
Lovastatin Differentially Regulates α7 and α4 Neuronal Nicotinic Acetylcholine Receptor Levels in Rat Hippocampal Neurons


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
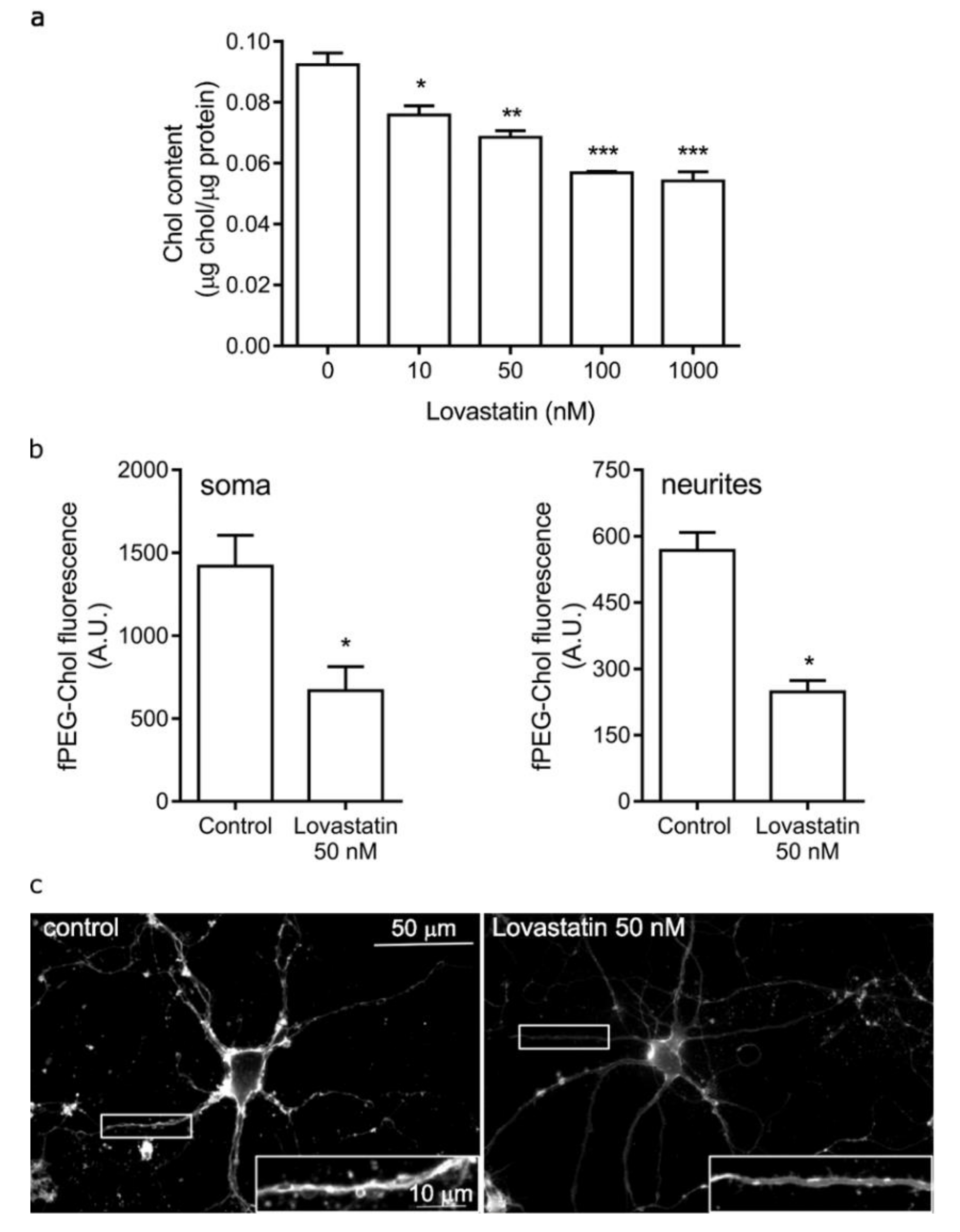
2.1. Chronic Lovastatin Treatment Reduces Cholesterol Levels in Cultured Hippocampal Neurons
2.2. Chronic Lovastatin Treatment Increases α7 nAChR Levels in Cultured Hippocampal Neurons
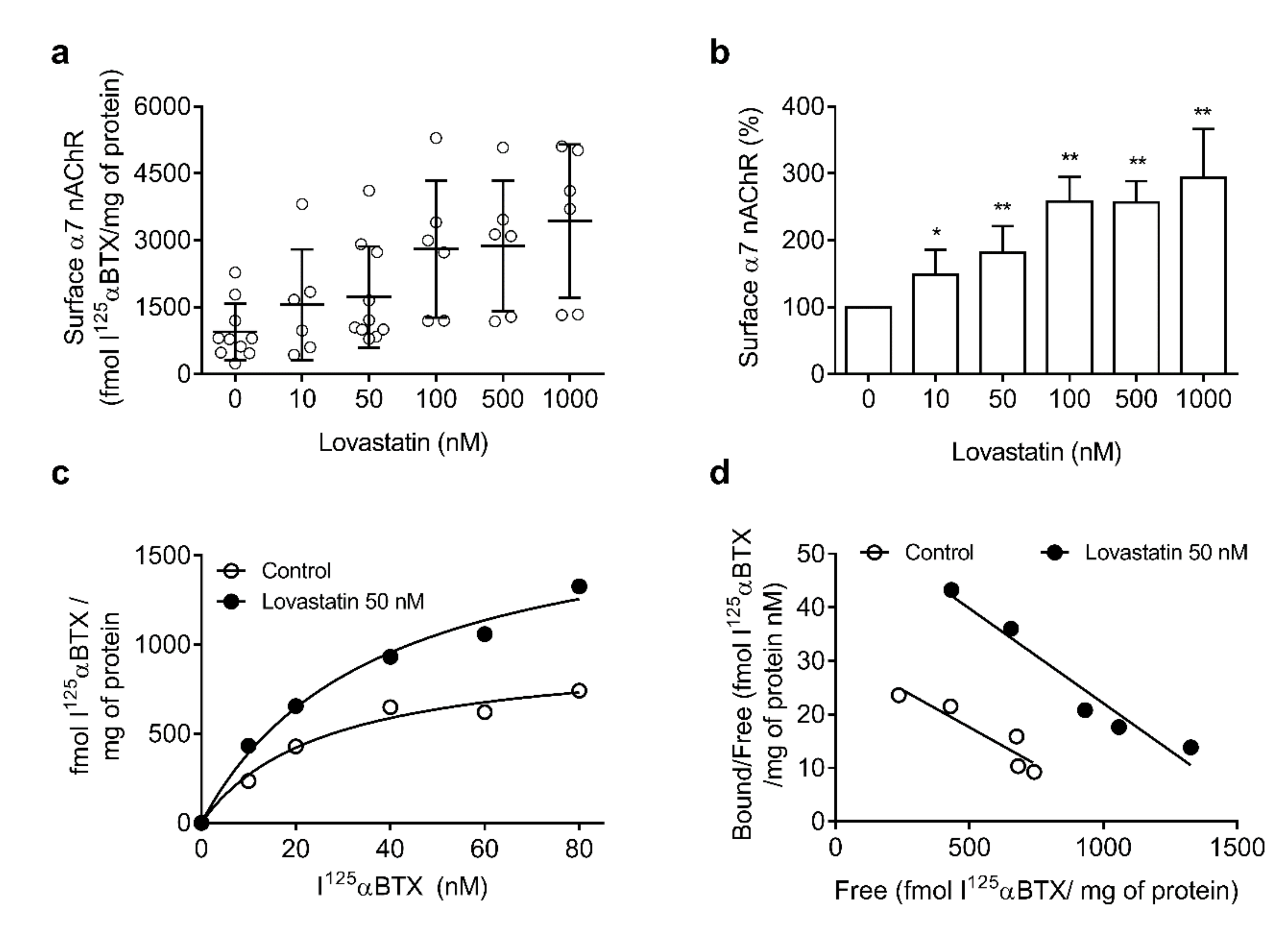
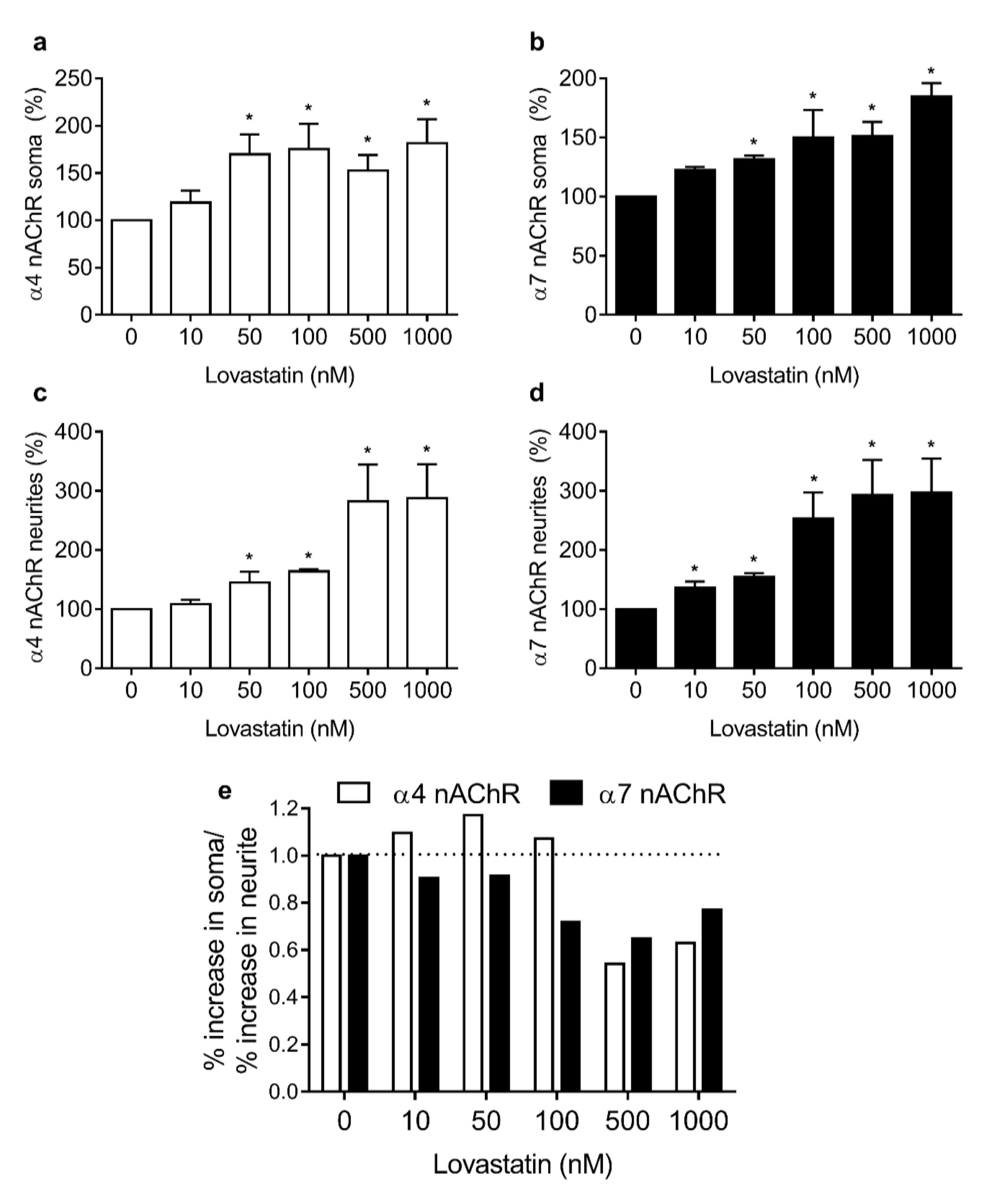
2.3. Chronic Lovastatin Treatment Increases Surface α7 and α4 nAChR Levels Differentially in Soma and Neurites of Cultured Hippocampal Neurons
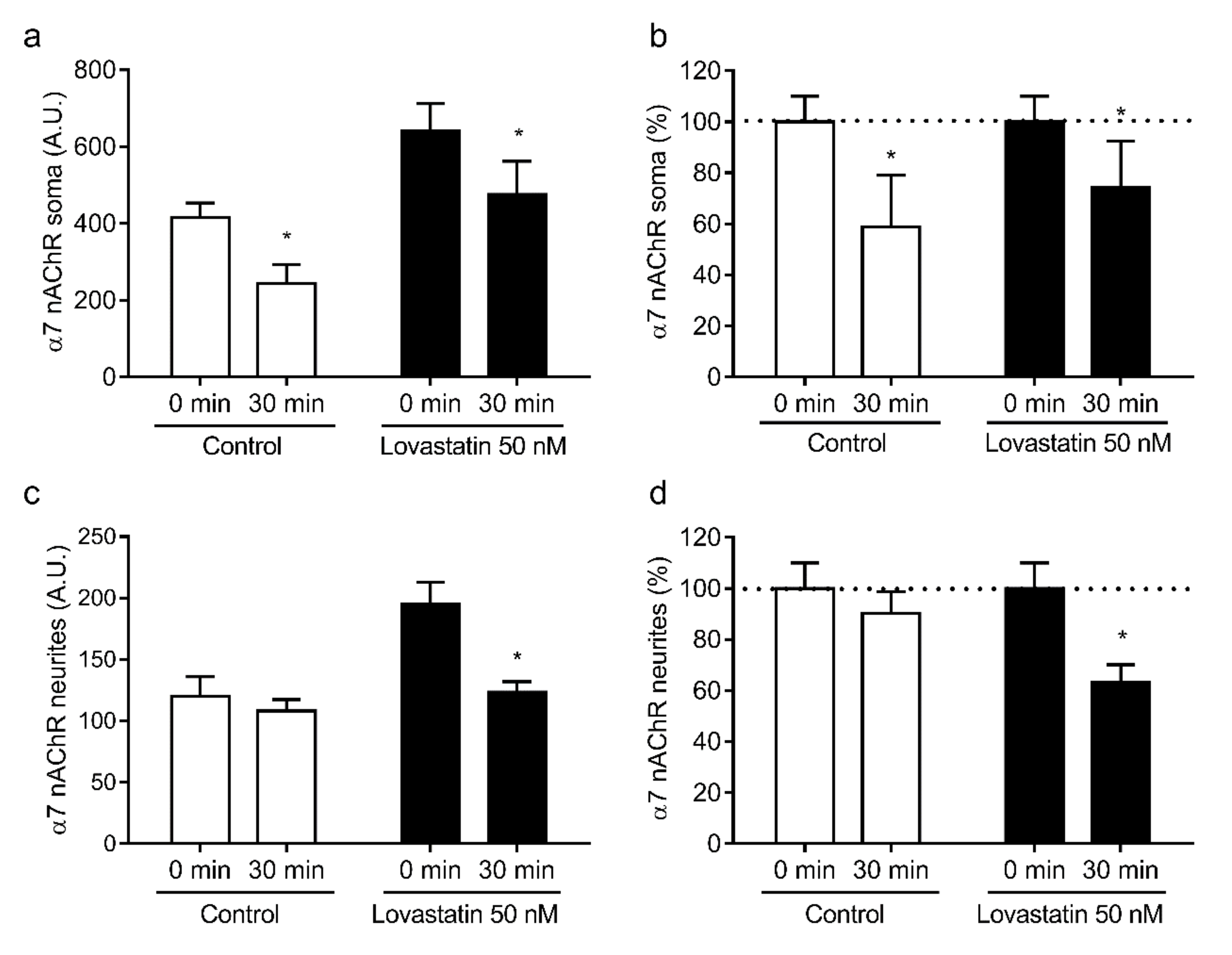
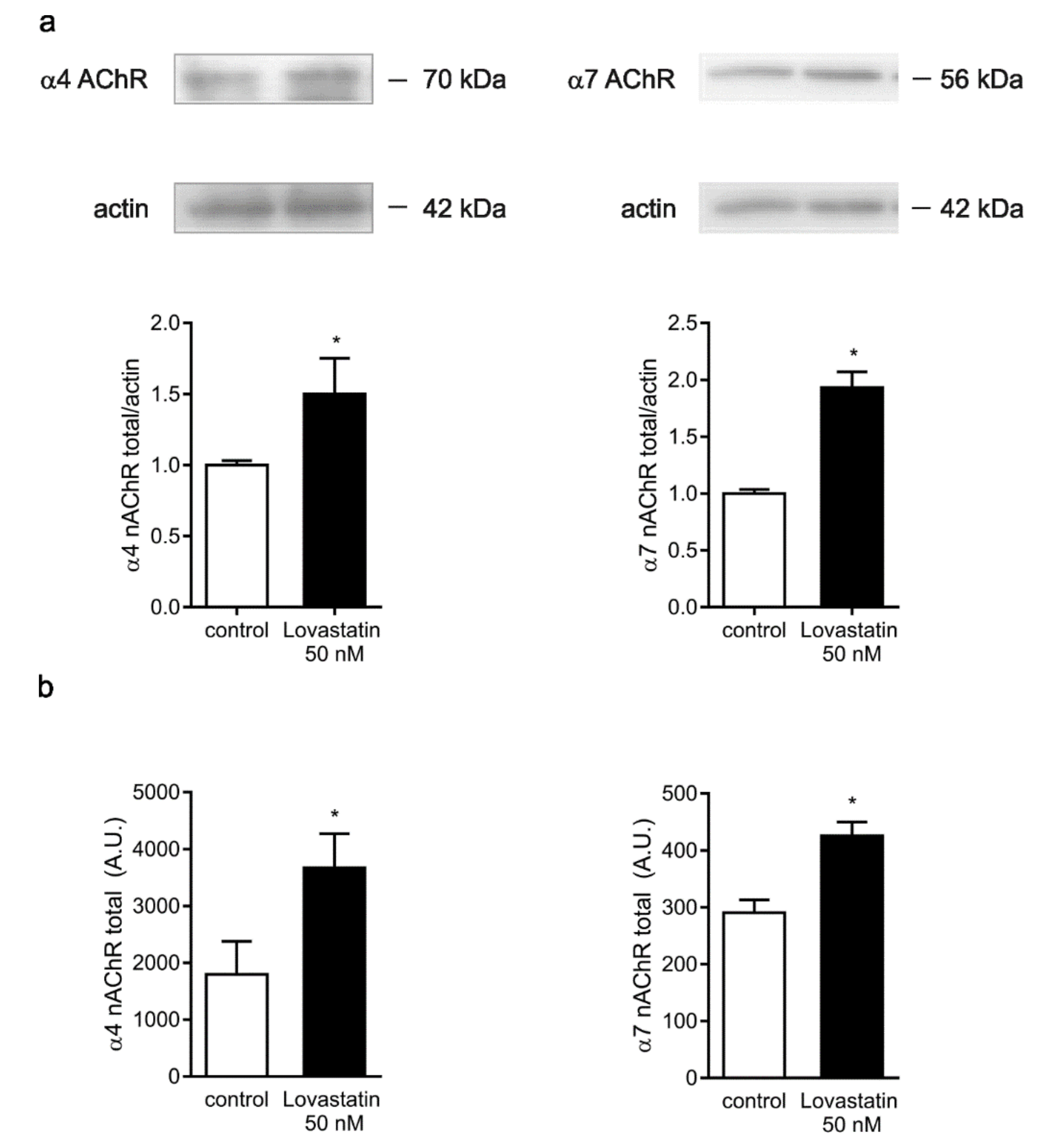
2.4. Chronic Lovastatin Treatment Increases α4 and α7 nAChR Total Levels in Cultured Hippocampal Neurons
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Hippocampal Cultures
4.3. Protein Content
4.4. Cholesterol Determination
4.5. αBTX Radioiodination
4.6. Equilibrium [125I]-αBTX Binding Studies
4.7. Cell-Surface α7 and α4 nAChR Labeling
4.8. Internalization Assay
4.9. Fluorescence Microscopy
4.10. Quantitative Image Analysis
4.11. Western Blotting
4.12. Viability Assays
4.13. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barrantes, F.J. Cholesterol effects on nicotinic acetylcholine receptor. J. Neurochem. 2007, 103, 72–80. [Google Scholar] [CrossRef]
- Karlin, A. Emerging structure of the Nicotinic Acetylcholine receptors. Nat. Rev. Neurosci. 2002, 3, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Whiting, P.; Lindstrom, J. Characterization of bovine and human neuronal nicotinic acetylcholine receptors using monoclonal antibodies. J. Neurosci. 1988, 8, 3395–3404. [Google Scholar] [CrossRef] [PubMed]
