
Salinosporamide A, a Marine-Derived Proteasome Inhibitor, Inhibits T Cell Activation through Regulating Proliferation and the Cell Cycle

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Result
2.1. High Concentrations of Salinosporamide are Cytotoxic to T Cells and Leads to Apoptosis
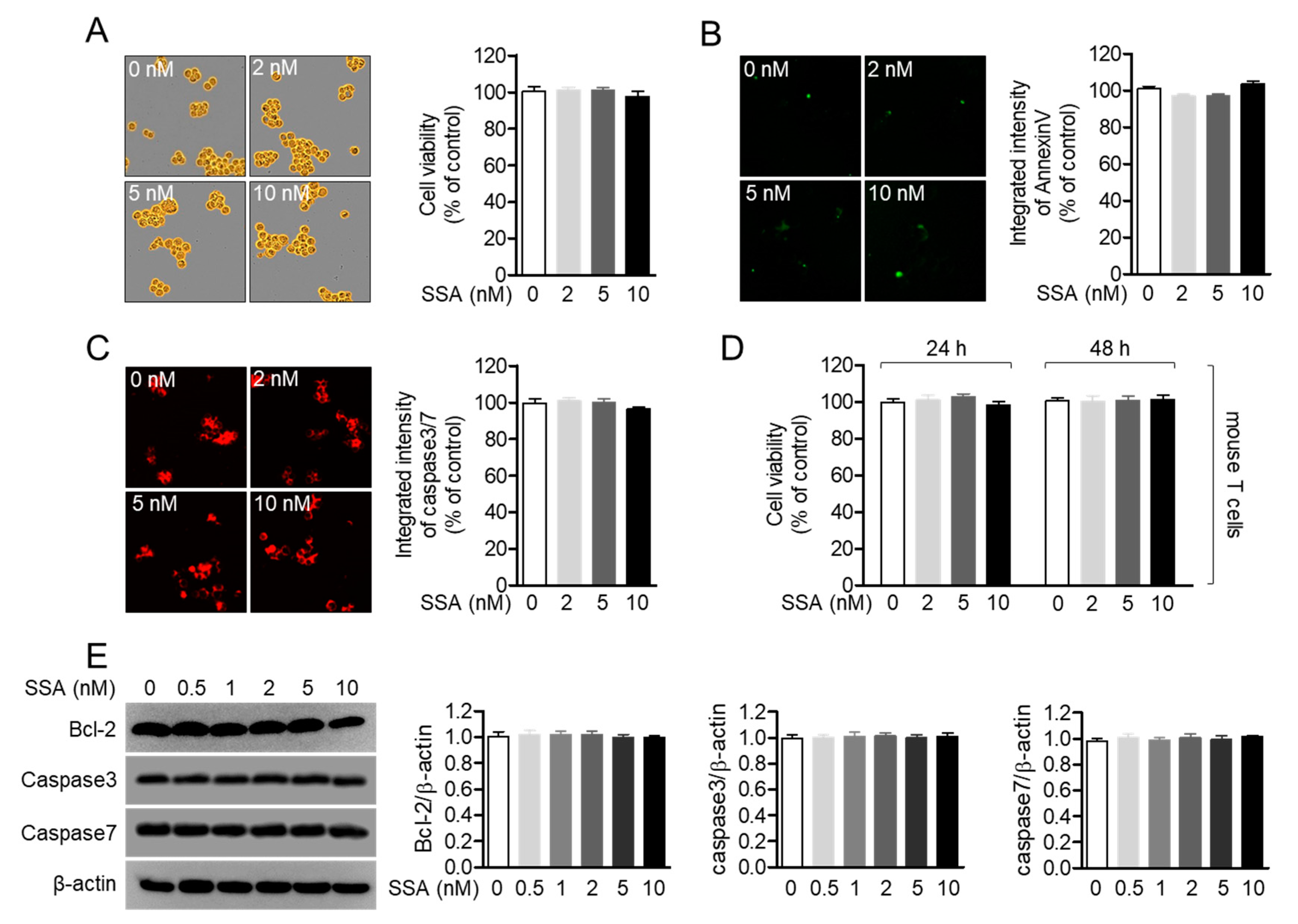
2.2. Treatment with 10 nM Salinosporamide A is not Cytotoxic to T Cells
2.3. Treatment with 10 nM Salinosporamide A Reduces T Cell Proliferation
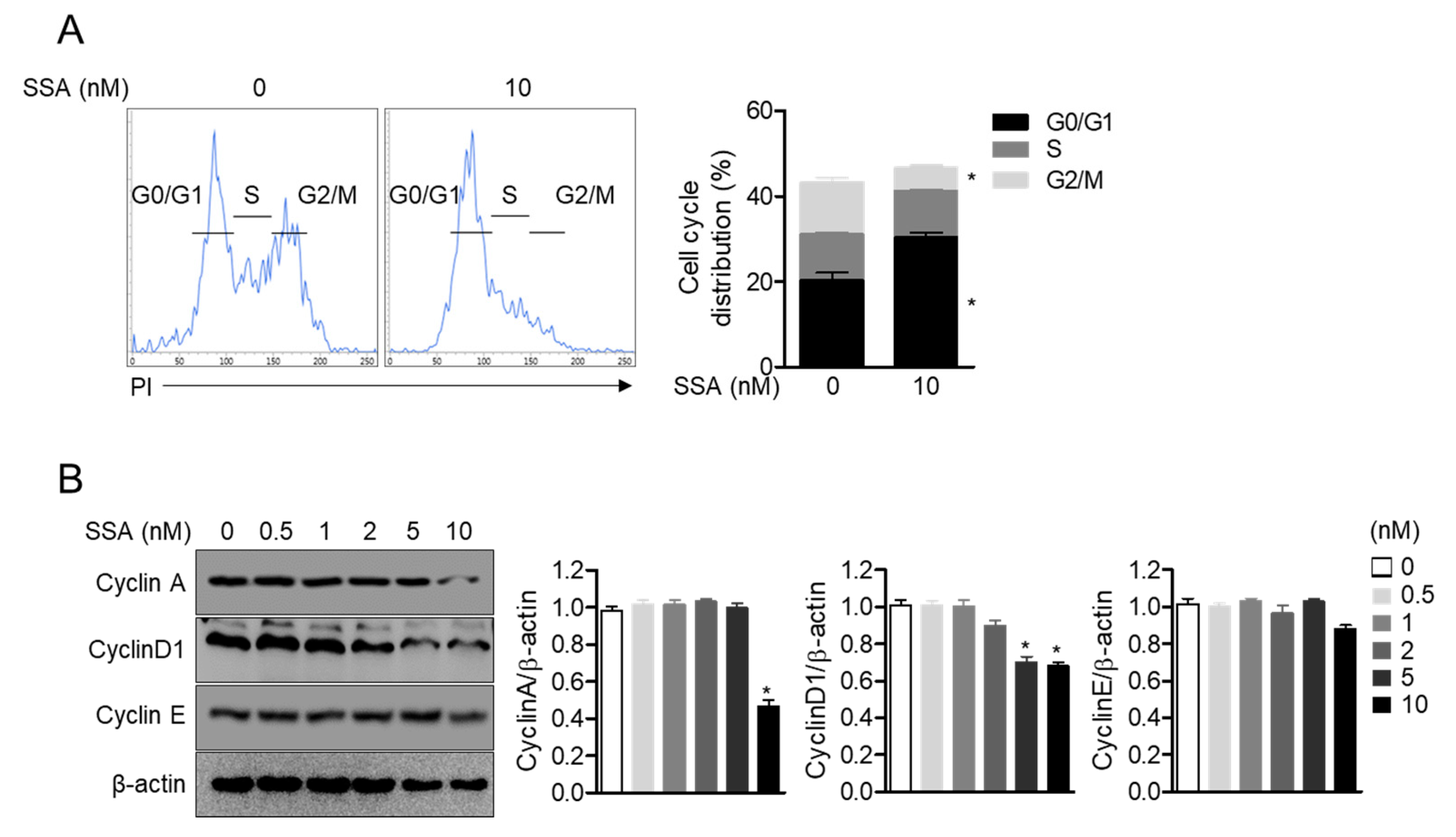
