
Target-Guided Isolation of O-tigloylcyclovirobuxeine-B from Buxus sempervirens L. by Centrifugal Partition Chromatography



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Target-Guided Isolation of O-tigloylcyclovirobuxeine-B (1)
2.1.1. Extraction and Alkaloid-Enrichment of B. sempervirens L. Leaves
2.1.2. Selection of Suitable Two-Phase Solvent Systems
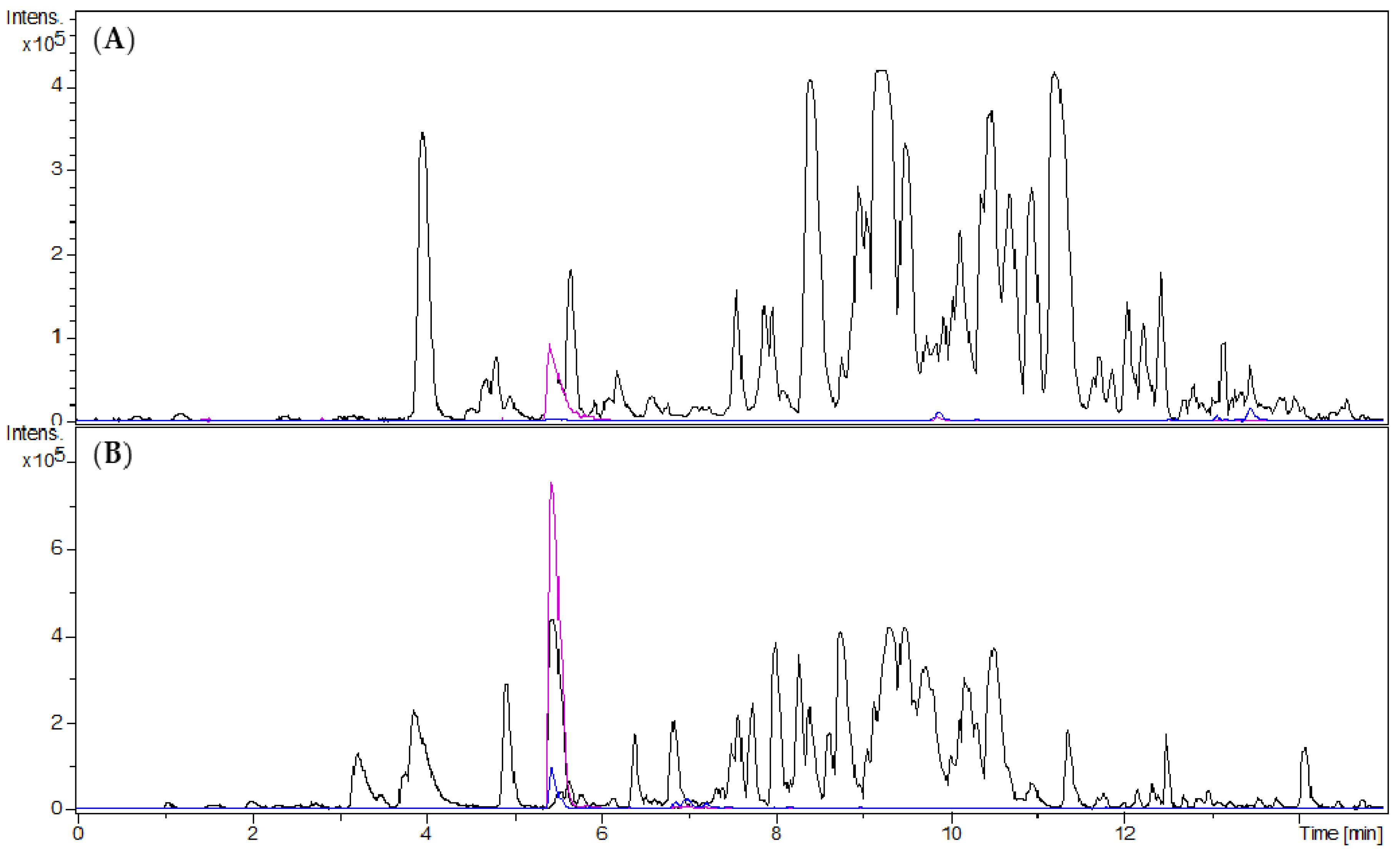