- Jones, C. α7 Nicotinic Acetylcholine Receptor: A Potential Target in Treating Cognitive Decline in Schizophrenia. J. Clin. Psychopharmacol. 2018, 38, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V.; Callahan, P.M. Nicotinic Acetylcholine Receptor Ligands, Cognitive Function, and Preclinical Approaches to Drug Discovery. Nicotine Tob. Res. 2019, 21, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.-G.; Qian, Y.-H. Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer’s disease. Neuropeptides 2019, 73, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Russo, P.; Del Bufalo, A.; Frustaci, A.; Fini, M.; Cesario, A. Beyond acetylcholinesterase inhibitors for treating Alzheimer’s disease: α7-nAChR agonists in human clinical trials. Curr. Pharm. Des. 2014, 20, 6014–6021. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Shahzadi, S.; Raza, H.; Abbasi, M.A.; Alashwal, H.; Zaki, N.; Moustafa, A.A.; Seo, S.-Y. Computational investigation of mechanistic insights of Aβ42 interactions against extracellular domain of nAChRα7 in Alzheimer’s disease. Int. J. Neurosci. 2019, 129, 666–680. [Google Scholar] [CrossRef]
- Nagele, R.G.; D’Andrea, M.R.; Anderson, W.J.; Wang, H.-Y. Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 2002, 110, 199–211. [Google Scholar] [CrossRef]
- Dineley, K.T. Beta-amyloid peptide--nicotinic acetylcholine receptor interaction: The two faces of health and disease. Front. Biosci. 2007, 12, 5030–5038. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wu, J. Nicotinic Cholinergic Mechanisms in Alzheimer’s Disease. Int. Rev. Neurobiol. 2015, 124, 275–292. [Google Scholar] [CrossRef]
- Kamerbeek, C.B.; Borroni, V.; Pediconi, M.F.; Sato, S.B.; Kobayashi, T.; Barrantes, F.J. Antibody-induced acetylcholine receptor clusters inhabit liquid-ordered and liquid-disordered domains. Biophys. J. 2013, 105, 1601–1611. [Google Scholar] [CrossRef] [Green Version]