2.4. Treatment with 10 nM Salinosporamide A Leads to Cell Cycle Arrest and Regulates Cyclin-Dependent Kinase Expression in T Cells
2.5. Treatment with 10 nM Salinosporamide A Inhibits T-Cell Activity Following TCR-Mediated and PMA/A23187 Stimulation
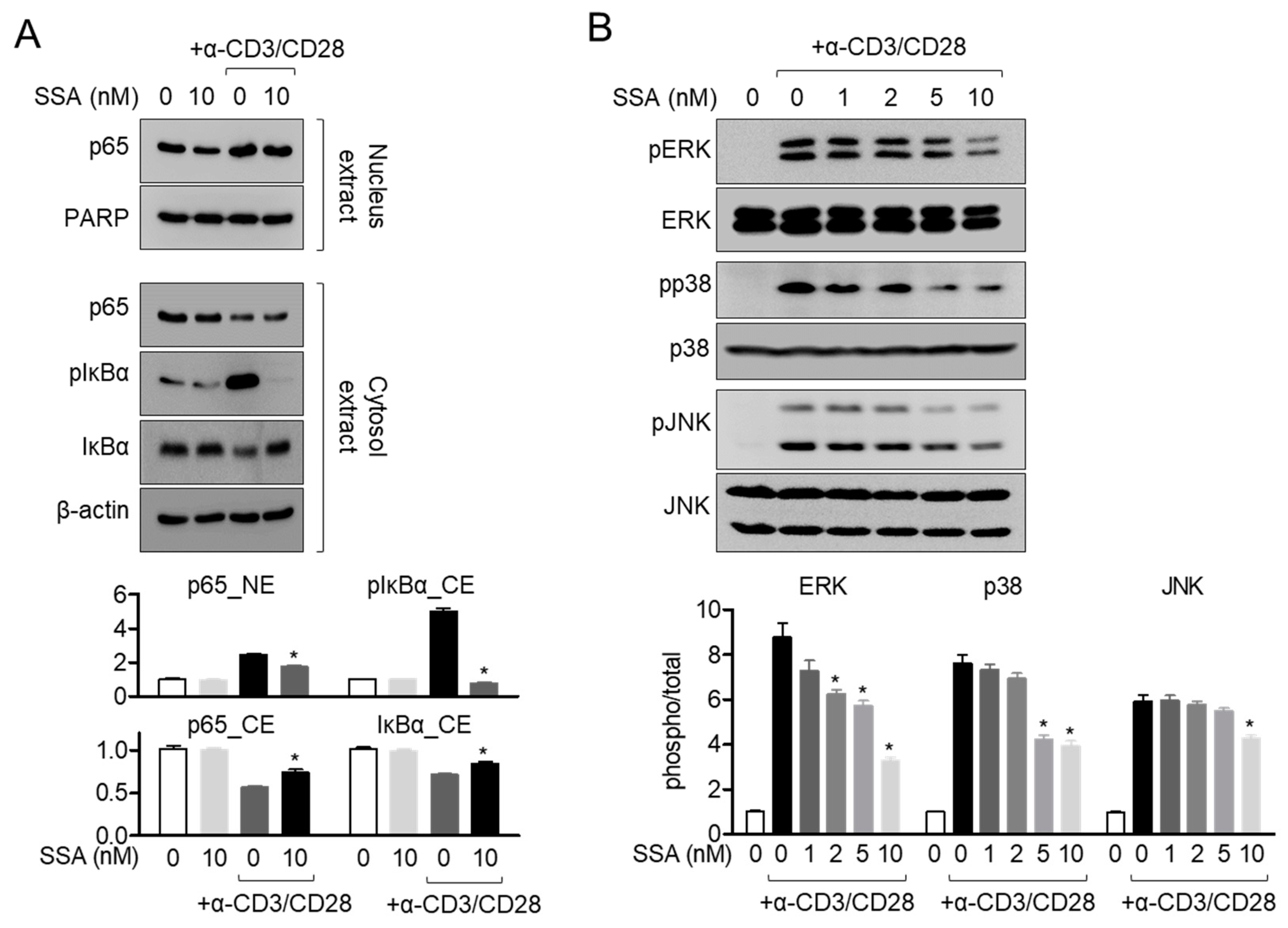
2.6. Treatment with 10 nM Salinosporamide A Dampens the MAPK Pathway in Activated T Cells
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Reagents and Antibodies
4.3. MTT Assay
4.4. Assessment of the Expressions of AnnexinV and Caspase3/7 by IncuCyte
4.5. AnnexinV/PI Apoptosis Assay
4.6. Isolation of Mouse T Cells
4.7. Western Blot Assay
4.8. Proliferation Assay
4.9. Cell Cycle Arrest Assay
4.10. Real-Time Quantitative PCR
4.11. Statistics
Author Contributions
Funding
Conflicts of Interest
References
- Charles, A.; Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. T Cell-mediated immunity. In Immunobiology: The immune System in Health and Diseases; Garland Science: New York, NY, USA, 2001. Available online: https://www.ncbi.nlm.nih.gov/books/NBK10762/ (accessed on 29 October 2020).
