
From Phenotypic Hit to Chemical Probe: Chemical Biology Approaches to Elucidate Small Molecule Action in Complex Biological Systems


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Identification of Bioactive Molecules
2.1. Target-Based Approaches
2.2. Phenotypic Screens
2.3. In-Cell Fragment-Based Ligand Discovery
3. To Label or Not to Label?
4. Label-Based Approaches
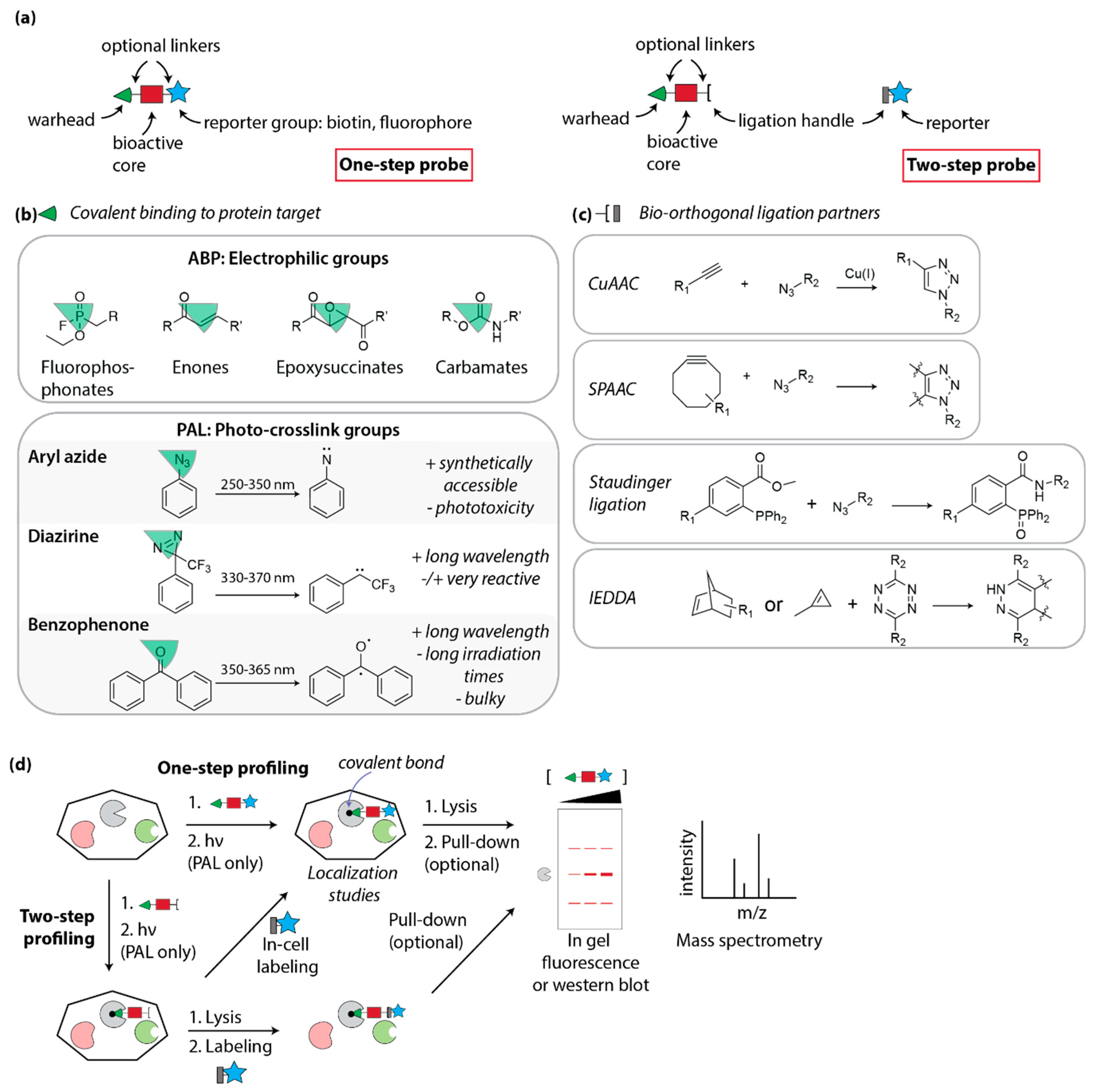
4.1. Design of Covalent Probes: Activity- and Affinity-Based Probes
4.2. Activity-Based Profiling
4.3. Photo-Affinity Labeling
4.3.1. Photocrosslink Groups
4.3.2. Minimalist Linkers
4.3.3. PAL for Phenotypic Hit Target Identification
4.4. FITGE
5. Label-Free Approaches
5.1. Mass-Spectrometry Based Methods
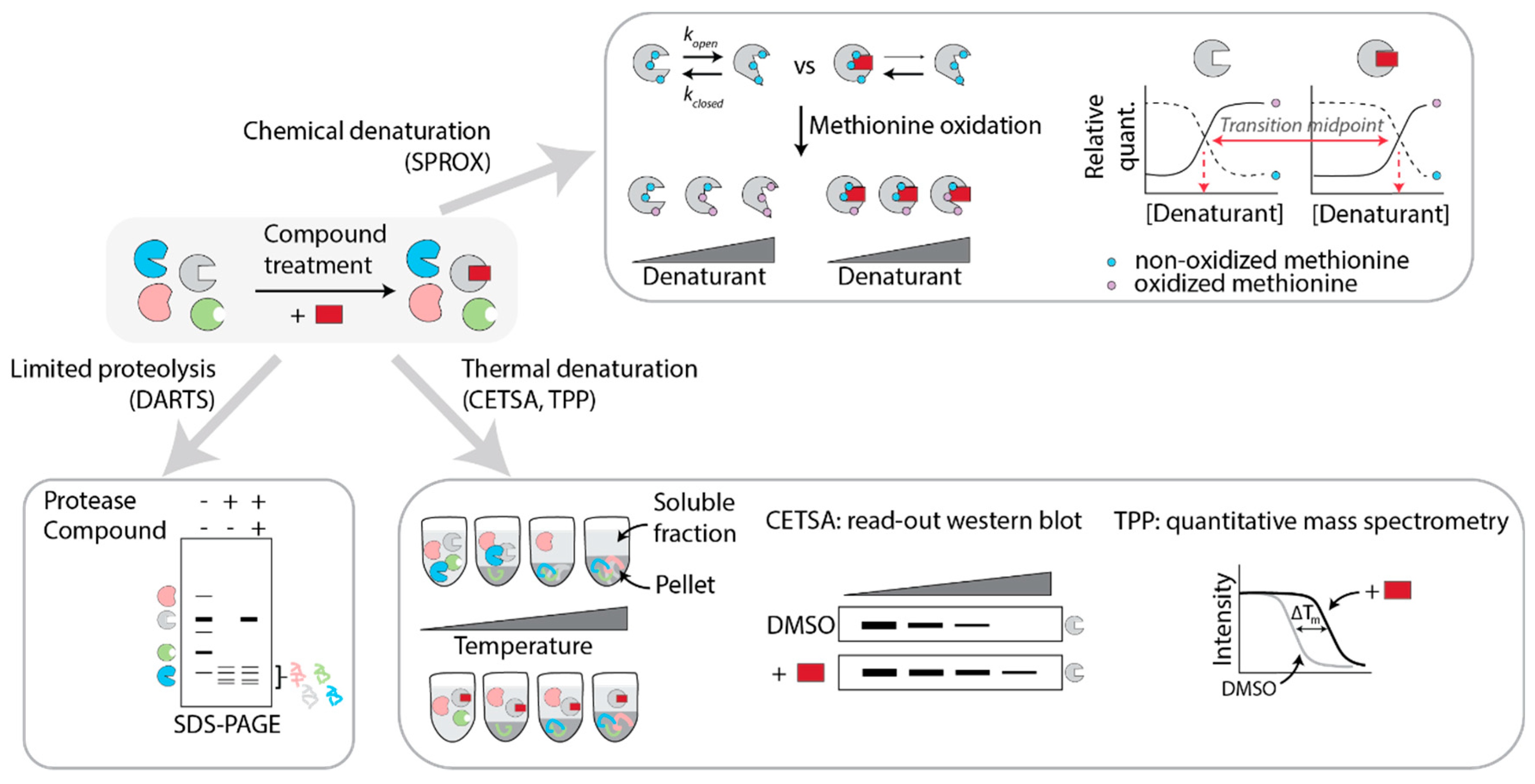
5.1.1. DARTS
5.1.2. SPROX
5.1.3. CETSA
5.1.4. FITExP
5.1.5. Large-Scale Proteomics
5.2. Genetic Approaches
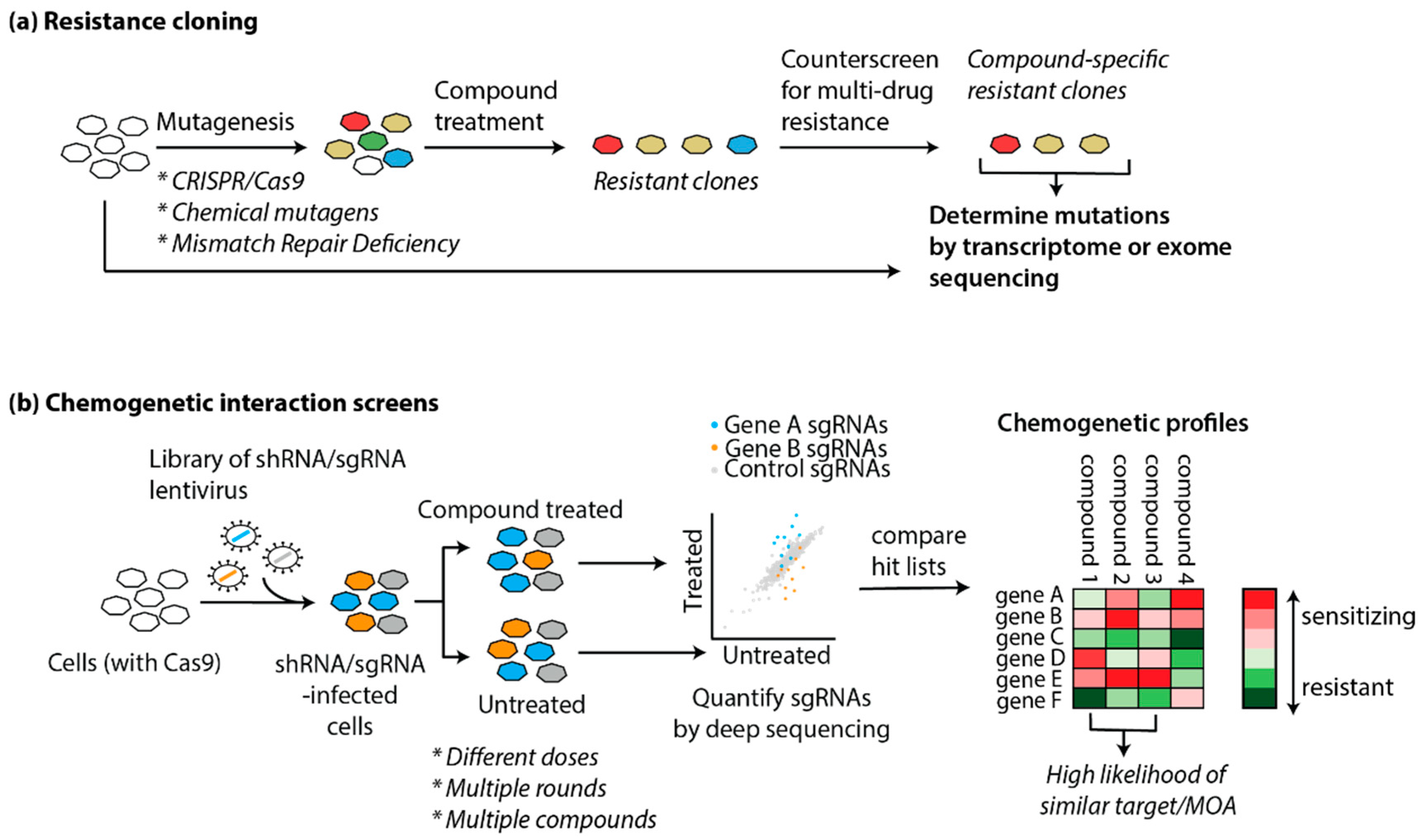
5.2.1. Resistance Cloning
5.2.2. Genetic Screens
5.2.3. “Chemogenomics”: Examples of Gene-Drug Interaction Screens
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Blagg, J.; Workman, P. Choose and Use Your Chemical Probe Wisely to Explore Cancer Biology. Cancer Cell 2017, 32, 9–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrowsmith, C.H.; Audia, J.E.; Austin, C.; Baell, J.; Bennett, J.; Blagg, J.; Bountra, C.; Brennan, P.E.; Brown, P.J.; Bunnage, M.E.; et al. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaraj, N.K. The Future of Bioorthogonal Chemistry. ACS Cent. Sci. 2018, 4, 952–959. [Google Scholar] [CrossRef] [Green Version]
- Schenone, M.; Dančík, V.; Wagner, B.K.; Clemons, P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Jost, M.; Weissman, J.S. CRISPR Approaches to Small Molecule Target Identification. ACS Chem. Biol. 2018, 13, 366–375. [Google Scholar] [CrossRef]
