
Study on the Influence of Chirality in the Threading of Calix[6]arene Hosts with Dialkylammonium Axles

 ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
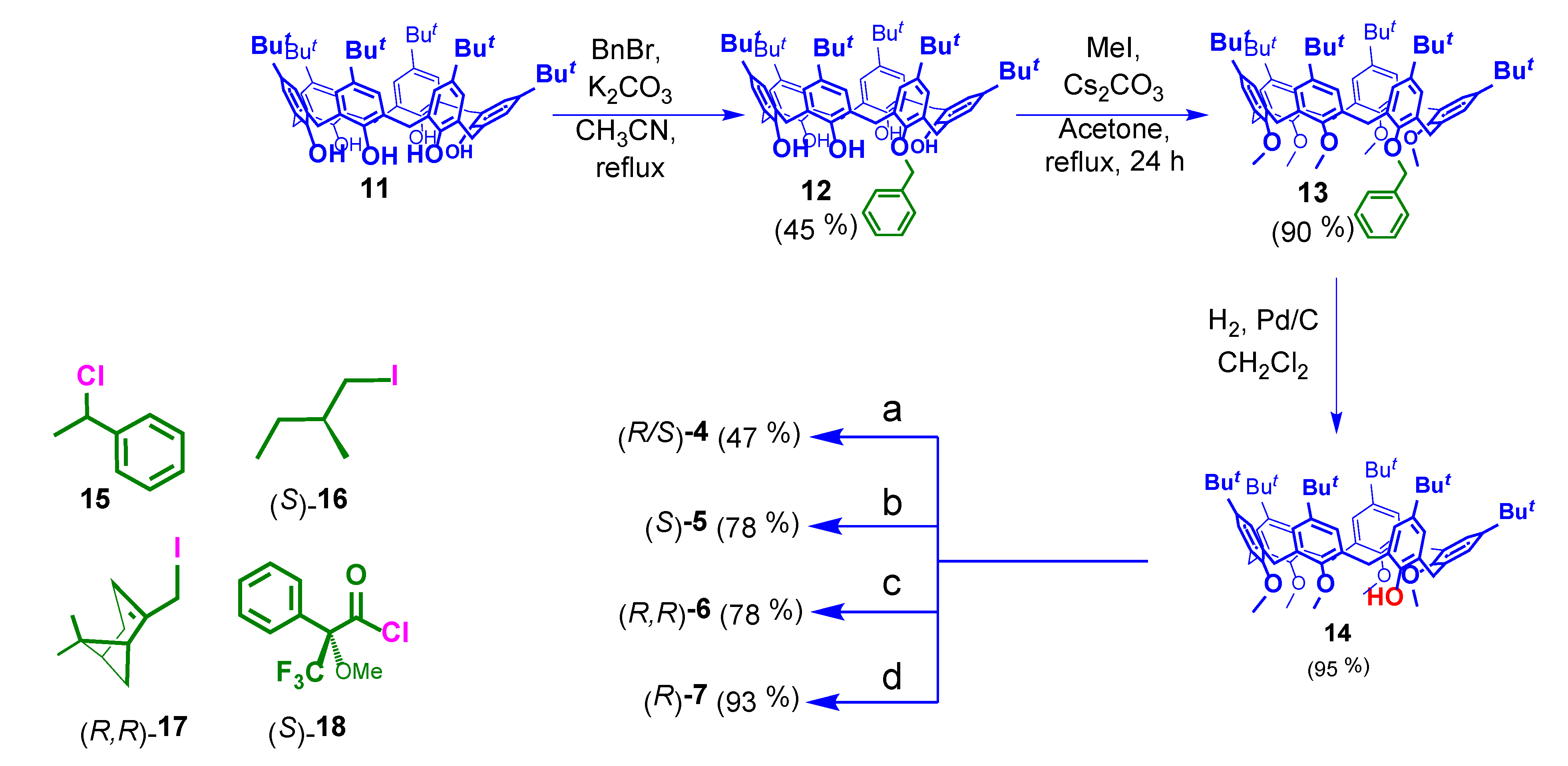
2.1. Synthesis of Chiral Calixarenes
