3.1.1. General
Chemicals, reagents, and solvents were purchased from commercial suppliers (Sigma-Aldrich Corporation, St. Louis, MO, USA; TCI, Tokyo, Japan and Alfa Aesar, Haverhill, MA, USA) and used without further purification. Melting points (Mp) were determined on an SRS Optimelt digital apparatus (Stanford Research Systems Inc., Sunnyvale, CA, USA) and are uncorrected. For MW-assisted syntheses, a CEM Discover SP (CEM Corporation, Matthews, NC, USA) laboratory MW reactor was used with a maximum power of 200 W (running a dynamic control program). Elementary analysis data were obtained with a PerkinElmer CHN analyzer model 2400 (PerkinElmer Inc, Waltham, MA, USA). The transformations were monitored by TLC using 0.25 mm thick Kieselgel-G plates (Si 254 F, Merck KGaA, Darmstadt, Germany). The compound spots were detected by spraying with 5% phosphomolybdic acid in 50% aqueous phosphoric acid. Flash chromatographic purifications were carried out on silica gel 60, 40–63 μm (Merck KGaA, Darmstadt, Germany). NMR spectra were recorded with a Bruker DRX 500 (Bruker, Billerica, MA, USA) instrument at room temperature in CDCl3 or DMSO-d6 using residual solvent signals as an internal reference. Chemical shifts are reported in ppm (δ scale), and coupling constants (J) are given in Hz. Multiplicities of the 1H signals are indicated as a singlet (s), a broad singlet (bs), a doublet (d), a triplet (t), or a multiplet (m). 13C NMR spectra are 1H-decoupled and the J-MOD pulse sequence was used for multiplicity editing. In this spin-echo type experiment, the signal intensity is modulated by the different coupling constants J of carbons depending on the number of attached protons. Both protonated and unprotonated carbons can be detected (CH3 and CH carbons appear as positive signals, while CH2 and C carbons as negative signals).
Automated flow injection analyses were performed with an HPLC/MSD system. System accessories: a micro-well plate autoinjector, an Agilent 1100 micro vacuum degasser (Agilent Technologies, Santa Clara, CA, USA), a quaternary pump, and a 1946A MSD equipped with an electrospray ion source (ESI) operated in positive ion mode. ESI parameters were: nebulizing gas N2, at 35 psi; drying gas N2, at 350 °C and 12 L/min; capillary voltage 3000 V; fragmentor voltage 70 V. The MSD was operated with a mass range of m/z 60−620 in scan mode. Samples (0.2 μL) were injected directly into the solvent flow (0.3 mL/min) of acetonitrile/H2O = 70:30 (v/v) with the simultaneous addition of 0.1% formic acid with an automated needle wash. Agilent LC/MSD Chemstation (C.01.08, Agilent Technologies Inc., Santa Clara, CA, USA) was used as software to control the system.
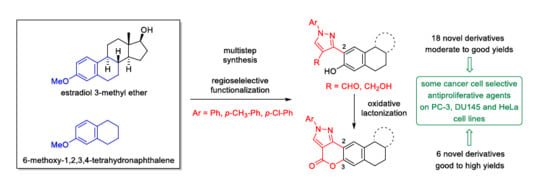
3.1.2. General Procedure for the Friedel-Crafts Acetylation/Demethylation of Estradiol 3-Methyl Ether (2) or 6-Methoxy-1,2,3,4-Tetrahydronaphthalene (14)
Anhydrous AlCl3 (10.5 g, 78.6 mmol) was suspended in dry CH2Cl2 (75 mL) under N2 atmosphere, and the mixture was cooled to 0 °C. Acetyl chloride (3.1 mL, 43.6 mmol) was added slowly, then the mixture was stirred for 15 min. A solution of 2 (5 g, 17.5 mmol) or 14 (2.6g, 17.6 mmol) in dry CH2Cl2 (25 mL) was added dropwise to the mixture over a period of 10 min, then stirred for 15 min at 0 °C, after which it was allowed to warm to room temperature and stirred for another 3 h. The reaction mixture was poured onto crushed ice and stirred vigorously for 10 min. The organic layer was separated, and the remaining aqueous layer was extracted with CH2Cl2 (2 × 50 mL). The combined organic layers were washed with brine, dried with anhydrous Na2SO4, and concentrated in vacuo.
2-Acetyl-estra-1,3,5(10)-triene-3,17β-diol-17-acetate (
4). The crude product was recrystallized from MeOH. Yield: 4.95 g (80%, white solid); Mp 200–202 °C (198–200 °C [
37]); Anal. Calcd. for C
22H
28O
4 (356.46) C, 74.13; H, 7.92; O, 17.95. Found C 74.25; H 7.86.
1H NMR (CDCl
3, 500 MHz):
δ 0.84 (s, 3H, 18-CH
3), 1.27–1.58 (overlapping m, 7H), 1.76 (m, 1H), 1.87–1.92 (overlapping m, 2H), 2.07 (s, 3H, AcO-CH
3), 2.15–2.32 (overlapping m, 3H), 2.60 (s, 3H, 2-Ac-CH
3), 2.87 (m, 2H, 6-H
2), 4.70 (t, 1H,
J = 8.5 Hz, 17-H), 6.69 (s, 1H, 4-H), 7.59 (s, 1H, 1-H), 12.04 (s, 1H, OH);
13C NMR (CDCl
3, 125 MHz):
δ 12.2 (C-18), 21.4 (AcO-CH
3), 23.4 (CH
2), 26.3 (CH
2), 26.7 (2-Ac-CH
3), 26.9 (CH
2), 27.7 (CH
2), 30.0 (CH
2), 36.8 (CH
2), 38.4 (CH), 43.0 (C-13), 43.5 (CH), 49.9 (CH), 82.7 (C-17), 117.7 (C-4), 118.0 (C-2), 127.4 (C-1), 131.5 (C-10), 147.3 (C-5), 160.2 (C-3), 171.4 (AcO-CO), 204.2 (2-Ac-CO); ESI-MS 357 [M + H]
+.
3-Acetyl-5,6,7,8-tetrahydro-2-naphthol (
15). The crude product was purified by column chromatography with hexane/CH
2Cl
2 = 1:1. Yield: 2.29 g (69%, white solid); Mp 70−72 °C (71−72 °C [
42]); Anal. Calcd. for C
12H
14O
2 (190.24) C 75.76; H 7.42. Found C 75.62; H 7.35.
1H NMR (CDCl
3, 500 MHz):
δ 1.78 (t-like m, 4H, 6-H
2 and 7-H
2), 2.59 (s, 3H, Ac-CH
3), 2.71 (m, 2H) and 2.76 (m, 2H): 5-H
2 and 8-H
2, 6.67 (s, 1H, 1-H), 7.40 (s, 1H, 4-H), 11.97 (s, 1H, OH);
13C NMR (CDCl
3, 125 MHz):
δ 22.8 and 23.3: C-6 and C-7, 26.7 (Ac-CH
3), 28.7 and 30.1: C-5 and C-8, 117.7 (C-1), 118.1 (C-3), 128.0 (C-4a), 131.0 (C-4), 147.6 (C-8a), 160.0 (C-2), 204.1 (Ac-CO); ESI-MS 191 [M + H]
+.