- Borroni, V.; Barrantes, F.J. Cholesterol modulates the rate and mechanism of acetylcholine receptor internalization. J. Biol. Chem. 2011, 286, 17122–17132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borroni, V.; Borroni, V.; Baier, C.J.; Lang, T.; Bonini, I.; White, M.M.; Garbus, I.; Barrantes, F.J. Cholesterol depletion activates rapid internalization of submicron-sized acetylcholine receptor domains at the cell membrane. Mol. Membr. Biol. 2007, 24, 1–15. [Google Scholar] [CrossRef]
- Kumari, S.; Borroni, V.; Chaudhry, A.; Chanda, B.; Massol, R.; Mayor, S.; Barrantes, F.J. Nicotinic acetylcholine receptor is internalized via a Rac-dependent, dynamin-independent endocytic pathway. J. Cell Biol. 2008, 181, 1179–1193. [Google Scholar] [CrossRef] [Green Version]
- Pato, C.; Stetzkowski-Marden, F.; Gaus, K.; Recouvreur, M.; Cartaud, A.; Cartaud, J. Role of lipid rafts in agrin-elicited acetylcholine receptor clustering. Chem. -Biol. Interact. 2008, 175, 64–67. [Google Scholar] [CrossRef]
- Stetzkowski-Marden, F.; Gaus, K.; Recouvreur, M.; Cartaud, A.; Cartaud, J. Agrin elicits membrane lipid condensation at sites of acetylcholine receptor clusters in C2C12 myotubes. J. Lipid Res. 2006, 47, 2121–2133. [Google Scholar] [CrossRef] [Green Version]
- Pediconi, M.F.; Gallegos, C.E.; De Los Santos, E.B.; Barrantes, F.J. Metabolic cholesterol depletion hinders cell-surface trafficking of the nicotinic acetylcholine receptor. Neuroscience 2004, 128, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, Y.; Zhang, T.; Zhang, B.; Chen, L.; Zhao, L.; Chen, L. Simvastatin Enhances Activity and Trafficking of α7 Nicotinic Acetylcholine Receptor in Hippocampal Neurons Through PKC and CaMKII Signaling Pathways. Front. Pharm. 2018, 9. [Google Scholar] [CrossRef]