2.2. Syntheses of Chiral Axles
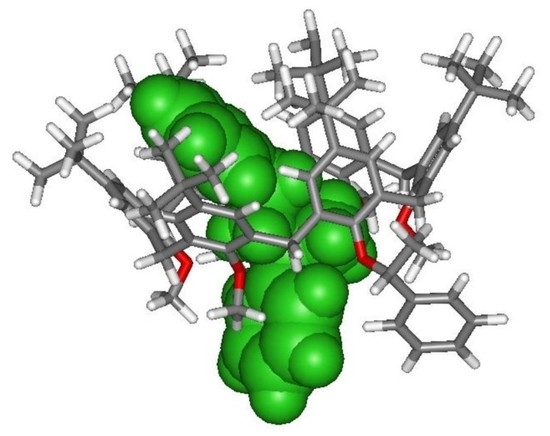
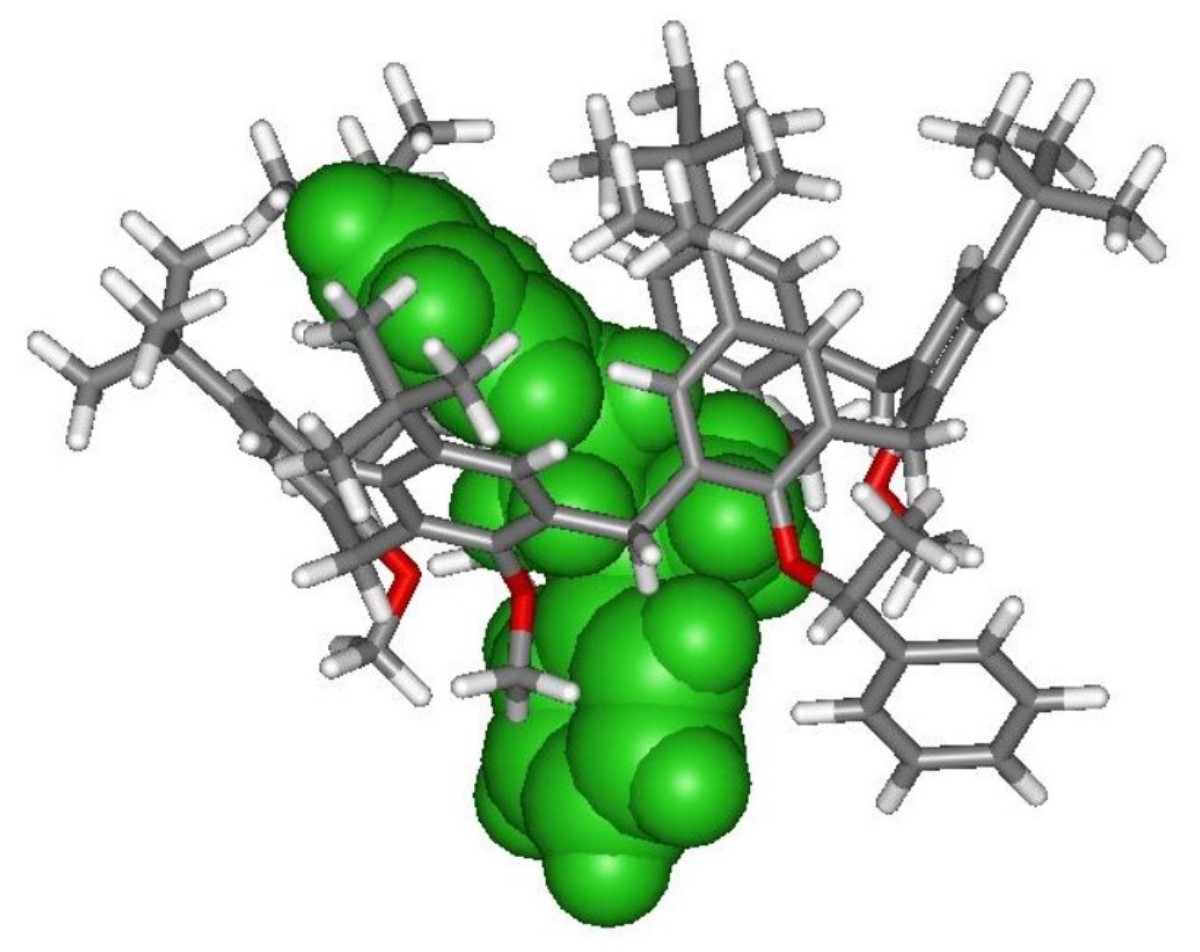
2.3. NMR Threading Studies of Chiral Calixarene (R/S)-4
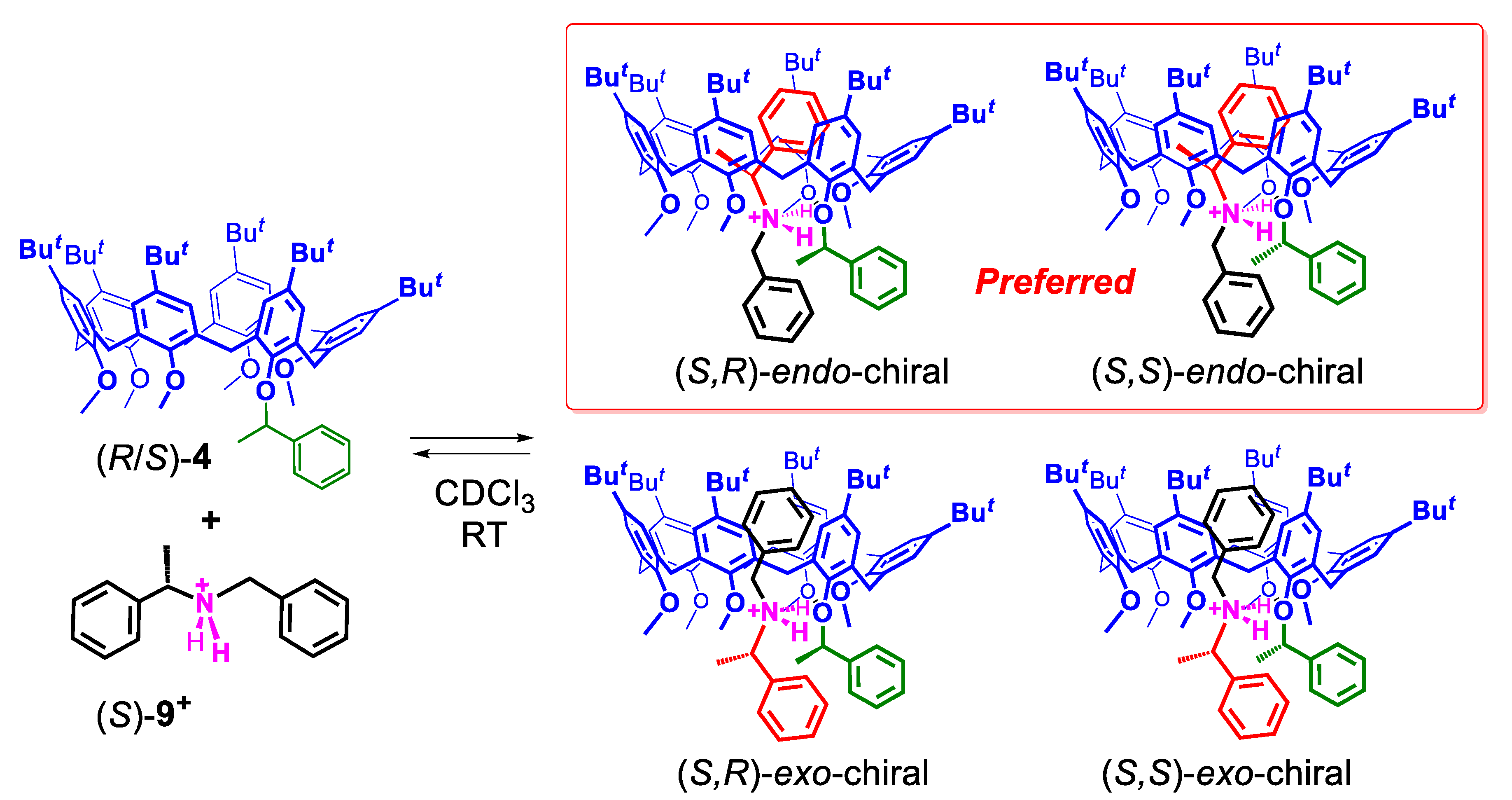
2.3.1. Threading of Racemic Calix[6]arene-wheel (R/S)-4 with Dibenzylammonium Axles 8+ and 9+
2.3.2. Threading of Racemic Calix[6]arene-wheel (R/S)-4 with Isopropylbenzylammonium Axle (S)-10+
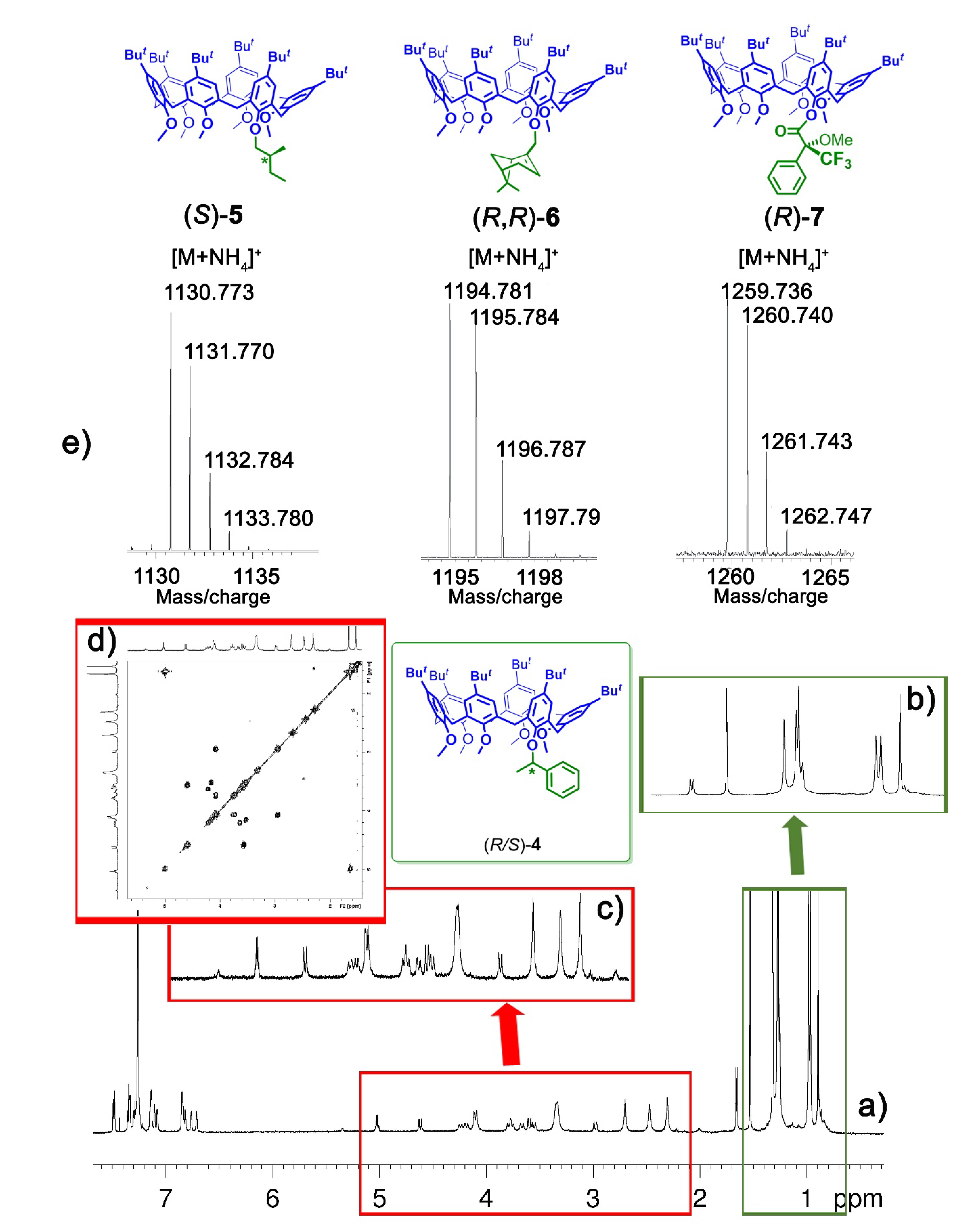
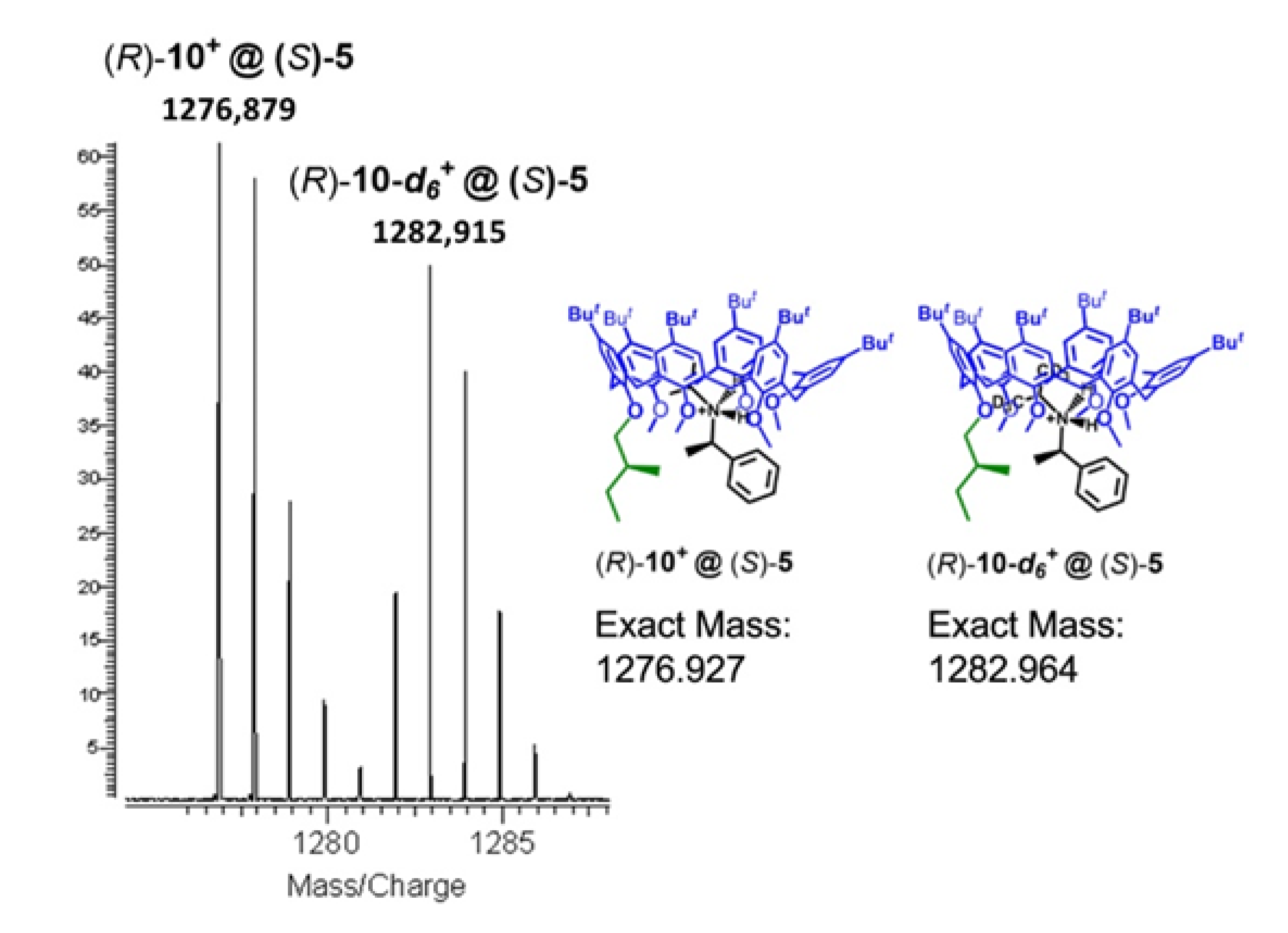
2.4. MS Experiments of the Threading of Chiral Calixarene (S)-5, (R)-6, and (R>)-7
- IS/IR-d6> 1 means that a given chiral host binds more strongly the (S)-enantiomer (the larger the IS/IR-d6ratio value, and higher the degree of chiral discrimination by the host).