3.1.3. General Procedure for the MW-Assisted Synthesis of Arylhydrazones (6a–c) and Their Conversion to Steroidal 4-Formylpyrazoles (7a–c) under Vilsmeier–Haack Conditions
To a solution of 4 (713 mg, 2.0 mmol) or 15 (380 mg, 2.0 mmol) in EtOH (5 mL), anhydrous NaOAc (246 mg, 3.0 mmol) and (p-substituted) phenylhydrazine hydrochloride (3.0 mmol) were added, and the mixture was irradiated at 100 °C for 30 min in a closed tube. Hydrazones 6a–c can be obtained by chilling the reaction vessel, filtering off the yellow precipitate and washing it with ice-cold methanol. 16a–c could not be isolated; thus, the reaction mixture was evaporated to dryness and was used as-is in the next step. POCl3 (10.73 mmol, 1.0 mL) was added to DMF (10 mL) cooled to 0 °C. After stirring for 15 min, a solution of 6a–c or 16a–c (obtained in the previous step) in DMF (5 mL) was added dropwise to the reaction mixture and was then heated to 60 °C and stirred for another 3 h (7a–c) or 16 h (17a–c). The mixture was then poured onto crushed ice, extracted with EtOAc (3 × 25 mL), and the combined organic phases were washed with brine, dried with anhydrous Na2SO4, and concentrated in vacuo. The crude products were purified by column chromatography with EtOAc/CH2Cl2 = 1:99 (7a–c) or hexane/CH2Cl2 = 2:8 (17a–c).
2-(4′-Formyl-1′-phenyl-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-triene-3,17β-diol-17-acetate (7a)
According to the general procedure, phenylhydrazine hydrochloride (435 mg) was used. Yield of the phenylhydrazone (6a, yellow solid): 678 mg (76%). Yield of 7a (yellow solid): 550 mg (75%). Mp 230−232 °C; Anal. Calcd. for C30H32N2O4 (484.60) C 74.36; H 6.66. Found C 74.48; H 6.52. 1H NMR (CDCl3, 500 MHz): δ 0.85 (s, 3H, 18-CH3), 1.26–1.61 (overlapping m, 7H), 1.76 (m, 1H), 1.92 (m, 2H), 2.07 (s, 3H, Ac-CH3), 2.20–2.30 (overlapping m, 2H), 2.39 (m, 1H), 2.90 (m, 2H, 6-H2), 4.69 (t, 1H, J = 8.4 Hz, 17-H), 6.83 (s, 1H, 4-H), 7.43 (t-like m, 1H, 4″-H), 7.54 (t-like m, 2H, 3″-H and 5″-H), 7.88 (d, 2H, J = 7.9 Hz, 2″-H and 6″-H), 7.88 (s, 1H, 1-H), 8.56 (s, 1H, 5′-H), 9.84 (bs, 1H, 3-OH), 10.19 (s, 1H, CHO); 13C NMR (CDCl3, 125 MHz): δ 12.2 (C-18), 21.4 (Ac-CH3), 23.4 (CH2), 26.5 (CH2), 27.3 (CH2), 27.8 (CH2), 29.7 (CH2), 36.9 (CH2), 38.6 (CH), 43.0 (C-13), 43.9 (CH), 49.9 (CH), 82.9 (C-17), 113.2 (C-4′), 117.1 (C-4), 119.7 (2C, C-2″ and C-6″), 123.1 (C-2), 126.9 (C-1), 128.5 (C-4″), 130.0 (2C, C-3″ and C-5″), 132.2 (C-10), 132.7 (C-5′), 138.4 (C-1″), 140.7 (C-5), 153.2 (C-3′), 153.8 (C-3), 171.4 (Ac-CO), 184.5 (CHO); ESI-MS 485 [M + H]+.
2-(4′-Formyl-1′-(4″-tolyl)-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-triene-3,17β-diol-17-acetate (7b)
According to the general procedure, 4-tolylhydrazine hydrochloride (475 mg) was used. Yield of the 4-tolylhydrazone (6b, yellow solid): 717 mg (78%). Yield of 7b (white solid): 607 mg (78%). Mp 229−231 °C; Anal. Calcd. for C31H34N2O4 (498.62) C 74.67; H 6.87. Found C 74.53; H 6.99. 1H NMR (CDCl3, 500 MHz): δ 0.85 (s, 3H, 18-CH3), 1.27–1.59 (overlapping m, 7H), 1.76 (m, 1H), 1.91 (m, 2H), 2.07 (s, 3H, Ac-CH3), 2.20–2.29 (overlapping m, 2H), 2.39 (m, 1H), 2.43 (s, 3H, 4″-CH3), 2.90 (m, 2H, 6-H2), 4.69 (t, 1H, J = 8.4 Hz, 17-H), 6.82 (s, 1H, 4-H), 7.32 (d, 2H, J = 8.4 Hz, 3″-H and 5″-H), 7.59 (d, 2H, J = 8.8 Hz, 2″-H and 6″-H), 7.88 (s, 1H, 1-H), 8.51 (s, 1H, 5′-H), 9.89 (bs, 1H, 3-OH), 10.17 (s, 1H, CHO); 13C NMR (CDCl3, 125 MHz): δ 12.2 (C-18), 21.2 (4″-CH3), 21.4 (Ac-CH3), 23.4 (CH2), 26.4 (CH2), 27.3 (CH2), 27.8 (CH2), 29.7 (CH2), 36.9 (CH2), 38.6 (CH), 43.0 (C-13), 43.9 (CH), 49.9 (CH), 82.9 (C-17), 113.2 (C-4′), 117.1 (C-4), 119.6 (2C, C-2″ and C-6″), 122.9 (C-2), 126.9 (C-1), 130.5 (2C, C-3″ and C-5″), 132.1 (C-10), 132.6 (C-5′), 136.1 (C-4″), 138.6 (C-1″), 140.6 (C-5), 153.0 (C-3′), 153.8 (C-3), 171.4 (Ac-CO), 184.5 (CHO); ESI-MS 499 [M + H]+.
2-(1′-(4″-Chlorophenyl)-4′-formyl-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-triene-3,17β-diol-17-acetate (7c)
According to the general procedure, 4-chlorophenylhydrazine hydrochloride (539 mg) was used. Yield of the 4-chlorophenylhydrazone (6c, white solid): 722 mg (75%). Yield of 7c (white solid): 560 mg (72%). Mp 268–270 °C; Anal. Calcd. for C30H31ClN2O4 (519.04) C 69.42; H 6.02. Found C 69.29; H 6.11. 1H NMR (CDCl3, 500 MHz): δ 0.85 (s, 3H, 18-CH3), 1.26–1.59 (overlapping m, 7H), 1.76 (m, 1H), 1.92 (m, 2H), 2.07 (s, 3H, Ac-CH3), 2.20–2.29 (overlapping m, 2H), 2.37 (m, 1H), 2.90 (m, 2H, 6-H2), 4.69 (t, 1H, J = 8.4 Hz, 17-H), 6.83 (s, 1H, 4-H), 7.51 (d, 2H, J = 8.8 Hz, 3″-H and 5″-H), 7.67 (d, 2H, J = 8.8 Hz, 2″-H and 6″-H), 7.84 (s, 1H, 1-H), 8.53 (s, 1H, 5′-H), 9.65 (bs, 1H, 3-OH), 10.18 (s, 1H, CHO); 13C NMR (CDCl3, 125 MHz): δ 12.2 (C-18), 21.4 (Ac-CH3), 23.4 (CH2), 26.5 (CH2), 27.2 (CH2), 27.8 (CH2), 29.7 (CH2), 36.9 (CH2), 38.6 (CH), 43.0 (C-13), 43.8 (CH), 49.9 (CH), 82.9 (C-17), 113.0 (C-4′), 117.2 (C-4), 120.9 (2C, C-2″ and C-6″), 123.3 (C-2), 126.9 (C-1), 130.2 (2C, C-3″ and C-5″), 132.3 (C-10), 132.6 (C-5′), 134.3 (C-4″), 136.9 (C-1″), 140.9 (C-5), 153.4 (C-3′), 153.7 (C-3), 171.4 (Ac-CO), 184.5 (CHO); ESI-MS 519 [M + H]+.