- Báez-Pagán, C.A.; del Hoyo-Rivera, N.; Quesada, O.; Otero-Cruz, J.D.; Lasalde-Dominicci, J.A. Heterogeneous Inhibition in Macroscopic Current Responses of Four Nicotinic Acetylcholine Receptor Subtypes by Cholesterol Enrichment. J. Membr Biol. 2016, 249, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Barthold, D.; Joyce, G.; Brinton, R.D.; Wharton, W.; Kehoe, P.G.; Zissimopoulos, J. Association of combination statin and antihypertensive therapy with reduced Alzheimer’s disease and related dementia risk. PLoS ONE 2020, 15, e0229541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrantes, F.J.; Borroni, V.; Vallés, S. Neuronal nicotinic acetylcholine receptor-cholesterol crosstalk in Alzheimer’s disease. Febs Lett. 2010, 584, 1856–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, K.; Zhao, T.; Qu, G.; Liu, H.; Chen, X.; Sun, Y. The efficacy of statins in the treatment of Alzheimer’s disease: A meta-analysis of randomized controlled trial. Neurol. Sci. 2020, 41, 1391–1404. [Google Scholar] [CrossRef]
- Jick, H.; Zornberg, G.L.; Jick, S.S.; Seshadri, S.; Drachman, D.A. Statins and the risk of dementia. Lancet 2000, 356, 1627–1631. [Google Scholar] [CrossRef]
- Wolozin, B. Cholesterol, statins and dementia. Curr. Opin. Lipidol. 2004, 15, 667–672. [Google Scholar] [CrossRef]
- Johnson-Anuna, L.N.; Eckert, G.P.; Keller, J.H.; Igbavboa, U.; Franke, C.; Fechner, T.; Schubert-Zsilavecz, M.; Karas, M.; Müller, W.E.; Wood, W.G. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J. Pharmacol. Exp. Ther. 2005, 312, 786–793. [Google Scholar] [CrossRef] [Green Version]