- IS/IR-d6 < 1 means that a given chiral host binds more strongly the (R)-labelled guest.
- IS/IR-d6= 1.0 ± 0.1 means that a given chiral host cannot differentiate the chirality of a given guest.
2.4.1. Concentration Effect
2.4.2. Isotopic Effects
2.4.3. Chiral Discrimination
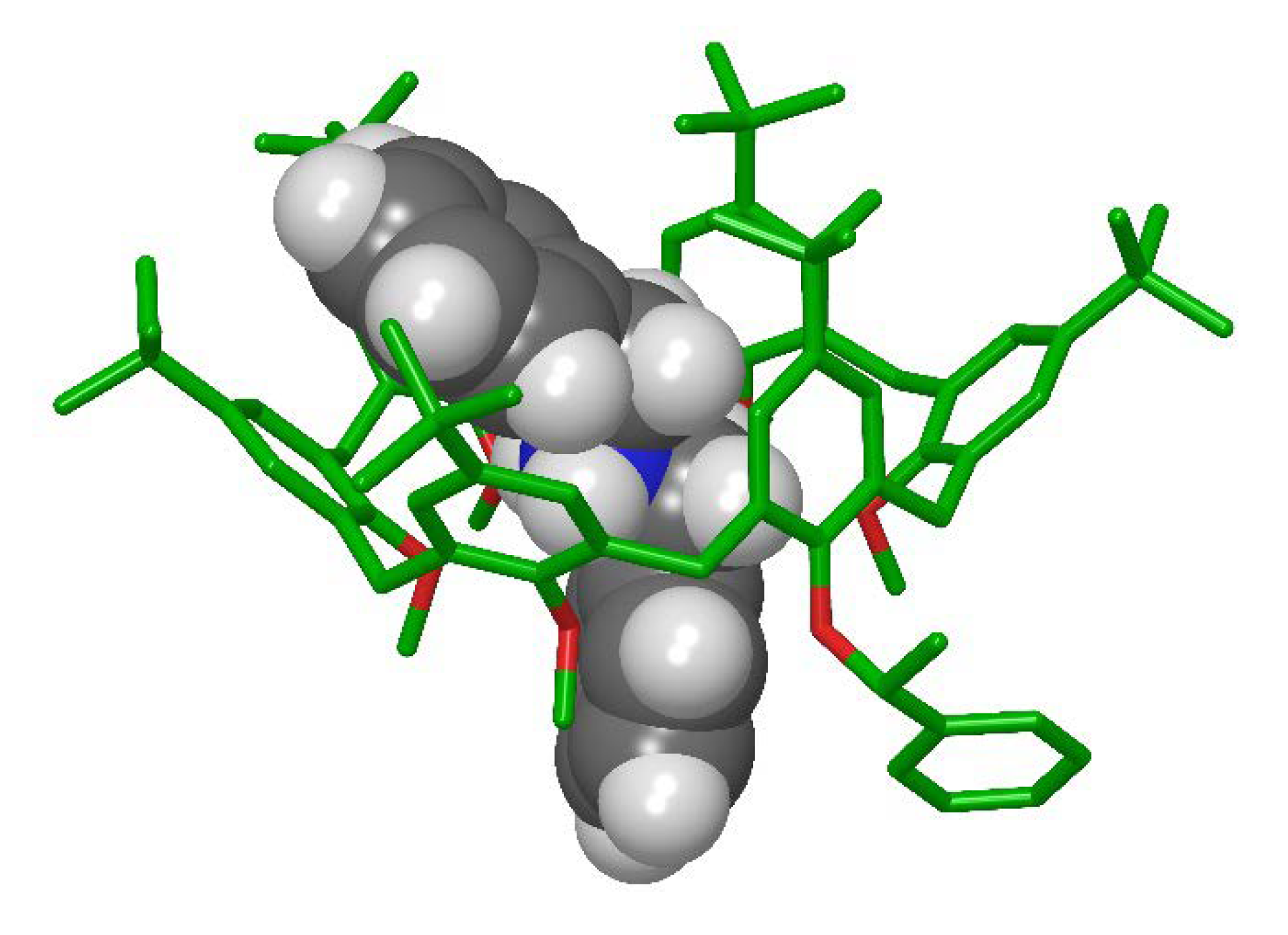
2.5. Possible Rationales
3. Conclusions
4. Experimental Section
4.1. General Procedure for MS Experiments (Isotopic Effect)
4.1.1. Sample Preparation
4.1.2. MS Conditions
4.1.3. IR/IR-dn Evaluation
4.2. General Procedure for MS Experiments (Chiral Recognition Effect)
4.3. DFT Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bruns, C.J.; Stoddart, J.F. The Nature of the Mechanical Bond: From Molecules to Machines, 1st ed.; John Wiley & Sons: New York, NY, USA, 2017. [Google Scholar]
- Feringa, B.L. The Art of Building Small: From Molecular Switches to Motors. Angew. Chem. Int. Ed. 2017, 56, 11060–11078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvage, J.-P. From Chemical Topology to Molecular Machines. Angew. Chem. Int. Ed. 2017, 56, 11080–11093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoddart, J.F. Mechanically Interlocked Molecules (MIMs)-Molecular Shuttles, Switches, and Machines. Angew. Chem. Int. Ed. 2017, 56, 11094–11125. [Google Scholar] [CrossRef]
- Yu, H.; Luo, Y.; Beverly, K.; Stoddart, J.F.; Tseng, H.; Heath, J.R. The Molecule–Electrode Interface in Single-Molecule Transistors. Angew. Chem. Int. Ed. 2003, 42, 5706–5711. [Google Scholar] [CrossRef] [PubMed]
- Coskun, A.; Spruell, J.M.; Barin, G.; Dichtel, W.R.; Flood, A.H.; Botrosghi, Y.Y.; Stoddart, J.F. High hopes: Can molecular electronics realise its potential? Chem. Soc. Rev. 2012, 41, 4827–4859. [Google Scholar] [CrossRef]
- Mendes, P.M.; Flood, A.H.; Stoddart, J.F. Nanoelectronic devices from self-organized molecular switches. Appl. Phys. A 2005, 80, 1197–1209. [Google Scholar] [CrossRef]
- Kassem, S.; Lee, A.T.L.; Leigh, D.A.; Marcos, V.; Palmer, L.I.; Pisano, S. Stereodivergent synthesis with a programmable molecular machine. Nature 2017, 549, 374–378. [Google Scholar] [CrossRef]
- Erbas-Cakmak, S.; Leigh, D.A.; McTernan, C.T.; Nussbaumer, A.L. Artificial molecular machines. Chem. Rev. 2015, 115, 10081–10206. [Google Scholar] [CrossRef] [Green Version]
- Balzani, V.; Credi, A.; Venturi, M. Molecular Devices and Machines, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Beswick, J.; Blanco, V.; De Bo, G.; Leigh, D.A.; Lewandowska, U.; Lewandowski, B.; Mishiro, K. Selecting reactions and reactants using a switchable rotaxane organocatalyst with two different active sites. Chem. Sci. 2015, 6, 140–143. [Google Scholar] [CrossRef] [Green Version]
- Blanco, V.; Leigh, D.A.; Marcos, V. Artificial switchable catalysts. Chem. Soc. Rev. 2015, 44, 5341–5370. [Google Scholar] [CrossRef] [Green Version]