3′-(3-Hydroxy-5,6,7,8-tetrahydronaphthalen-2-yl)-1′-phenyl-1′H-pyrazole-4′-carbaldehyde (17a)
According to the general procedure, phenylhydrazine hydrochloride (435 mg) was used. Phenylhydrazone (16a) could not be isolated. Yield of 17a (white solid): 242 mg (38%). Mp 137–138 °C; Anal. Calcd. for C20H18N2O2 (318.38) C 75.45; H 5.70. Found C 75.32; H 5.55. 1H NMR (CDCl3, 500 MHz): δ 1.82 (t-like m, 4H, 6-H2 and 7-H2), 2.79 (d-like m, 5-H2 and 8-H2), 6.83 (s, 1H, 4-H), 7.42 (t-like m, 1H, 4″-H), 7.53 (m, 3H, 1-H, 3″-H and 5″-H), 7.72 (d, 2H, J = 7.7 Hz, 2″-H and 6″-H), 8.56 (s, 1H, 5′-H), 9.68 (bs, 1H, 3-OH), 10.20 (s, 1H, CHO); 13C NMR (CDCl3, 125 MHz): δ 23.1 and 23.5: C-6 and C-7, 28.8 and 29.6: C-5 and C-8, 113.3 (C-4′), 117.2 (C-4), 119.7 (2C, C-2″ and C-6″), 123.0 (C-2), 128.5 (C-1), 128.8 (C-8a), 130.0 (2C, C-3″ and C-5″), 130.1 (C-4″), 132.0 (C-5′), 138.4 (C-1″), 140.7 (C-4a), 153.1 (C-3′), 153.6 (C-3), 184.9 (CHO); ESI-MS 319 [M + H]+.
3′-(3-Hydroxy-5,6,7,8-tetrahydronaphthalen-2-yl)-1′-(4″-tolyl)-1′H-pyrazole-4′-carbaldehyde (17b)
According to the general procedure, 4-tolylhydrazine hydrochloride (475 mg) was used. 4-tolylhydrazone (16b) could not be isolated. Yield of 17b (white solid): 273 mg (41%). Mp 151–153 °C; Anal. Calcd. for C21H20N2O2 (332.40) C 75.88; H 6.06. Found C 76.02; H 5.95. 1H NMR (CDCl3, 500 MHz): δ 1.82 (t-like m, 4H, 6-H2 and 7-H2), 2.42 (s, 3H, 4″-CH3), 2.79 (d-like m, 5-H2 and 8-H2), 6.82 (s, 1H, 4-H), 7.32 (d, 2H, J = 8.5 Hz, 3″-H and 5″-H), 7.55 (s, 1H, 1-H), 7.59 (d, 2H, J = 8.9 Hz, 2″-H and 6″-H), 8.50 (s, 1H, 5′-H), 9.69 (bs, 1H, 3-OH), 10.19 (s, 1H, CHO); 13C NMR (CDCl3, 125 MHz): δ (4″-CH3), 23.2 and 23.6: C-6 and C-7, 28.8 and 29.7: C-5 and C-8, 113.5 (C-4′), 117.2 (C-4), 119.7 (2C, C-2″ and C-6″), 122.9 (C-2), 128.8 (C-8a), 130.1 (C-1), 130.5 (2C, C-3″ and C-5″), 131.9 (C-5′), 136.2 (C-4″), 138.6 (C-1″), 140.9 (C-4a), 153.0 (C-3′), 153.7 (C-3), 184.8 (CHO); ESI-MS 333 [M + H]+.
1′-(4″-Chlorophenyl)-3′-(3-hydroxy-5,6,7,8-tetrahydronaphthalen-2-yl)-1′H-pyrazole-4″-carbaldehyde (17c)
According to the general procedure, 4-chlorophenylhydrazine hydrochloride (540 mg) was used. 4-chlorophenylhydrazone (16c) could not be isolated. Yield of 17c (white solid): 240 mg (34%). Mp 182–184 °C; Anal. Calcd. for C20H17ClN2O2 (352.82) C 68.09; H 4.86. Found C 67.95; H 4.95. 1H NMR (CDCl3, 500 MHz): δ 1.82 (t-like m, 4H, 6-H2 and 7-H2), 2.79 (d-like m, 5-H2 and 8-H2), 6.82 (s, 1H, 4-H), 7.49 (d, 2H, J = 8.9 Hz, 3″-H and 5″-H), 7.51 (s, 1H, 1-H), 7.67 (d, 2H, J = 8.9 Hz, 2″-H and 6″-H), 8.52 (s, 1H, 5′-H), 9.45 (bs, 1H, 3-OH), 10.18 (s, 1H, CHO); 13C NMR (CDCl3, 125 MHz): δ 23.1 and 23.5: C-6 and C-7, 28.8 and 29.7: C-5 and C-8, 113.3 (C-4′), 117.3 (C-4), 120.9 (2C, C-2″ and C-6″), 123.4 (C-2), 129.0 (C-8a), 130.1 (C-1), 130.2 (2C, C-3″ and C-5″), 132.0 (C-5′), 134.3 (C-4″), 137.0 (C-1″), 141.3 (C-4a), 153.4 (C-3′), 153.6 (C-3), 184.8 (CHO); ESI-MS 353 [M + H]+.
3.1.4. General Procedure for the Reduction of Steroidal and Non-Steroidal 4-Formylpyrazoles (7a–c and 17a–c)
4-formylpyrazole (7a–c, 0.30 mmol or 17a–c, 0.50 mmol) was suspended in EtOH (10 mL), and NaBH4 (45 mg, 1.20 mmol) was added. The solution was stirred at room temperature for 2 h, then poured into water and neutralized with diluted HCl. The resulting precipitate was filtered, washed with water, dried, and purified by flash chromatography if necessary.
3-(4′-(Hydroxymethyl)-1′-phenyl-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-triene-3,17β-diol-17-acetate (10a)
According to the general procedure, compound 7a (145 mg) was used. After purification with EtOAc/CH2Cl2 = 5:95 as eluent, 10a was obtained as a white solid (127 mg, 87%). Mp 257–259 °C; Anal. Calcd. for C30H34N2O4 (486.61) C 74.05; H 7.04. Found C 73.91; H 7.18. 1H NMR (CDCl3, 500 MHz): δ 0.84 (s, 3H, 18-CH3), 1.28–1.57 (overlapping m, 7H), 1.76 (m, 1H), 1.90 (m, 2H), 2.07 (s, 3H, Ac-CH3), 2.20–2.27 (overlapping m, 2H), 2.37 (m, 1H), 2.87 (m, 2H, 6-H2), 4.69 (t, 1H, J = 8.4 Hz, 17-H), 4.87 (s, 2H, CH2-OH), 6.80 (s, 1H, 4-H), 7.31 (t-like m, 1H, 4″-H), 7.47 (t-like m, 2H, 3″-H and 5″-H), 7.65 (d, 2H, J = 8.0 Hz, 2″-H and 6″-H), 7.68 (s, 1H, 1-H), 8.02 (s, 1H, 5′-H), 10.56 (3-OH); 13C NMR (CDCl3, 125 MHz): δ 12.2 (C-18), 21.4 (Ac-CH3), 23.4 (CH2), 26.4 (CH2), 27.4 (CH2), 27.7 (CH2), 29.6 (CH2), 37.1 (CH2), 38.7 (CH), 43.0 (C-13), 44.0 (CH), 49.9 (CH), 56.7 (CH2-OH), 82.9 (C-17), 114.4 (C-2), 116.8 (C-4), 118.7 (2C, C-2″ and C-6″), 120.9 (C-4′), 125.0 (C-4″), 126.9 (C-1), 127.9 (C-5′), 129.8 (2C, C-3″ and C-5″), 131.6 (C-10), 138.8 (C-5), 139.1 (C-1″), 150.6 (C-3′), 153.9 (C-3), 171.4 (Ac-CO); ESI-MS 487 [M + H]+.