- Sodero, A.O.; Barrantes, F.J. Pleiotropic effects of statins on brain cells. Biochim. Biophys. Acta Biophys. Incl. Photosynth. 2020, 1862, 183340. [Google Scholar] [CrossRef]
- Samuel, N.; Wonnacott, S.; Lindstrom, J.; Futerman, A.H. Parallel increases in [α-125I]bungarotoxin binding and α7 nicotinic subunit immunoreactivity during the development of rat hippocampal neurons in culture. Neurosci. Lett. 1997, 222, 179–182. [Google Scholar] [CrossRef]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.B.; Korade, Z.; Mirnics, K. Cholesterol Biosynthesis and Uptake in Developing Neurons. ACS Chem. Neurosci. 2019, 10, 3671–3681. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, C.; Eckert, G.P.; Mueller, W.E. Statin effects on cholesterol micro-domains in brain plasma membranes. Biochem. Pharmacol. 2003, 65, 843–856. [Google Scholar] [CrossRef]
- Eckert, G.P.; Kirsch, C.; Mueller, W.E. Differential effects of lovastatin treatment on brain cholesterol levels in normal and apoE-deficient mice. Neuroreport 2001, 12, 883–887. [Google Scholar] [CrossRef]
- Sebastião, A.M.; Colino-Oliveira, M.; Assaife-Lopes, N.; Dias, R.B.; Ribeiro, J.A. Lipid rafts, synaptic transmission and plasticity: Impact in age-related neurodegenerative diseases. Neuropharmacology 2013, 64, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Dason, J.S.; Smith, A.J.; Marin, L.; Charlton, M.P. Vesicular sterols are essential for synaptic vesicle cycling. J. Neurosci. 2010, 30, 15856–15865. [Google Scholar] [CrossRef]
- Renner, M.; Choquet, D.; Triller, A. Control of the postsynaptic membrane viscosity. J. Neurosci. 2009, 29, 2926–2937. [Google Scholar] [CrossRef]
- Brachet, A.; Norwood, S.; Brouwers, J.F.; Palomer, E.; Helms, J.B.; Dotti, C.G.; Esteban, J.A. LTP-triggered cholesterol redistribution activates Cdc42 and drives AMPA receptor synaptic delivery. J. Cell Biol. 2015, 208, 791–806. [Google Scholar] [CrossRef] [Green Version]