- Blanco, V.; Leigh, D.A.; Lewandowska, U.; Lewandowski, B.; Marcos, V. Goldberg Active Template Synthesis of a [2]Rotaxane Ligand for Asymmetric Transition-Metal Catalysis. J. Am. Chem. Soc. 2014, 136, 15775–15780. [Google Scholar] [CrossRef] [PubMed]
- Molecular Catenanes, Rotaxanes and Knots: A Journey Through the World of Molecular Topology; Sauvage, J.P.; Dietrich-Buchecker, C. (Eds.) Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Jamieson, E.M.G.; Modicom, F.; Goldup, S.M. Chirality in rotaxanes and catenanes. Chem. Soc. Rev. 2018, 47, 5266–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, N.H. Chiral Catenanes and Rotaxanes: Fundamentals and Emerging Applications. Chem. Eur. J. 2018, 24, 3101–3112. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.C.; Marques, I.; Félix, V.; Beer, P.D. Enantioselective Anion Recognition by Chiral Halogen-Bonding [2]Rotaxanes. J. Am. Chem. Soc. 2017, 139, 12228–12239. [Google Scholar] [CrossRef]
- Heard, A.W.; Goldup, S.M. Synthesis of a Mechanically Planar Chiral Rotaxane Ligand for Enantioselective Catalysis. Chem 2020, 6, 994–1006. [Google Scholar] [CrossRef]
- Blanco, V.; Leigh, D.A.; Marcos, V.; Morales-Serna, J.A.; Nussbaumer, A.L. A Switchable [2]Rotaxane Asymmetric Organocatalyst That Utilizes an Acyclic Chiral Secondary Amine. J. Am. Chem. Soc. 2014, 136, 4905–4908. [Google Scholar] [CrossRef]
- Cakmak, Y.; Erbas-Cakmak, S.; Leigh, D.A. Asymmetric Catalysis with a Mechanically Point-Chiral Rotaxane. J. Am. Chem. Soc. 2016, 138, 1749–1751. [Google Scholar] [CrossRef] [Green Version]
- Leigh, D.A.; Wong, J.K.Y.; Dehez, F.; Zerbetto, F. Unidirectional rotation in a mechanically interlocked molecular rotor. Nature 2003, 424, 174–179. [Google Scholar] [CrossRef]
- Lewandowski, B.; De Bo, G.W.; Ward, J.; Papmeyer, M.; Kuschel, S.; Aldegunde, M.J.; Gramlich, P.M.E.; Heckmann, D.; Goldup, S.M.; D’Souza, D.M.; et al. Sequence-specific peptide synthesis by an artificial small-molecule machine. Science 2013, 339, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.R.; Solà, J.; Carlone, A.; Goldup, S.M.; Lebrasseur, N.; Leigh, D.A. An autonomous chemically fuelled small-molecule motor. Nature 2016, 534, 235–240. [Google Scholar] [CrossRef] [Green Version]
- Ashton, P.R.; Iriepa, I.; Reddington, M.V.; Spencer, N.; Slawin, A.M.Z.; Stoddart, J.F.; Williams, D.J. An optically-active [2]catenane made to order. Tetrahedron Lett. 1994, 35, 4835–4838. [Google Scholar] [CrossRef]
- Ashton, P.R.; Heiss, A.M.; Pasini, D.; Raymo, F.M.; Shipway, A.N.; Stoddart, J.F.; Spencer, N. Diastereoselective Self-Assembly of [2]Catenanes. Eur. J. Org. Chem. 1999, 995–1004. [Google Scholar] [CrossRef]
- Yamamoto, C.; Okamoto, Y.; Schmidt, T.; Jäger, R.; Vögtle, F. Enantiomeric resolution of cycloenantiomeric rotaxane, topologically chiral catenane, and pretzelshaped molecules: Observation of pronounced circular dichroism. J. Am. Chem. Soc. 1997, 119, 10547–10548. [Google Scholar] [CrossRef]