3-(4′-(Hydroxymethyl)-1′-(4″-tolyl)-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-triene-3,17β-diol-17-acetate (10b)
According to the general procedure, compound 7b (150 mg) was used. After purification with EtOAc/CH2Cl2 = 5:95 as eluent, 10b was obtained as a white solid (128 mg, 85%). Mp 254–256 °C; Anal. Calcd. for C31H36N2O4 (500.64) C 74.37; H 7.25. Found C 74.50; H 7.32. 1H NMR (CDCl3, 500 MHz): δ 0.84 (s, 3H, 18-CH3), 1.26–1.57 (overlapping m, 7H), 1.74 (m, 1H), 1.89 (m, 2H), 2.06 (s, 3H, Ac-CH3), 2.16–2.25 (m, 2H), 2.36 (m, 1H), 2.39 (s, 3H, 4″-CH3), 2.87 (m, 2H, 6-H2), 4.68 (t, 1H, J = 8.5 Hz, 17-H), 4.84 (d-like m, 2H, CH2OH), 6.79 (s, 1H, 4-H), 7.26 (d, 2H, J = 8.4 Hz, 3″-H and 5″-H), 7.52 (d, 2H, J = 8.4 Hz, 2″-H and 6″-H), 7.67 (s, 1H, 1-H), 7.96 (s, 1H, 5′-H); 13C NMR (CDCl3, 125 MHz): δ 12.2 (C-18), 21.1 (4″-CH3), 21.4 (Ac-CH3), 23.4 (CH2), 26.3 (CH2), 27.3 (CH2), 27.7 (CH2), 29.6 (CH2), 37.0 (CH2), 38.7 (CH), 43.0 (C-13), 44.0 (CH), 49.9 (CH), 56.7 (CH2-OH), 82.9 (C-17), 114.5 (C-2), 116.8 (C-4), 118.7 (2C, C-2″ and C-6″), 120.6 (C-4′), 125.0 (C-1), 127.8 (C-5′), 130.2 (2C, C-3″ and C-5″), 131.6 (C-4″), 136.8 (C-10), 136.9 (C-1″), 138.7 (C-5), 150.2 (C-3′), 153.8 (C-3), 171.4 (Ac-CO); ESI-MS 501 [M + H]+.
3-(1′-(4″-chlorophenyl)-4′-(hydroxymethyl)-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-triene-3,17β-diol-17-acetate (10c)
According to the general procedure, compound 7c (156 mg) was used. After purification with EtOAc/CH2Cl2 = 5:95 as eluent, 10c was obtained as a white solid (120 mg, 77%). Mp 288–290 °C; Anal. Calcd. for C30H33ClN2O4 C 69.15; H 6.38. Found C 69.03; H 6.27. 1H NMR (DMSO-d6, 500 MHz): δ 0.79 (s, 3H, 18-CH3), 1.28–1.43 (overlapping m, 6H), 1.49 (m, 1H), 1.69 (m, 1H), 1.77 (m, 1H), 1.82 (m, 1H), 2.01 (s, 3H, Ac-CH3), 2.11 (m, 1H), 2.19 (m, 1H), 2.30 (m, 1H), 2.79 (m, 2H, 6-H2), 4.51 (d-like m, 2H, CH2OH), 4.61 (t, 1H, J = 8.4 Hz, 17-H), 5.15 (t-like m, 1H, CH2OH), 6.65 (s, 1H, 4-H), 7.56 (s, 1H, 1-H), 7.57 (d, 2H, J = 8.9 Hz, 3″-H and 5″-H), 7.87 (d, 2H, J = 8.9 Hz, 2″-H and 6″-H), 8.54 (s, 1H, 5′-H), 9.93 (3-OH); 13C NMR (DMSO-d6, 125 MHz): δ 12.0 (C-18), 20.9 (Ac-CH3), 22.8 (CH2), 25.8 (CH2), 26.8 (CH2), 27.2 (CH2), 28.9 (CH2), 36.5 (CH2), 38.2 (CH), 42.5 (C-13), 43.3 (CH), 49.1 (CH), 54.7 (CH2-OH), 81.9 (C-17), 115.5 (C-2), 115.8 (C-4), 119.6 (2C, C-2″ and C-6″), 123.2 (C-4′), 126.5 (C-1), 128.3 (C-5′), 129.6 (2C, C-3″ and C-5″), 130.2 and 130.8: C-4″ and C-10, 137.9 (2C): C-5 and C-1″, 150.0 (C-3′), 152.9 (C-3), 171.4 (Ac-CO); ESI-MS 521 [M + H]+.
3-(4′-(Hydroxymethyl)-1′-phenyl-1′H-pyrazol-3′-yl)-5,6,7,8-tetrahydro-2-naphthol (18a)
According to the general procedure, compound 17a (159 mg) was used. After purification with EtOAc/CH2Cl2 = 2:98 as eluent, 18a was obtained as a white solid (133 mg, 83%). Mp 135–137 °C; Anal. Calcd. for C20H20N2O2 (320.39) C 74.98; H 6.29. Found C 75.12; H 6.16. 1H NMR (CDCl3, 500 MHz): δ 1.80 (m, 4H, 6-H2 and 7-H2), 2.76 (d-like m, 4H, 5-H2 and 8-H2), 4.87 (s, 2H, CH2-OH), 6.79 (s, 1H, 1-H), 7.31 (t-like m, 1H, 4″-H), 7.37 (s, 1H, 4-H), 7.46 (t-like m, 2H, 3″-H and 5″-H), 7.64 (d, 2H, J = 7.9 Hz, 2″-H and 6″-H), 8.00 (s, 1H, 5′-H); 13C NMR (CDCl3, 125 MHz): δ 23.3 and 23.6: C-6 and C-7, 29.0 and 29.5: C-5 and C-8, 56.8 (CH2-OH), 114.6 (C-3), 116.9 (C-1), 118.7 (2C, C-2″ and C-6″), 121.1 (C-4′), 126.9 (C-4), 127.8 (C-5′), 128.1 (C-4″), 128.2 (C-4a), 129.7 (2C, C-3″ and C-5″), 139.1 (C-1″), 139.2 (C-8a), 150.1 (C-3′), 153.7 (C-2); ESI-MS 321 [M + H]+.