2.1.3. Separation of the Alkaloid-Enriched Fraction by Centrifugal Partition Chromatography
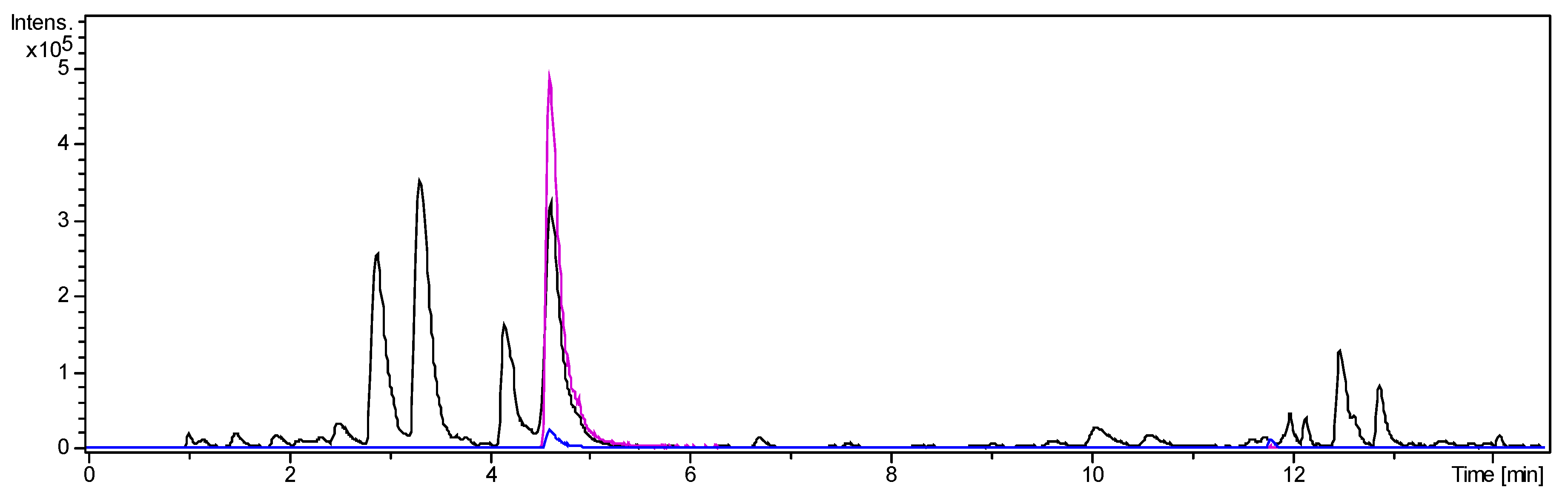
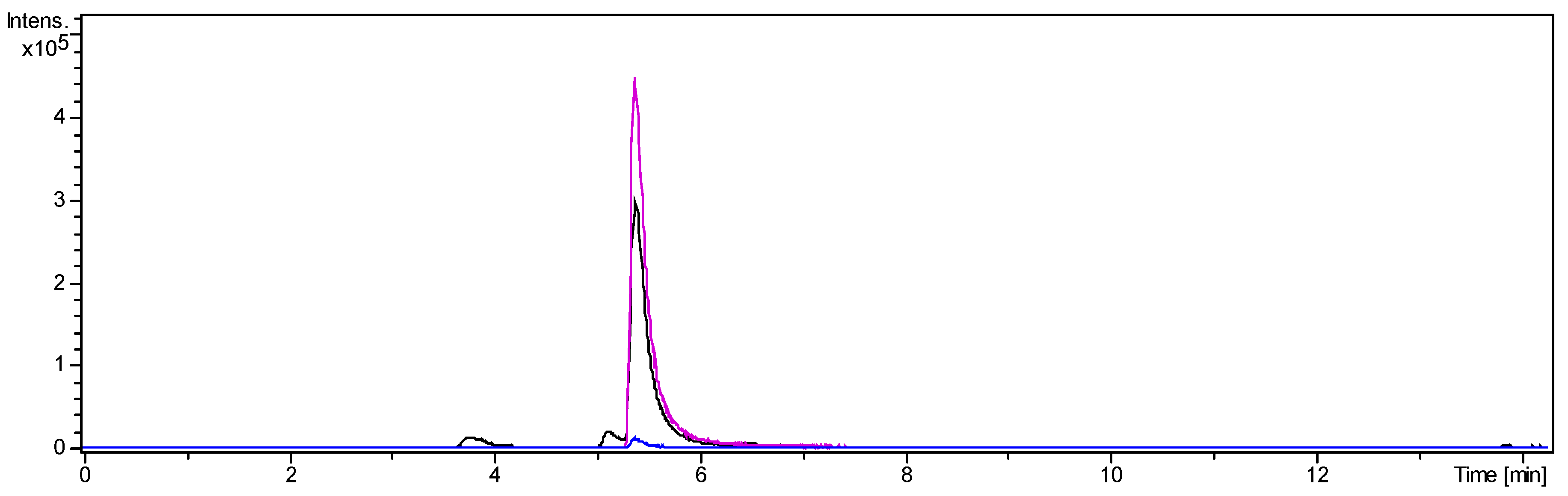
2.1.4. Separation of Fraction 2 by Centrifugal Partition Chromatography
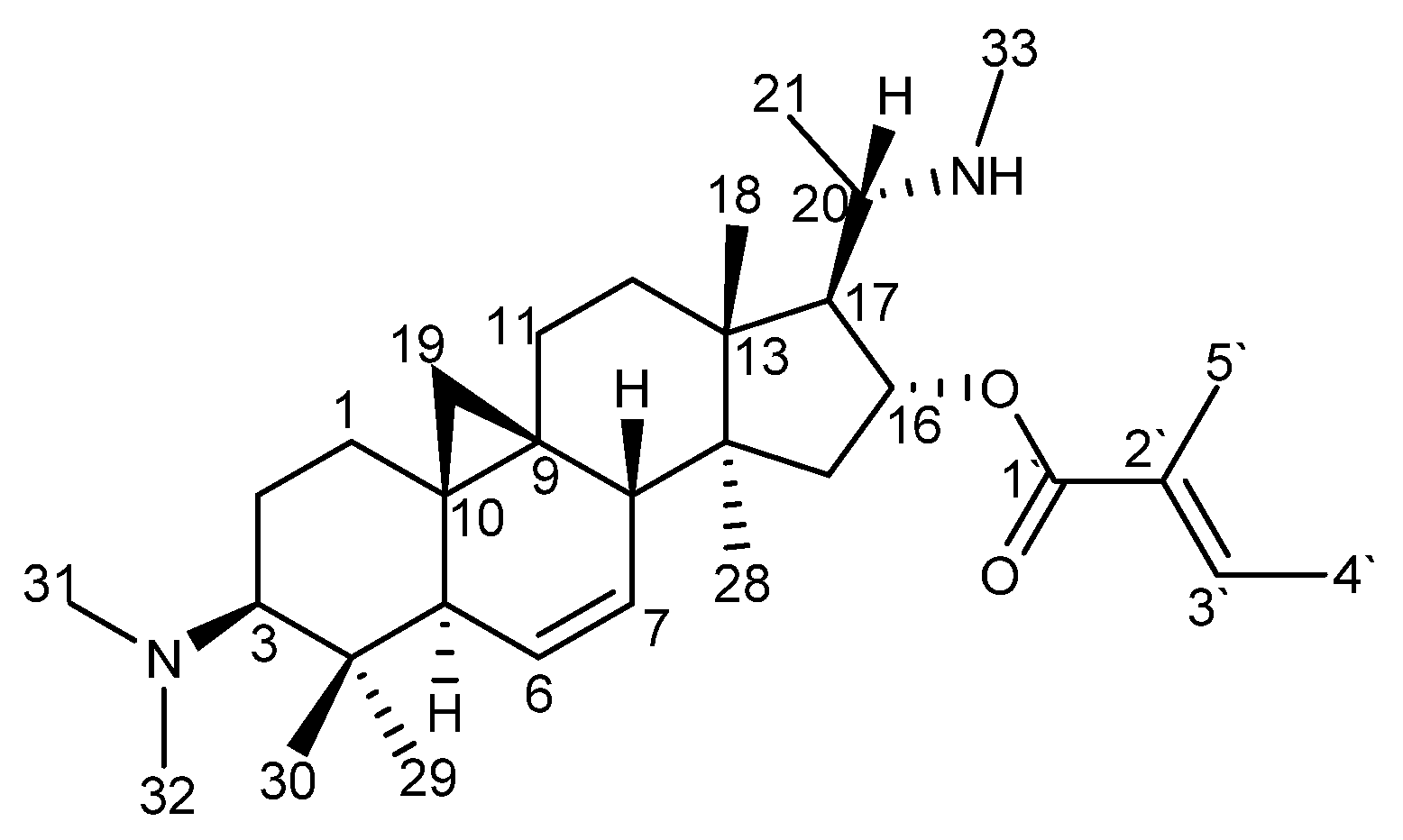
2.2. Identification of O-Tigloylcyclovirobuxeine-B (1)
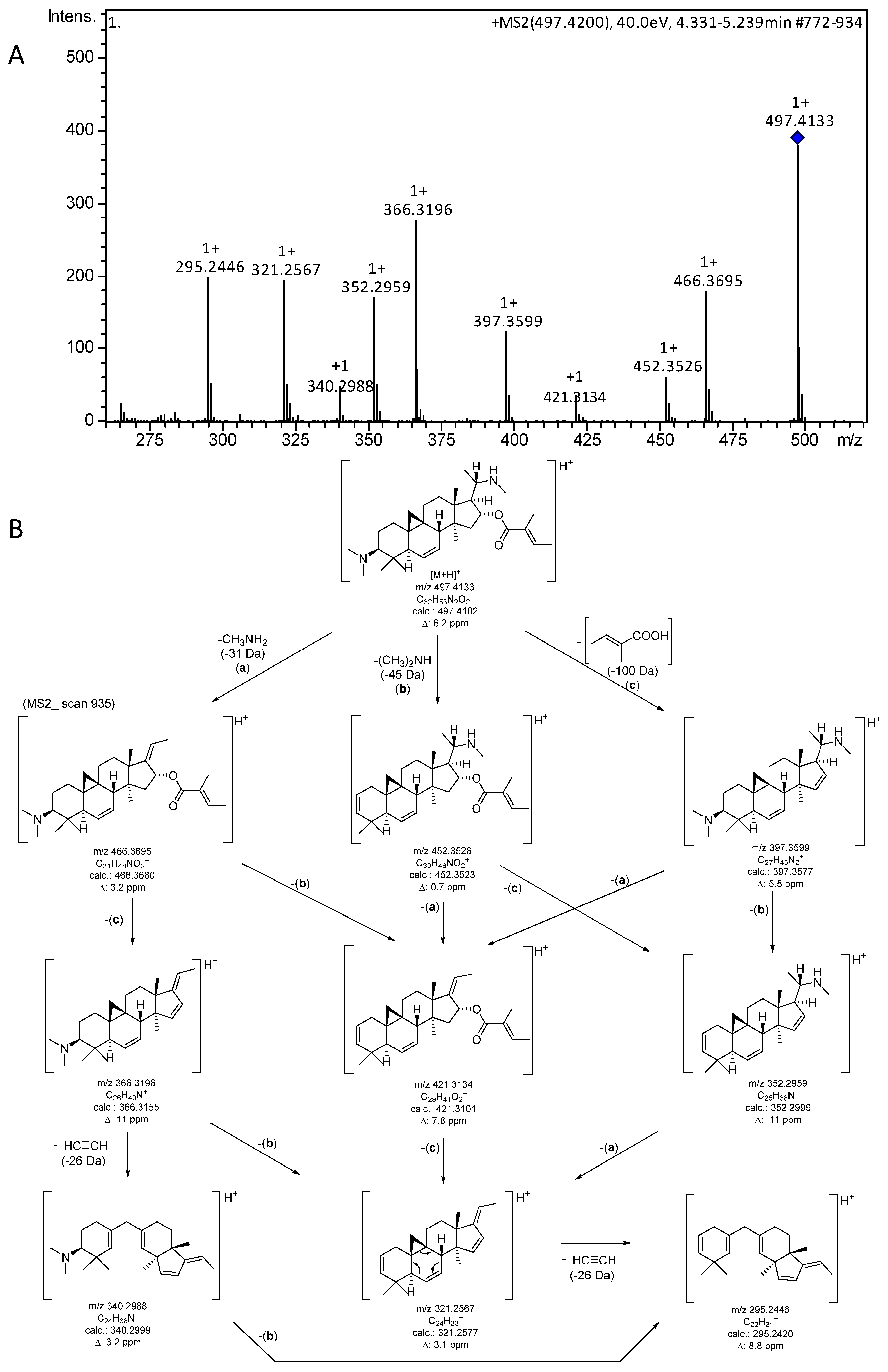