- Kaida, Y.; Okamoto, Y.; Chambron, J.-C.; Mitchell, D.K.; Sauvage, J.-P. The separation of optically active copper (I) catenates. Tetrahedron Lett. 1993, 34, 1019–1022. [Google Scholar] [CrossRef]
- Bordoli, J.R.; Goldup, S.M. An Efficient Approach to Mechanically Planar Chiral Rotaxanes. J. Am. Chem. Soc. 2014, 136, 4817–4820. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, Y.; Ikeyatsu, K.; Mutoh, Y.; Hosoya, S.; Saito, S. Synthesis of mechanically planar chiral rac-[2]rotaxanes by partitioning of an achiral [2]rotaxane: Stereoinversion induced by shuttling. Org. Lett. 2017, 19, 4347–4350. [Google Scholar] [CrossRef]
- Goldup, S.M. Mechanical chirality: A chiral catalyst with a ring to it. Nat. Chem. 2016, 8, 404–406. [Google Scholar] [CrossRef] [Green Version]
- Gaeta, C.; Talotta, C.; De Rosa, M.; Soriente, A.; Neri, P. Calixarenes and Beyond; Neri, P., Sessler, J.L., Wang, M.-X., Eds.; Springer: Dordrecht, The Netherlands, 2016; pp. 783–809. [Google Scholar]
- Gaeta, C.; Troisi, F.; Neri, P. endo-Cavity Complexation and Through-the-Annulus Threading of Large Calixarenes Induced by Very Loose Alkylammonium Ion Pairs. Org. Lett. 2010, 12, 2092–2095. [Google Scholar] [CrossRef]
- Gaeta, C.; Talotta, C.; Margarucci, L.; Casapullo, A.; Neri, P. Through-the-Annulus Threading of the Larger Calix[8]arene Macrocycle. J. Org. Chem. 2013, 78, 7627–7638. [Google Scholar] [CrossRef]
- Gaeta, C.; Talotta, C.; Farina, F.; Camalli, M.; Campi, G.; Neri, P. Conformational Features and Recognition Properties of a Conformationally Blocked Calix[7]arene Derivative. Chem. Eur. J. 2012, 18, 1219–1230. [Google Scholar] [CrossRef]
- Arduini, A.; Orlandini, G.; Secchi, A.; Credi, A.; Silvi, S.; Venturi, M. Calixarenes and Beyond; Neri, P., Sessler, J.L., Wang, M.-X., Eds.; Springer: Dordrecht, The Netherlands, 2016; pp. 761–781. [Google Scholar]
- Talotta, C.; Gaeta, C.; Qi, Z.; Schalley, C.A.; Neri, P. Pseudorotaxanes with Self-Sorted Sequence and Stereochemical Orientation. Angew. Chem. Int. Ed. 2013, 52, 7437–7441. [Google Scholar] [CrossRef] [PubMed]
- Talotta, C.; Gaeta, C.; Neri, P. Stereoprogrammed Direct Synthesis of Calixarene-Based [3]Rotaxanes. Org. Lett. 2012, 14, 3104–3107. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, C.; Talotta, C.; Mirra, S.; Margarucci, L.; Casapullo, A.; Neri, P. Catenation of Calixarene Annulus. Org. Lett. 2013, 15, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Talotta, C.; De Simone, N.A.; Gaeta, C.; Neri, P. Calix[6]arene threading with weakly interacting tertiary ammonium axles: Generation of chiral pseudorotaxane architectures. Org. Lett. 2015, 17, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.G.; Verboom, W.; Reinhoudt, D.N.; Casnati, A.; Freriks, M.; Pochini, A.; Ugozzoli, F.; Ungaro, R.; Nieto, P.M.; Carramolino, M.; et al. Procedures for the Selective Alkylation of Calix[6]arenes at the Lower Rim. Synthesis 1993, 4, 380–386. [Google Scholar] [CrossRef] [Green Version]