3-(4′-(Hydroxymethyl)-1′-(4″-tolyl)-1′H-pyrazol-3-yl)-5,6,7,8-tetrahydro-2-naphthol (18b)
According to the general procedure, compound 17b (166 mg) was used. After purification with EtOAc/CH2Cl2 = 2:98 as eluent, 18b was obtained as a white solid (139 mg, 83%). Mp 163–165 °C; Anal. Calcd. for C21H22N2O2 (334.42) C 75.42; H 6.63. Found C 75.55; H 6.51. 1H NMR (CDCl3, 500 MHz): δ 1.80 (m, 4H, 6-H2 and 7-H2), 2.39 (s, 3H, 4″-CH3), 2.76 (d-like m, 4H, 5-H2 and 8-H2), 4.86 (s, 2H, CH2-OH), 6.79 (s, 1H, 1-H), 7.26 (d, 2H, J = 8.5 Hz, 3″-H and 5″-H), 7.37 (s, 1H, 4-H), 7.52 (d, 2H, J = 8.5 Hz, 2″-H and 6″-H), 7.96 (s, 1H, 5′-H); 13C NMR (CDCl3, 125 MHz): δ 21.1 (4″-CH3), 23.3 and 23.7: C-6 and C-7, 29.0 and 29.6: C-5 and C-8, 56.8 (CH2-OH), 114.8 (C-3), 116.9 (C-1), 118.8 (2C, C-2″ and C-6″), 120.8 (C-4′), 127.8 (C-4), 128.1 (C-5′), 128.2 (C-4a), 130.2 (2C, C-3″ and C-5″), 136.8 (C-4″), 137.0 (C-1″), 139.1 (C-8a), 149.9 (C-3′), 153.8 (C-2); ESI-MS 335 [M + H]+.
3-(1′-(4″-Chlorophenyl)-4′-(hydroxymethyl)-1′H-pyrazol-3′-yl)-5,6,7,8-tetrahydro-2-naphthol (18c)
According to the general procedure, compound 17c (176 mg) was used. After purification with EtOAc/CH2Cl2 = 2:98 as eluent, 18c was obtained as a white solid (117 mg, 66%). Mp 154–156 °C; Anal. Calcd. for C20H19ClN2O2 (354.83) C 67.70; H 5.40. Found C 67.82; H 5.27. 1H NMR (CDCl3, 500 MHz): δ 1.80 (m, 4H, 6-H2 and 7-H2), 2.76 (d-like m, 4H, 5-H2 and 8-H2), 4.86 (s, 2H, CH2-OH), 6.79 (s, 1H, 1-H), 7.34 (s, 1H, 4-H), 7.43 (d, 2H, J = 9.0 Hz, 3″-H and 5″-H), 7.57 (d, 2H, J = 9.0 Hz, 2″-H and 6″-H), 7.97 (s, 1H, 5′-H); 13C NMR (CDCl3, 125 MHz): δ 23.3 and 23.7: C-6 and C-7, 29.0 and 29.6: C-5 and C-8, 56.8 (CH2-OH), 114.5 (C-3), 117.0 (C-1), 119.9 (2C, C-2″ and C-6″), 121.6 (C-4′), 127.7 (C-4), 128.2 (C-5′), 128.4 (C-4a), 129.9 (2C, C-3″ and C-5″), 132.4 (C-4″), 137.8 (C-1″), 139.5 (C-8a), 150.4 (C-3′), 153.8 (C-2); ESI-MS 355 [M + H]+.
3.1.5. General Procedure for the Oxidative Lactonization of Steroidal and Non-steroidal 4-Formylpyrazoles (7a–c and 17a–c)
4-formylpyrazole (7a–c, 0.30 mmol or 17a–c, 0.50 mmol) was suspended in acetone (10 mL) and Jones reagent was added dropwise into the solution until its color remained. The mixture was then stirred at room temperature for 10 min (7a–c) or kept at reflux temperature for 30 min (17a–c). After the given reaction time, the solution was diluted with water (15 mL). The precipitate that formed was extracted with EtOAc (3 × 10 mL), and the combined organic phases were washed with brine (10 mL), then dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product (9a–c or 19a–c) was purified by column chromatography with EtOAc/hexane = 20:80.
11β-Acetoxy-2-phenylpyrazolo [3,4:4,5]pyrano [2,3:3,2]estra-1,3,5(10)-triene-4(2H)-one (9a)
According to the general procedure, compound 7a (145 mg) was used. Compound 9a was obtained as a white solid (106 mg, 73%). Mp 198–200 °C; Anal. Calcd. for C30H30N2O4 (482.58) C 74.67; H 6.27. Found C 74.80; H 6.18. 1H NMR (CDCl3, 500 MHz): δ 0.86 (s, 3H, 11a-CH3), 1.28–1.64 (overlapping m, 7H), 1.78 (m, 1H), 1.95 (m, 2H), 2.07 (s, 3H, 11-OAc-CH3), 2.19–2.35 (overlapping m, 2H), 2.51 (m, 1H), 2.97 (m, 2H, 7-H2), 4.71 (t, 1H, J = 8.5 Hz, 11-H), 7.10 (s, 1H, 6-H), 7.44 (t-like m, 1H, 4′-H), 7.55 (t-like m, 2H, 3′-H and 5′-H), 7.84 (d, 2H, J = 8.0 Hz, 2′-H and 6′-H), 8.05 (s, 1H, 14-H), 8.65 (s, 1H, 3-H); 13C NMR (CDCl3, 125 MHz): δ 12.2 (11a-CH3), 21.4 (11-OAc-CH3), 23.4 (CH2), 26.4 (CH2), 27.1 (CH2), 27.7 (CH2), 29.9 (CH2), 36.9 (CH2), 38.3 (CH), 43.0 (C-11a), 44.1 (CH), 50.0 (CH), 82.8 (C-11), 109.4 (C-3a), 112.1 (C-14a), 117.3 (C-6), 119.6 (C-4′), 120.6 (2C, C-2′ and C-6′), 128.6 (C-14), 129.6 (C-3), 130.0 (2C, C-3′ and C-5′), 137.5 (C-13b), 139.4 (C-1′), 140.7 (C-6a), 150.4 and 151.3 (C-5a, C-14b), 158.5 (C-4), 171.4 (11a-OAc-CO); ESI-MS 483 [M + H]+.
11β-Acetoxy-2-(4′-tolyl)-pyrazolo[3,4:4,5]pyrano[2,3:3,2]estra-1,3,5(10)-triene-4(2H)-one (9b)
According to the general procedure, compound 7b (150 mg) was used. Compound 9b was obtained as a white solid (103 mg, 69%). Mp 264–266 °C; Anal. Calcd. for C31H32N2O4 (496.61) C 74.98; H 6.50. Found C 75.12; H 6.38. 1H NMR (CDCl3, 500 MHz): δ 0.86 (s, 3H, 11a-CH3), 1.28–1.65 (overlapping m, 7H), 1.77 (m, 1H), 1.95 (m, 2H), 2.07 (s, 3H, 11-OAc-CH3), 2.20–2.33 (overlapping m, 2H), 2.43 (s, 3H, 4′-CH3), 2.50 (m, 1H), 2.97 (m, 2H, 7-H2), 4.71 (t, 1H, J = 8.5 Hz, 11-H), 7.09 (s, 1H, 6-H), 7.34 (d, 2H, J = 8.4 Hz, 3′-H and 5′-H), 7.71 (d, 2H, J = 8.4 Hz, 2′-H and 6′-H), 8.04 (s, 1H, 14-H), 8.60 (s, 1H, 3-H); 13C NMR (CDCl3, 125 MHz): 12.2 (11a-CH3), 21.2 (4′-CH3), 21.4 (11-OAc-CH3), 23.4 (CH2), 26.4 (CH2), 27.1 (CH2), 27.7 (CH2), 29.9 (CH2), 36.9 (CH2), 38.3 (CH), 43.0 (C-11a), 44.1 (CH), 50.0 (CH), 82.8 (C-11), 109.2 (C-3a), 112.1 (C-14a), 117.2 (C-6), 119.6 (C-14), 120.4 (2C, C-2′ and C-6′), 129.4 (C-3), 130.4 (2C, C-3′ and C-5′), 137.1 (C-4′), 137.4 (C-13b), 138.8 (C-1′), 140.5 (C-6a), 150.3 and 151.3 (C-5a, C-14b), 158.5 (C-4), 171.4 (11a-OAc-CO); ESI-MS 497 [M + H]+.