- Skapenko, A.; Leipe, J.; Lipsky, P.E.; Schulze-Koops, H. The role of the T cell in autoimmune inflammation. Arthritis Res. Ther. 2005, 7, S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilli, D.; Zou, A.; Tea, F.; Dale, R.C.; Brilot, F. Expanding role of T cells in human autoimmune diseases of the central nervous system. Front. Immunol. 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugamura, K.; Asao, H.; Kondo, M.; Tanaka, N.; Ishii, N.; Nakamura, M.; Takeshita, T. The common γ-chain for multiple cytokine receptors. Adv. Immunol. 1995, 59, 225–277. [Google Scholar] [PubMed]
- Shi, M.; Lin, T.H.; Appell, K.C.; Berg, L.J. Cell Cycle Progression following Naive T Cell Activation Is Independent of Jak3/Common γ-Chain Cytokine Signals. J. Immunol. 2009, 183, 4493–4501. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuka, S.; Ogawa, S.; Wakamatsu, E.; Abe, R. Cell cycle arrest caused by MEK/ERK signaling is a mechanism for suppressing growth of antigen-hyperstimulated effector T cells. Int. Immunol. 2016, 28, 547–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, C.J. G1 phase progression: Cycling on cue. Cell 1994, 79, 551–555. [Google Scholar] [CrossRef]
- Robb, R.J.; Lin, Y. T-Cell Growth Factor. In Thymic Hormones and Lymphokines; Springer: Boston, MA, USA, 1984; pp. 247–256. [Google Scholar]
- Morgan, D.A.; Ruscetti, F.W.; Gallo, R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 1976, 193, 1007–1008. [Google Scholar] [CrossRef]
- Depper, J.M.; Leonard, W.J.; Robb, R.J.; Waldmann, T.A.; Greene, W.C. Blockade of the interleukin-2 receptor by anti-Tac antibody: Inhibition of human lymphocyte activation. J. Immunol. 1983, 131, 690–696. [Google Scholar]
- Wedel, J.; Hottenrott, M.C.; Stamellou, E.; Breedijk, A.; Tsagogiorgas, C.; Hillebrands, J.-L.; Yard, B.A. N-Octanoyl dopamine transiently inhibits T cell proliferation via G1 cell-cycle arrest and inhibition of redox-dependent transcription factors. J. Leukoc. Biol. 2014, 96, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Berges, C.; Haberstock, H.; Fuchs, D.; Miltz, M.; Sadeghi, M.; Opelz, G.; Daniel, V.; Naujokat, C. Proteasome inhibition suppresses essential immune functions of human CD4 + T cells. Immunology 2008, 124, 234–246. [Google Scholar] [CrossRef]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew. Chem. Int. Ed. 2003, 42, 355–357. [Google Scholar] [CrossRef]
- Harer, S.L.; Bhatia, M.S.; Bhatia, N.M. Proteasome inhibitors mechanism; Source for design of newer therapeutic agents. J. Antibiot. (Tokyo) 2012, 65, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Fribley, A.; Wang, C.Y. Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol. Ther. 2006, 5, 745–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, N.; Peng, Z. MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia. Pac. J. Clin. Oncol. 2013, 9, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Laphanuwat, P.; Jirawatnotai, S. Immunomodulatory Roles of Cell Cycle Regulators. Front. Cell Dev. Biol. 2019, 7, 23. [Google Scholar]
- Paul, S.; Schaefer, B.C. A new look at T cell receptor signaling to nuclear factor-κB. Trends Immunol. 2013, 34, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Wang, X.; Luo, H.; Chen, H.; Duguid, W. Proliferation Role of Proteasomes in T Cell Activation and proliferation. J. Immunol. 1998, 160, 788–801. [Google Scholar]
- Seufert, W.; Futcher, B.; Jentsch, S. Role of a ubiquitin-conjugating enzyme in degradation of S- and M-phase cyclins. Nature 1995, 373, 78–81. [Google Scholar] [CrossRef]