- Korinek, M.; Gonzalez-Gonzalez, I.M.; Smejkalova, T.; Hajdukovic, D.; Skrenkova, K.; Krusek, J.; Horak, M.; Vyklicky, L. Cholesterol modulates presynaptic and postsynaptic properties of excitatory synaptic transmission. Sci. Rep. 2020, 10, 12651. [Google Scholar] [CrossRef]
- Martín-Segura, A.; Ahmed, T.; Casadomé-Perales, Á.; Palomares-Perez, I.; Palomer, E.; Kerstens, A.; Munck, S.; Balschun, D.; Dotti, C.G. Age-associated cholesterol reduction triggers brain insulin resistance by facilitating ligand-independent receptor activation and pathway desensitization. Aging Cell 2019, 18, e12932. [Google Scholar] [CrossRef]
- Sodero, A.O.; Weissmann, C.; Ledesma, M.D.; Dotti, C.G. Cellular stress from excitatory neurotransmission contributes to cholesterol loss in hippocampal neurons aging in vitro. Neurobiol. Aging 2011, 32, 1043–1053. [Google Scholar] [CrossRef]
- Thorvaldsson, V.; Skoog, I.; Johansson, B. Cholesterol and cognitive aging: Between-person and within-person associations in a population-based representative sample not on lipid-lowering medication. Psychol. Aging 2020, 35, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.G.; Ahmed, T.; Korovaichuk, A.; Venero, C.; Menchón, S.A.; Salas, I.; Munck, S.; Herreras, O.; Balschun, D.; Dotti, C.G. Constitutive hippocampal cholesterol loss underlies poor cognition in old rodents. EMBO Mol. Med. 2014, 6, 902–917. [Google Scholar] [CrossRef] [PubMed]
- Egawa, J.; Pearn, M.L.; Lemkuil, B.P.; Patel, P.M.; Head, B.P. Membrane lipid rafts and neurobiology: Age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 2016, 594, 4565–4579. [Google Scholar] [CrossRef] [Green Version]