11β-Acetoxy-2-(4′-chlorophenyl)-pyrazolo[3,4:4,5]pyrano[2,3-3,2]estra-1,3,5(10)-triene-4(2H)-one (9c)
According to the general procedure, compound 7c (156 mg) was used. Compound 9c was obtained as a white solid (112 mg, 72%). Mp 283–285 °C; Anal. Calcd. for C30H29ClN2O4 (517.02) C 69.69; H 5.65. Found C 69.81; H 5.52. 1H NMR (CDCl3, 500 MHz): δ 0.86 (s, 3H, 11a-CH3), 1.27–1.65 (overlapping m, 7H), 1.78 (m, 1H), 1.95 (m, 2H), 2.06 (s, 3H, 11-OAc-CH3), 2.21–2.34 (overlapping m, 2H), 2.49 (m, 1H), 2.97 (m, 2H, 7-H2), 4.71 (t, 1H, J = 8.5 Hz, 11-H), 7.09 (s, 1H, 6-H), 7.52 (d, 2H, J = 8.8 Hz, 3′-H and 5′-H), 7.79 (d, 2H, J = 8.8 Hz, 2′-H and 6′-H), 8.03 (s, 1H, 14-H), 8.62 (s, 1H, 3-H); 13C NMR (CDCl3, 125 MHz): δ 12.2 (11a-CH3), 21.4 (11-OAc-CH3), 23.4 (CH2), 26.4 (CH2), 27.0 (CH2), 27.7 (CH2), 29.9 (CH2), 36.9 (CH2), 38.3 (CH), 43.0 (C-11a), 44.1 (CH), 50.0 (CH), 82.7 (C-11), 109.4 (C-3a), 111.8 (C-14a), 117.3 (C-6), 119.6 (C-14), 121.6 (2C, C-2′ and C-6′), 129.5 (C-3), 130.1 (2C, C-3′ and C-5′), 134.3 (C-4′), 137.6 (C-13b), 137.9 (C-1′), 140.9 (C-6a), 150.6 and 151.3 (C-5a, C-14b), 158.2 (C-4), 171.4 (11a-OAc-CO); ESI-MS 518 [M + H]+.
2-Phenyl-7,8,9,10-tetrahydrobenzo[6,7]chromeno[4,3-c]pyrazol-4(2H)-one (19a)
According to the general procedure, compound 17a (159 mg) was used. Compound 19a was obtained as a white solid (101 mg, 64%). Mp 221–223 °C; Anal. Calcd. for C20H16N2O2 (316.36) C 75.93; H 5.10. Found C 76.09; H 5.19. 1H NMR (CDCl3, 500 MHz): δ 1.84 (m, 4H, 8-H2 and 9-H2), 2.87 (d-like m, 4H, 7-H2 and 10-H2), 7.09 (s, 1H, 6-H), 7.44 (t-like m, 1H, 4′-H), 7.55 (t-like m, 2H, 3′-H and 5′-H), 7.83 (d, 2H, J = 7.9 Hz, 2′-H and 6′-H), 7.85 (s, 1H, 11-H), 8.64 (s, 1H, 3-H); 13C NMR (CDCl3, 125 MHz): δ 22.9 and 23.2: C-8 and C-9, 29.0 and 29.9: C-7 and C-10, 109.5 (C-3a), 112.0 (C-11a), 117.4 (C-6), 117.4 (2C, C-2′ and C-6′), 122.8 (C-4′), 128.6 (C-11), 129.6 (C-3), 129.9 (2C, C-3′ and C-5′), 134.1 (C-10a), 139.4 (C-1′), 140.9 (C-6a), 150.3 (C-5a), 151.2 (C-11b), 158.5 (C-4); ESI-MS 317 [M + H]+.
2-(4′-Tolyl)-7,8,9,10-tetrahydrobenzo[6,7]chromeno[4,3-c]pyrazol-4(2H)-one (19b)
According to the general procedure, compound 17b (166 mg) was used. Compound 19b was obtained as a white solid (89 mg, 54%). Mp 224–226 °C; Anal. Calcd. for C21H18N2O2 (330.39) C 76.34; H 5.49. Found C 76.48; H 5.36. 1H NMR (CDCl3, 500 MHz): δ1.84 (m, 4H, 8-H2 and 9-H2), 2.43 (s, 3H, 4′-CH3), 2.86 (d-like m, 4H, 7-H2 and 10-H2), 7.08 (s, 1H, 6-H), 7.33 (d, 2H, J = 8.5 Hz, 3′-H and 5′-H), 7.69 (d, 2H, J = 8.5 Hz, 2′-H and 6′-H), 7.84 (s, 1H, 11-H), 8.59 (s, 1H, 3-H); 13C NMR (CDCl3, 125 MHz): δ21.2 (4′-CH3), 23.0 and 23.2: C-8 and C-9, 29.0 and 29.9: C-7 and C-10, 109.3 (C-3a), 112.2 (C-11a), 117.3 (C-6), 120.4 (2C, C-2′ and C-6′), 122.8 (C-11), 129.4 (C-3), 130.4 (2C, C-3′ and C-5′), 134.0 (C-10a), 137.2 (C-4′), 138.7 (C-1′), 140.8 (C-6a), 150.2 (C-5a), 151.2 (C-11b), 158.5 (C-4); ESI-MS 331 [M + H]+.
2-(4′-Chlorophenyl)-7,8,9,10-tetrahydrobenzo[6,7]chromeno[4,3-c]pyrazol-4(2H)-one (19c)
According to the general procedure, compound 17c (176 mg) was used. Compound 19c was obtained as a white solid (89 mg, 51%). Mp 291–293 °C; Anal. Calcd. for C20H15ClN2O2 (350.80) C 68.48; H 4.31. Found C 68.60; H 4.22. 1H NMR (CDCl3, 500 MHz): δ1.85 (m, 4H, 8-H2 and 9-H2), 2.87 (d-like m, 4H, 7-H2 and 10-H2), 7.09 (s, 1H, 6-H), 7.52 (d, 2H, J = 9.0 Hz, 3′-H and 5′-H), 7.79 (d, 2H, J = 9.0 Hz, 2′-H and 6′-H), 7.83 (s, 1H, 11-H), 8.61 (s, 1H, 3-H); 13C NMR (CDCl3, 125 MHz): δ 22.9 and 23.2: C-8 and C-9, 29.0 and 30.0: C-7 and C-10, 109.9 (C-3a), 111.9 (C-11a), 117.4 (C-6), 121.6 (2C, C-2′ and C-6′), 122.9 (C-11), 129.5 (C-3), 130.1 (2C, C-3′ and C-5′), 134.2 (C-10a), 134.4 (C-4′), 138.0 (C-1′), 141.2 (C-6a), 150.5 (C-5a), 151.3 (C-11b), 158.3 (C-4); ESI-MS 351 [M + H]+.