- Park, H.; Ha, J.; Park, S.B. Label-free target identification in drug discovery via phenotypic screening. Curr. Opin. Chem. Biol. 2019, 50, 66–72. [Google Scholar] [CrossRef]
- Kubota, K.; Funabashi, M.; Ogura, Y. Target deconvolution from phenotype-based drug discovery by using chemical proteomics approaches. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2019, 1867, 22–27. [Google Scholar] [CrossRef]
- Garbaccio, R.M.; Parmee, E.R. The Impact of Chemical Probes in Drug Discovery: A Pharmaceutical Industry Perspective. Cell. Chem. Biol. 2016, 23, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Swinney, D.C. Phenotypic vs. Target-Based Drug Discovery for First-in-Class Medicines. Clin. Pharmacol. Ther. 2013, 93, 299–301. [Google Scholar] [CrossRef]
- Croston, G.E. The utility of target-based discovery. Expert. Opin. Drug Discov. 2017, 12, 427–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstenberger, B.S.; Ambler, C.; Arnold, E.P.; Banker, M.-E.; Brown, M.F.; Clark, J.D.; Dermenci, A.; Dowty, M.E.; Fensome, A.; Fish, S.; et al. Discovery of Tyrosine Kinase 2 (TYK2) Inhibitor (PF-06826647) for the Treatment of Autoimmune Diseases. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Keserű, G.M.; Erlanson, D.A.; Ferenczy, G.G.; Hann, M.M.; Murray, C.W.; Pickett, S.D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59, 8189–8206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, B.-S.; Perera, S.A.; Piesvaux, J.A.; Presland, J.P.; Schroeder, G.K.; Cumming, J.N.; Trotter, B.W.; Altman, M.D.; Buevich, A.V.; Cash, B.; et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 2020, 369. [Google Scholar] [CrossRef]