2.3. +ESI-QqTOF MS/MS fragmentation of O-tigloylcyclovirobuxeine-B (1)
3. Materials and Methods
3.1. Plant Material
3.2. Extraction and Preparation of the Alklaloid-Enriched Fraction
3.3. Isolation of O-Tigloylcyclovirobuxeine-B (1)
3.3.1. Selection of a Two-Phase Solvent System
3.3.2. Fractionation of the Alkaloid Fraction (ALOF) by Centrifugal Partition Chromatography
3.3.3. Subfractionation of Fraction 2 by Centrifugal Partition Chromatography
3.4. Spectroscopic Analysis of O-tigloylcyclovirobuxeine-B (1)
3.4.1. NMR Spectroscopy
3.4.2. UHPLC/+ESI-QqTOF-mass Spectrometry
3.5. Spectral Data of O-tigloylcyclovirobuxeine-B (1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Neves, J.M.; Matos, C.M.; Moutinho, C.; Queiroz, G.; Gomes, L.R. Ethnopharmacological Notes about Ancient Uses of Medicinal Plants in Trás-Os-Montes (northern of Portugal). J. Ethnopharmacol. 2009, 124, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Leporatti, M.L.; Pavesi, A.; Posocco, E. Phytotherapy in the Valnerina Marche (Central Italy). J. Ethnopharmacol. 1985, 14, 53–63. [Google Scholar] [CrossRef]
- World Health Organization. World-Malaria-Report-2019. Available online: https://www.who.int/publications/i/item/9789241565721 (accessed on 13 August 2020).