3.1.6. General Procedure for the Deacetylation of Compounds 4, 7a–c and 10a–c
Compound 4 (0.50 mmol), 7a–c or 10a–c (0.30 mmol) was dissolved in MeOH (10 mL), and KOH (50 mg, 0.89 mmol) was added. The solution was stirred at room temperature for 2 h (for 4 and 7a–c) or 40 °C for 3 h (for 10a–c), then poured into water and neutralized with diluted HCl. The resulting precipitate was filtered off, washed with water, and dried.
2-Acetyl-estra-1,3,5(10)-triene-3,17β-diol (5)
According to the general procedure, compound
4 (178 mg) was used. Yield (
5): 141 mg (90%, white solid); Mp 189–191 °C (192–195 °C [
38]); Anal. Calcd. for C
20H
26O
3 (314.43) C 76.40; H 8.34. Found C 76.56; H 8.25.
1H NMR (CDCl
3, 500 MHz):
δ 0.79 (s, 3H, 18-CH
3), 1.17–1.59 (overlapping m, 7H), 1.71 (m, 1H), 1.89 (m, 1H), 1.99 (m, 1H), 2.10–2.19 (overlapping m, 2H), 2.34 (m, 1H), 2.60 (s, 3H, Ac-CH
3), 2.85 (m, 2H, 6-H
2), 3.74 (t, 1H,
J = 8.6 Hz, 17-H), 6.69 (s, 1H, 4-H), 7.61 (s, 1H, 1-H), 12.04 (s, 1H, OH);
13C NMR (CDCl
3, 125 MHz): δ 11.2 (C-18), 23.2 (CH
2), 26.4 (CH
2), 26.6 (Ac-CH
3), 26.9 (CH
2), 30.1 (CH
2), 30.7 (CH
2), 36.7 (CH
2), 38.7 (CH), 43.3 (C-13), 43.6 (CH), 50.2 (CH), 81.9 (C-17), 117.7 (C-4), 118.0 (C-2), 127.4 (C-1), 131.7 (C-10), 147.4 (C-5), 160.1 (C-3), 204.2 (Ac-CO); ESI-MS 315 [M + H]
+.
2-(4′-Formyl-1′-phenyl-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-trien-3,17β-diol (8a)
According to the general procedure, compound 7a (145 mg) was used. Yield (8a): 118 mg (89%, white solid); Mp 172–174 °C; Anal. Calcd. for C28H30N2O3 (442.56) C 75.99; H 6.83. Found C 76.08; H 6.74. 1H NMR (CDCl3, 500 MHz): δ 0.81 (s, 3H, 18-CH3), 1.30–1.43 (overlapping m, 4H), 1.45–1.63 (overlapping m, 3H), 1.72 (m, 1H), 1.92 (m, 1H), 1.99 (m, 1H), 2.13 (m, 1H), 2.25 (m, 1H), 2.43 (m, 1H), 2.90 (m, 2H, 6-H2), 3.76 (t, 1H, J = 8.5 Hz, 17-H), 6.83 (s, 1H, 4-H), 7.43 (t-like m, 1H, 4″-H), 7.55 (t-like m, 2H, 3″-H and 5″-H), 7.73 (d, 2H, J = 7.9 Hz, 2″-H and 6″-H), 7.90 (s, 1H, 1-H), 8.57 (s, 1H, 5′-H), 10.20 (s, 1H, CHO); 13C NMR (CDCl3, 125 MHz): δ 11.1 (C-18), 23.2 (CH2), 26.5 (CH2), 27.2 (CH2), 29.6 (CH2), 30.6 (CH2), 36.7 (CH2), 38.8 (CH), 43.3 (C-13), 43.9 (CH), 50.1 (CH), 81.9 (C-17), 113.0 (C-4′), 117.0 (C-4), 119.6 (2C, C-2″ and C-6″), 123.0 (C-2), 126.8 (C-1), 128.4 (C-4″), 129.9 (2C, C-3″ and C-5″), 132.2 (C-10), 132.7 (C-5′), 138.2 (C-1″), 140.7 (C-5), 153.1 (C-3′), 153.7 (C-3), 184.4 (CHO); ESI-MS 443 [M + H]+.
2-(4′-Formyl-1′-(4″-tolyl)-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-trien-3,17β-diol (8b)
According to the general procedure, compound 7b (150 mg) was used. Yield (8b): 123 mg (91%, white solid); Mp > 190 °C (decomp.); Anal. Calcd. for C29H32N2O3 (456.59) C 76.29; H 7.06. Found C 76.43; H 6.95. 1H NMR (CDCl3, 500 MHz): δ 0.80 (s, 3H, 18-CH3), 1.30–1.43 (overlapping m, 4H), 1.44–1.62 (overlapping m, 3H), 1.71 (m, 1H), 1.91 (m, 1H), 1.99 (m, 1H), 2.13 (m, 1H), 2.25 (m, 1H), 2.42 (m, 1H), 2.43 (s, 3H, 4″-CH3), 2.90 (m, 2H, 6-H2), 3.75 (t, 1H, J = 8.5 Hz, 17-H), 6.83 (s, 1H, 4-H), 7.32 (d, 2H, J = 8.5 Hz, 3″-H and 5″-H), 7.59 (d, 2H, J = 8.5 Hz, 2″-H and 6″-H), 7.90 (s, 1H, 1-H), 8.52 (s, 1H, 5′-H), 9.90 (bs, 1H, 3-OH), 10.20 (s, 1H, CHO); 13C NMR (CDCl3, 125 MHz): δ 11.2 (C-18), 21.2 (4″-CH3), 23.3 (CH2), 26.6 (CH2), 27.3 (CH2), 29.7 (CH2), 30.7 (CH2), 36.8 (CH2), 38.9 (CH), 43.4 (C-13), 44.0 (CH), 50.2 (CH), 82.0 (C-17), 113.2 (C-4′), 117.1 (C-4), 119.6 (2C, C-2″ and C-6″), 122.9 (C-2), 126.9 (C-1), 130.5 (2C, C-3″ and C-5″), 132.2 (C-10), 132.6 (C-5′), 136.1 (C-4″), 138.7 (C-1″), 140.7 (C-5), 153.1 (C-3′), 153.8 (C-3), 184.6 (CHO); ESI-MS 457 [M + H]+.
2-(1′-(4″-Chlorophenyl)-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-trien-3,17β-diol (8c)
According to the general procedure, compound 7c (156 mg) was used. Yield (8c): 124 mg (87%, white solid); Mp > 190 °C (decomp.); Anal. Calcd. for C28H29ClN2O3 (477.00) C 70.50; H 6.13. Found C 70.62; H 6.04. 1H NMR (CDCl3, 500 MHz): δ 0.80 (s, 3H, 18-CH3), 1.30–1.43 (overlapping m, 4H), 1.44–1.62 (overlapping m, 3H), 1.72 (m, 1H), 1.91 (m, 1H), 1.99 (m, 1H), 2.13 (m, 1H), 2.24 (m, 1H), 2.41 (m, 1H), 2.90 (m, 2H, 6-H2), 3.75 (t, 1H, J = 8.5 Hz, 17-H), 6.83 (s, 1H, 4-H), 7.50 (d, 2H, J = 8.9 Hz, 3″-H and 5″-H), 7.66 (d, 2H, J = 8.9 Hz, 2″-H and 6″-H), 7.85 (s, 1H, 1-H), 8.53 (s, 1H, 5′-H), 9.66 (bs, 1H, 3-OH), 10.18 (s, 1H, CHO); 13C NMR (CDCl3, 125 MHz): δ 11.2 (C-18), 23.3 (CH2), 26.6 (CH2), 27.3 (CH2), 29.7 (CH2), 30.7 (CH2), 36.8 (CH2), 38.9 (CH), 43.4 (C-13), 44.0 (CH), 50.2 (CH), 82.0 (C-17), 113.0 (C-4′), 117.2 (C-4), 120.8 (2C, C-2″ and C-6″), 123.3 (C-2), 126.9 (C-1), 130.2 (2C, C-3″ and C-5″), 132.4 (C-10), 132.7 (C-5′), 134.2 (C-4″), 136.9 (C-1″), 141.0 (C-5), 153.5 (C-3′), 153.7 (C-3), 184.5 (CHO); ESI-MS 478 [M + H]+.