- Gamba, P.; Staurenghi, E.; Testa, G.; Giannelli, S.; Sottero, B.; Leonarduzzi, G. A Crosstalk Between Brain Cholesterol Oxidation and Glucose Metabolism in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 556. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Wang, F.; Yang, J.; Peng, H.; Li, Y.; Li, B.; Wang, S. Revealing a Novel Landscape of the Association Between Blood Lipid Levels and Alzheimer’s Disease: A Meta-Analysis of a Case-Control Study. Front. Aging Neurosci. 2019, 11, 370. [Google Scholar] [CrossRef]
- Jin, U.; Park, S.J.; Park, S.M. Cholesterol Metabolism in the Brain and Its Association with Parkinson’s Disease. Exp. Neurobiol. 2019, 28, 554–567. [Google Scholar] [CrossRef] [Green Version]
- González-Guevara, E.; Cárdenas, G.; Pérez-Severiano, F.; Martínez-Lazcano, J.C. Dysregulated Brain Cholesterol Metabolism Is Linked to Neuroinflammation in Huntington’s Disease. Mov. Disord. 2020, 35, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M.; Gabr, M.T. Multitarget therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2019, 14, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Roensch, J.; Crisby, M.; Nordberg, A.; Xiao, Y.; Zhang, L.-J.; Guan, Z.-Z. Effects of statins on α7 nicotinic receptor, cholinesterase and α-form of secreted amyloid precursor peptide in SH-SY5Y cells. Neurochem. Int. 2007, 50, 800–806. [Google Scholar] [CrossRef]
- Chen, T.; Wang, C.; Sha, S.; Zhou, L.; Chen, L.; Chen, L. Simvastatin Enhances Spatial Memory and Long-Term Potentiation in Hippocampal CA1 via Upregulation of α7 Nicotinic Acetylcholine Receptor. Mol. Neurobiol. 2016, 53, 4060–4072. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.; Mulhaupt, F.; Myit, S.; Mach, F. Statins as a newly recognized type of immunomodulator. Nat. Med. 2000, 6, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Taqueti, V.R.; Ridker, P.M. Lipid-Lowering and Anti-Inflammatory Benefits of Statin Therapy: More Than Meets the Plaque. Circ. Cardiovasc Imaging 2017, 10, e006676. [Google Scholar] [CrossRef] [Green Version]