- Mussari, C.P.; Dodd, D.S.; Sreekantha, R.K.; Pasunoori, L.; Wan, H.; Posy, S.L.; Critton, D.; Ruepp, S.; Subramanian, M.; Watson, A.; et al. Discovery of Potent and Orally Bioavailable Small Molecule Antagonists of Toll-like Receptors 7/8/9 (TLR7/8/9). ACS Med. Chem. Lett. 2020, 11, 1751–1758. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Szabo, M.; Svensson Akusjärvi, S.; Saxena, A.; Liu, J.; Chandrasekar Janebjer, G.; Kitambi, S.S. Cell and small animal models for phenotypic drug discovery. DDDT 2017, 11, 1957–1967. [Google Scholar] [CrossRef] [Green Version]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [Green Version]
- Ni, T.T.; Rellinger, E.J.; Mukherjee, A.; Xie, S.; Stephens, L.; Thorne, C.A.; Kim, K.; Hu, J.; Lee, E.; Marnett, L.; et al. Discovering Small Molecules that Promote Cardiomyocyte Generation by Modulating Wnt Signaling. Chem. Biol. 2011, 18, 1658–1668. [Google Scholar] [CrossRef] [Green Version]
- Gonsalves, F.C.; Klein, K.; Carson, B.B.; Katz, S.; Ekas, L.A.; Evans, S.; Nagourney, R.; Cardozo, T.; Brown, A.M.C.; DasGupta, R. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 5954–5963. [Google Scholar] [CrossRef] [Green Version]
- Koch, P.D.; Miller, H.R.; Yu, G.; Tallarico, J.A.; Sorger, P.K.; Wang, Y.; Feng, Y.; Thomas, J.R.; Ross, N.T.; Mitchison, T. A High Content Screen in Macrophages Identifies Small Molecule Modulators of STING-IRF3 and NFkB Signaling. ACS Chem. Biol. 2018, 13, 1066–1081. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, G.J.; Alicke, B.; Weinmann, L.; Januario, T.; West, K.; Modrusan, Z.; Burdick, D.; Goldsmith, R.; Robarge, K.; Sutherlin, D.; et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011, 71, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [Green Version]