- Pagano, M.; Tam, S.W.; Theodoras, A.M.; Beer-Romero, P.; Del Sal, G.; Chau, V.; Yew, P.R.; Draetta, G.F.; Rolfe, M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 1995, 269, 682–685. [Google Scholar] [CrossRef]
- Widjaja, C.E.; Olvera, J.G.; Metz, P.J.; Phan, A.T.; Savas, J.N.; De Bruin, G.; Leestemaker, Y.; Berkers, C.R.; De Jong, A.; Florea, B.I.; et al. Proteasome activity regulates CD8+ T lymphocyte metabolism and fate specification. J. Clin. Investig. 2017, 127, 3609–3623. [Google Scholar] [CrossRef]
- Moran, E.; Carbone, F.; Augusti, V.; Patrone, F.; Ballestrero, A.; Nencioni, A. Proteasome Inhibitors as Immunosuppressants: Biological Rationale and Clinical Experience. Semin. Hematol. 2012, 49, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The Proteasome Inhibitor PS-341 Inhibits Growth, Induces Apoptosis, and Overcomes Drug Resistance in Human Multiple Myeloma Cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar] [PubMed]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitinproteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef]
- Lee, J.; Kim, B.S.; Park, Y.; Lee, J.G.; Lim, B.J.; Jeong, H.J.; Kim, Y.S.; Huh, K.H. The effect of bortezomib on antibody-mediated rejection after kidney transplantation. Yonsei Med. J. 2015, 56, 1638–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbrugge, E.E.; Scheper, R.J.; Lems, W.F.; de Gruijl, T.D.; Jansen, G. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthr. Res. Ther. 2015, 17, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Welniak, L.A.; Panoskaltsis-Mortari, A.; O’Shaughnessy, M.J.; Liu, H.; Barao, I.; Riordan, W.; Sitcheran, R.; Wysocki, C.; Serody, J.S.; et al. Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitior bortezomib. Proc. Nat. Acad. Sci. USA 2004, 101, 8120–8125. [Google Scholar] [CrossRef] [Green Version]
- Vodanovic-Jankovic, S.; Hari, P.; Jacobs, P.; Komorowski, R.; Drobyski, W.R. NF-κB as a target for the prevention of graft-versus-host disease: Comparative efficacy of bortezomib and PS-1145. Blood 2006, 107, 827–834. [Google Scholar] [CrossRef]
- Lee, H.S.; Jeong, G.S. Chrysophanol mitigates T cell activation by regulating the expression of CD40 ligand in activated T cells. Int. J. Mol. Sci. 2020, 21, 6122. [Google Scholar] [CrossRef]







Sample Availability: Samples of the compounds are not available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-S.; Jeong, G.-S. Salinosporamide A, a Marine-Derived Proteasome Inhibitor, Inhibits T Cell Activation through Regulating Proliferation and the Cell Cycle. Molecules 2020, 25, 5031. https://doi.org/10.3390/molecules25215031
Lee H-S, Jeong G-S. Salinosporamide A, a Marine-Derived Proteasome Inhibitor, Inhibits T Cell Activation through Regulating Proliferation and the Cell Cycle. Molecules. 2020; 25(21):5031. https://doi.org/10.3390/molecules25215031
Chicago/Turabian StyleLee, Hyun-Su, and Gil-Saeng Jeong. 2020. "Salinosporamide A, a Marine-Derived Proteasome Inhibitor, Inhibits T Cell Activation through Regulating Proliferation and the Cell Cycle" Molecules 25, no. 21: 5031. https://doi.org/10.3390/molecules25215031
APA StyleLee, H. -S., & Jeong, G. -S. (2020). Salinosporamide A, a Marine-Derived Proteasome Inhibitor, Inhibits T Cell Activation through Regulating Proliferation and the Cell Cycle. Molecules, 25(21), 5031. https://doi.org/10.3390/molecules25215031