- De Mendoza, J.; Carramolino, M.; Cuevas, F.; Manule Nieto, P.; Reinhoudt, D.N.; Verboom, W.; Ungaro, R.; Casnati, A. Selective Functionalization of Calix[6]arenes at the Upper Rim. Synthesis 1994, 1, 47–50. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, M.; Soriente, A.; Concilio, G.; Talotta, C.; Gaeta, C.; Neri, P. Nucleophilic Functionalization of the Calix [6] arene Para- and Meta-Position via p-Bromodienone Route. J. Org. Chem. 2015, 80, 7295–7300. [Google Scholar] [CrossRef]
- Suezawa, H.; Ishihara, S.; Umezawa, Y.; Tsuboyama, S.; Nishio, M. The Aromatic CH/π Hydrogen Bond as an Important Factor in Determining the Relative Stability of Diastereomeric Salts Relevant to Enantiomeric Resolution−A Crystallographic Database Study. Eur. J. Org. Chem. 2004, 2004, 4816–4822. [Google Scholar] [CrossRef]
- Schug, K.A.; Maier, N.M.; Lindner, W. Deuterium isotope effects observed during competitive binding chiral recognition electrospray ionization—mass spectrometry of cinchona alkaloid-based systems. J. Mass Spectrom. 2006, 41, 157–161. [Google Scholar] [CrossRef]
- Behr, J.P.; Lehn, J.-M.; Moras, D.; Thierry, J.C. Chiral and functionalized face-discriminated and side-discriminated macrocyclic polyethers. Syntheses and crystal structures. J. Am. Chem. Soc. 1981, 103, 701–703. [Google Scholar] [CrossRef]
- Bako, P.; Fenichel, L.; Toke, L. The complexing ability of crown ethers incorporating glucose. J. Inclusion Phenom. Mol. Recognit. Chem. 1993, 16, 17–23. [Google Scholar] [CrossRef]
- Jung, Y.E.; Song, B.M.; Chang, S.K. Molecular recognition of alkyl- and arylakyl-amines in dischloromethane and chloroform by calix[4]-crown ethers. J. Chem. Soc. Perkin Trans. 1995, 2, 2031–2034. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talotta, C.; Concilio, G.; Della Sala, P.; Gaeta, C.; Schalley, C.A.; Neri, P. Study on the Influence of Chirality in the Threading of Calix[6]arene Hosts with Dialkylammonium Axles. Molecules 2020, 25, 5323. https://doi.org/10.3390/molecules25225323
Talotta C, Concilio G, Della Sala P, Gaeta C, Schalley CA, Neri P. Study on the Influence of Chirality in the Threading of Calix[6]arene Hosts with Dialkylammonium Axles. Molecules. 2020; 25(22):5323. https://doi.org/10.3390/molecules25225323
Chicago/Turabian StyleTalotta, Carmen, Gerardo Concilio, Paolo Della Sala, Carmine Gaeta, Christoph A. Schalley, and Placido Neri. 2020. "Study on the Influence of Chirality in the Threading of Calix[6]arene Hosts with Dialkylammonium Axles" Molecules 25, no. 22: 5323. https://doi.org/10.3390/molecules25225323
APA StyleTalotta, C., Concilio, G., Della Sala, P., Gaeta, C., Schalley, C. A., & Neri, P. (2020). Study on the Influence of Chirality in the Threading of Calix[6]arene Hosts with Dialkylammonium Axles. Molecules, 25(22), 5323. https://doi.org/10.3390/molecules25225323