- Orhan, I.E.; Erdem, S.A.; Senol, F.S.; Kartal, M.; Şener, B. Exploration of Cholinesterase and Tyrosinase Inhibitory, Antiprotozoal and Antioxidant Effects of Buxus Sempervirens L. (boxwood). Ind. Crop. Prod. 2012, 40, 116–121. [Google Scholar] [CrossRef]
- Althaus, J.B.; Jerz, G.; Winterhalter, P.; Kaiser, M.; Brun, R.; Schmidt, T.J. Antiprotozoal Activity of Buxus Sempervirens and Activity-Guided Isolation of O-Tigloylcyclovirobuxeine-B As the Main Constituent Active Against Plasmodium Falciparum. Molecules 2014, 19, 6184–6201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.; Risinger, A.L.; Nair, S.; Peng, J.; Anderson, T.J.C.; Du, L.; Powell, D.R.; Mooberry, S.L.; Cichewicz, R.H. Identification of Compounds With Efficacy Against Malaria Parasites from Common North American Plants. J. Nat. Prod. 2015, 79, 490–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, F.; Oka, H.; Ito, Y. Systematic Search for Suitable Two-Phase Solvent Systems for High-Speed Counter-Current Chromatography. J. Chromatogr. A 1991, 538, 99–108. [Google Scholar] [CrossRef]
- Ito, Y. Golden Rules and Pitfalls in Selecting Optimum Conditions for High-Speed Counter-Current Chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Marston, A.; Hostettmann, K. Counter-Current Chromatography As a Preparative Tool—Applications and Perspectives. J. Chromatogr. A 1994, 658, 315–341. [Google Scholar] [CrossRef]
- Conway, W.D. Counter-Current Chromatography: Simple Process and Confusing Terminology. J. Chromatogr. A 2011, 121, 6015–6023. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, Y.; Chen, F.; Zhang, L.; Yang, Y. Property Calculation and Prediction for Selecting Solvent Systems in CCC. J. Liq. Chromatogr. Relat. Technol. 2003, 26, 1397–1415. [Google Scholar] [CrossRef]
- Khodzhaev, B.U.; Shakirov, R.; Aripov, K.N.; Shakirov, T.T.; Yunusov, S.Y. Polybuffer Distribution of the Combined Alkaloids Of Buxus Sempervirens. Chem. Nat. Compd. 1975, 11, 126. [Google Scholar] [CrossRef]
- Althaus, J.B. Naturstoffe Mit Antiprotozoaler Wirkung: Alkamide ausgewählter Asteraceae Und Triterpenalkaloide Aus Buxus Sempervirens L. Ph.D. Thesis, Westfälische Wilhelms-Universität Münster, Münster, Germany, 2015. [Google Scholar]
- Nnadi, C.; Althaus, J.B.; Nwodo, N.J.; Schmidt, T.J. A 3D-QSAR Study on the Antitrypanosomal and Cytotoxic Activities of Steroid Alkaloids by Comparative Molecular Field Analysis. Molecules 2018, 23, 1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupchan, S.; Kennedy, R.; Schleigh, W.; Ohta, G. Buxus alkaloids—XII. Tetrahedron 1967, 23, 4563–4586. [Google Scholar] [CrossRef]





Sample Availability: Samples of compound 1 are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabó, L.U.; Schmidt, T.J. Target-Guided Isolation of O-tigloylcyclovirobuxeine-B from Buxus sempervirens L. by Centrifugal Partition Chromatography. Molecules 2020, 25, 4804. https://doi.org/10.3390/molecules25204804
Szabó LU, Schmidt TJ. Target-Guided Isolation of O-tigloylcyclovirobuxeine-B from Buxus sempervirens L. by Centrifugal Partition Chromatography. Molecules. 2020; 25(20):4804. https://doi.org/10.3390/molecules25204804
Chicago/Turabian StyleSzabó, Lara U., and Thomas J. Schmidt. 2020. "Target-Guided Isolation of O-tigloylcyclovirobuxeine-B from Buxus sempervirens L. by Centrifugal Partition Chromatography" Molecules 25, no. 20: 4804. https://doi.org/10.3390/molecules25204804
APA StyleSzabó, L. U., & Schmidt, T. J. (2020). Target-Guided Isolation of O-tigloylcyclovirobuxeine-B from Buxus sempervirens L. by Centrifugal Partition Chromatography. Molecules, 25(20), 4804. https://doi.org/10.3390/molecules25204804