- Mohammadkhani, N.; Gharbi, S.; Rajani, H.F.; Farzaneh, A.; Mahjoob, G.; Hoseinsalari, A.; Korsching, E. Statins: Complex outcomes but increasingly helpful treatment options for patients. Eur. J. Pharm. 2019, 863, 172704. [Google Scholar] [CrossRef]
- Scorticati, C.; Formoso, K.; Frasch, A.C. Neuronal glycoprotein M6a induces filopodia formation via association with cholesterol-rich lipid rafts. J. Neurochem. 2011, 119, 521–531. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borroni, V.; Kamerbeek, C.; Pediconi, M.F.; Barrantes, F.J. Lovastatin Differentially Regulates α7 and α4 Neuronal Nicotinic Acetylcholine Receptor Levels in Rat Hippocampal Neurons. Molecules 2020, 25, 4838. https://doi.org/10.3390/molecules25204838
Borroni V, Kamerbeek C, Pediconi MF, Barrantes FJ. Lovastatin Differentially Regulates α7 and α4 Neuronal Nicotinic Acetylcholine Receptor Levels in Rat Hippocampal Neurons. Molecules. 2020; 25(20):4838. https://doi.org/10.3390/molecules25204838
Chicago/Turabian StyleBorroni, Virginia, Constanza Kamerbeek, María F. Pediconi, and Francisco J. Barrantes. 2020. "Lovastatin Differentially Regulates α7 and α4 Neuronal Nicotinic Acetylcholine Receptor Levels in Rat Hippocampal Neurons" Molecules 25, no. 20: 4838. https://doi.org/10.3390/molecules25204838
APA StyleBorroni, V., Kamerbeek, C., Pediconi, M. F., & Barrantes, F. J. (2020). Lovastatin Differentially Regulates α7 and α4 Neuronal Nicotinic Acetylcholine Receptor Levels in Rat Hippocampal Neurons. Molecules, 25(20), 4838. https://doi.org/10.3390/molecules25204838