/molecules25143239