3-(4′-(Hydroxymethyl)-1′-phenyl-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-trien-3,17β-diol (11a)
According to the general procedure, compound 10a (146 mg) was used. Yield (11a): 121 mg (91%, white solid); Mp 141–143 °C; Anal. Calcd. for C28H32N2O3 (444.58) C 75.65; H 7.26. Found C 75.74; H 7.13. 1H NMR (DMSO-d6, 500 MHz): δ 0.68 (s, 3H, 18-CH3), 1.10–1.44 (overlapping m, 7H), 1.59 (m, 1H), 1.80–1.90 (overlapping m, 3H), 2.14 (m, 1H), 2.33 (m, 1H), 2.77 (m, 2H, 6-H2), 3.54 (m, 1H, 17-H), 4.51 (d, 1H, J = 4.8 Hz, 17-OH), 4.56 (d, 2H, J = 4.9 Hz, CH2-OH), 5.15 (t, 1H, J = 4.9 Hz, CH2-OH), 6.65 (s, 1H, 4-H), 7.31 (t-like m, 1H, 4″-H), 7.53 (t-like m, 2H, 3″-H and 5″-H), 7.65 (s, 1H, 1-H), 7.83 (d, 2H, J = 8.0 Hz, 2″-H and 6″-H), 8.60 (s, 1H, 5′-H), 10.14 (3-OH); 13C NMR (DMSO-d6, 125 MHz): δ 11.3 (C-18), 22.8 (CH2), 26.0 (CH2), 26.9 (CH2), 28.9 (CH2), 29.9 (CH2), 36.6 (CH2), 38.6 (CH), 42.8 (C-13), 43.6 (CH), 49.6 (CH), 54.7 (CH2-OH), 80.1 (C-17), 115.2 (C-2), 115.8 (C-4), 118.0 (2C, C-2″ and C-6″), 122.6 (C-4′), 126.2 (2C, C-4″ and C-1), 128.4 (C-5′), 129.7 (2C, C-3″ and C-5″), 131.1 (C-10), 137.9 (C-5), 139.0 (C-1″), 149.8 (C-3′), 152.9 (C-3); ESI-MS 445 [M + H]+.
3-(4′-(Hydroxymethyl)-1′-(4″-tolyl)-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-trien-3,17β-diol (11b)
According to the general procedure, compound 10b (150 mg) was used. Yield (11b): 122 mg (89%, white solid); Mp 201–203 °C; Anal. Calcd. for C29H34N2O3 (458.60) C 75.95; H 7.47. Found C 76.07; H 7.39. 1H NMR (DMSO-d6, 500 MHz): δ 0.69 (s, 3H, 18-CH3), 1.11–1.43 (overlapping m, 7H), 1.60 (m, 1H), 1.81–1.94 (overlapping m, 3H), 2.16 (m, 1H), 2.34 (m, 1H), 2.36 (s, 3H, 4″-CH3), 2.78 (m, 2H, 6-H2), 3.55 (m, 1H, 17-H), 4.52 (d, 1H, J = 4.9 Hz, 17-OH), 4.56 (d, 2H, J = 4.9 Hz, CH2OH), 5.18 (t, 1H, J = 4.9 Hz, CH2OH), 6.65 (s, 1H, 4-H), 7.34 (d, 2H, J = 8.5 Hz, 3″-H and 5″-H), 7.67 (s, 1H, 1-H), 7.72 (d, 2H, J = 8.5 Hz, 2″-H and 6″-H), 7.96 (s, 1H, 5′-H), 9.95 (3-OH); 13C NMR (DMSO-d6, 125 MHz): δ 11.3 (C-18), 20.4 (4″-CH3), 22.8 (CH2), 26.0 (CH2), 26.9 (CH2), 28.9 (CH2), 29.9 (CH2), 36.6 (CH2), 38.6 (CH), 42.8 (C-13), 43.6 (CH), 49.6 (CH), 54.7 (CH2-OH), 80.1 (C-17), 115.2 (C-2), 115.8 (C-4), 117.9 (2C, C-2″ and C-6″), 122.2 (C-4′), 126.1 (C-1), 128.3 (C-5′), 130.1 (2C, C-3″ and C-5″), 131.0 (C-4″), 135.6 (C-10), 136.8 (C-1″), 137.8 (C-5), 149.5 (C-3′), 152.9 (C-3); ESI-MS 459 [M + H]+.
3-(1′-(4″-Chlorophenyl)-4′-(hydroxymethyl)-1′H-pyrazol-3′-yl)-estra-1,3,5(10)-trien-3,17β-diol (11c)
According to the general procedure, compound 10c (156 mg) was used. Yield (11c): 126 mg (88%, white solid); Mp 240–242 °C; Anal. Calcd. for C28H31ClN2O3 (479.02) C 70.21; H 6.52. Found C 70.09; H 6.43. 1H NMR (DMSO-d6, 500 MHz): δ 0.68 (s, 3H, 18-CH3), 1.10–1.44 (overlapping m, 7H), 1.59 (m, 1H), 1.79–1.93 (overlapping m, 3H), 2.14 (m, 1H), 2.32 (m, 1H), 2.77 (m, 2H, 6-H2), 3.54 (m, 1H, 17-H), 4.50 (d, 1H, J = 4.9 Hz, 17-OH), 4.53 (d, 2H, J = 4.9 Hz, CH2OH), 5.18 (t, 1H, J = 4.9 Hz, CH2OH), 6.64 (s, 1H, 4-H), 7.57 (d, 2H, J = 9.0 Hz, 3″-H and 5″-H), 7.59 (s, 1H, 1-H), 7.87 (d, 2H, J = 8.9 Hz, 2″-H and 6″-H), 8.55 (s, 1H, 5′-H), 9.95 (3-OH); 13C NMR (DMSO-d6, 125 MHz): δ 11.3 (C-18), 22.8 (CH2), 26.0 (CH2), 26.9 (CH2), 28.9 (CH2), 29.9 (CH2), 36.6 (CH2), 38.6 (CH), 42.8 (C-13), 43.6 (CH), 49.6 (CH), 54.7 (CH2-OH), 80.1 (C-17), 115.3 (C-2), 115.8 (C-4), 119.6 (2C, C-2″ and C-6″), 123.1 (C-4′), 126.4 (C-1), 128.4 (C-5′), 129.6 (2C, C-3″ and C-5″), 130.2 and 131.1: C-4″ and C-10, 137.9 (C) and 138.0 (C): C-5 and C-1″, 150.0 (C-3′), 152.9 (C-3); ESI-MS 479 [M + H]+.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