- Mock, E.D.; Mustafa, M.; Gunduz-Cinar, O.; Cinar, R.; Petrie, G.N.; Kantae, V.; Di, X.; Ogasawara, D.; Varga, Z.V.; Paloczi, J.; et al. Discovery of a NAPE-PLD inhibitor that modulates emotional behavior in mice. Nat. Chem. Biol. 2020, 16, 667–675. [Google Scholar] [CrossRef]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Gao, J.; Mfuh, A.; Amako, Y.; Woo, C.M. Small Molecule Interactome Mapping by Photoaffinity Labeling Reveals Binding Site Hotspots for the NSAIDs. J. Am. Chem. Soc. 2018, 140, 4259–4268. [Google Scholar] [CrossRef]
- Bembenek, S.D.; Tounge, B.A.; Reynolds, C.H. Ligand efficiency and fragment-based drug discovery. Drug Discov. Today 2009, 14, 278–283. [Google Scholar] [CrossRef]
- Scott, D.E.; Coyne, A.G.; Hudson, S.A.; Abell, C. Fragment-Based Approaches in Drug Discovery and Chemical Biology. Biochemistry 2012, 51, 4990–5003. [Google Scholar] [CrossRef]
- Parker, C.G.; Galmozzi, A.; Wang, Y.; Correia, B.E.; Sasaki, K.; Joslyn, C.M.; Kim, A.S.; Cavallaro, C.L.; Lawrence, R.M.; Johnson, S.R.; et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 2017, 168, 527–541.e29. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dix, M.M.; Bianco, G.; Remsberg, J.R.; Lee, H.-Y.; Kalocsay, M.; Gygi, S.P.; Forli, S.; Vite, G.; Lawrence, R.M.; et al. Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat. Chem. 2019, 11, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Leslie, B.J.; Hergenrother, P.J. Identification of the cellular targets of bioactive small organic molecules using affinity reagents. Chem. Soc. Rev. 2008, 37, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.P.; Kallemeijn, W.W.; Debets, M.F.; Hansen, T.; Sobala, L.F.; Hakki, Z.; Williams, S.J.; Beenakker, T.J.M.; Aerts, J.M.F.G.; van der Marel, G.A.; et al. Spiro-epoxyglycosides as Activity-Based Probes for Glycoside Hydrolase Family 99 Endomannosidase/Endomannanase. Chem. Eur. J. 2018, 24, 9983–9992. [Google Scholar] [CrossRef] [PubMed]
- Hoogendoorn, S.; van Puijvelde, G.H.M.; Kuiper, J.; van der Marel, G.A.; Overkleeft, H.S. A Multivalent Ligand for the Mannose-6-Phosphate Receptor for Endolysosomal Targeting of an Activity-Based Probe. Angew. Chem. 2014, 126, 11155–11158. [Google Scholar] [CrossRef]
- Cognetta, A.B.; Niphakis, M.J.; Lee, H.-C.; Martini, M.L.; Hulce, J.J.; Cravatt, B.F. Selective N-Hydroxyhydantoin Carbamate Inhibitors of Mammalian Serine Hydrolases. Chem. Biol. 2015, 22, 928–937. [Google Scholar] [CrossRef] [Green Version]
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60, 839–885. [Google Scholar] [CrossRef]
- Willems, L.I.; van der Linden, W.A.; Li, N.; Li, K.-Y.; Liu, N.; Hoogendoorn, S.; van der Marel, G.A.; Florea, B.I.; Overkleeft, H.S. Bioorthogonal Chemistry: Applications in Activity-Based Protein Profiling. Acc. Chem. Res. 2011, 44, 718–729. [Google Scholar] [CrossRef]
- Nguyen, S.S.; Prescher, J.A. Developing bioorthogonal probes to span a spectrum of reactivities. Nat. Rev. Chem. 2020, 4, 476–489. [Google Scholar] [CrossRef]
- Smeenk, M.L.W.J.; Agramunt, J.; Bonger, K.M. Recent developments in bioorthogonal chemistry and the orthogonality within. Curr. Opin. Chem. Biol. 2021, 60, 79–88. [Google Scholar] [CrossRef]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A Strain-Promoted [3 + 2] Azide–Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Saxon, E.; Bertozzi, C.R. Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaraj, N.K.; Weissleder, R.; Hilderbrand, S.A. Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging. Bioconjugate Chem. 2008, 19, 2297–2299. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Lei, Q.; Wu, Y.; He, Y.; Li, W. Activity-based protein profiling: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2020, 191, 112151. [Google Scholar] [CrossRef] [PubMed]
- Speers, A.E.; Adam, G.C.; Cravatt, B.F. Activity-Based Protein Profiling in Vivo Using a Copper(I)-Catalyzed Azide-Alkyne [3 + 2] Cycloaddition. J. Am. Chem. Soc. 2003, 125, 4686–4687. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Patricelli, M.P.; Cravatt, B.F. Activity-based protein profiling: The serine hydrolases. Proc. Natl. Acad. Sci. USA 1999, 96, 14694–14699. [Google Scholar] [CrossRef] [Green Version]
- Bogyo, M.; McMaster, J.S.; Gaczynska, M.; Tortorella, D.; Goldberg, A.L.; Ploegh, H. Covalent modification of the active site threonine of proteasomal β subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proc. Natl. Acad. Sci. USA 1997, 94, 6629–6634. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, D.; Medzihradszky, K.F.; Burlingame, A.; Bogyo, M. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem. Biol. 2000, 7, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Witte, M.D.; Kallemeijn, W.W.; Aten, J.; Li, K.-Y.; Strijland, A.; Donker-Koopman, W.E.; van den Nieuwendijk, A.M.C.H.; Bleijlevens, B.; Kramer, G.; Florea, B.I.; et al. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat. Chem. Biol. 2010, 6, 907–913. [Google Scholar] [CrossRef] [Green Version]
- Benns, H.J.; Wincott, C.J.; Tate, E.W.; Child, M.A. Activity- and reactivity-based proteomics: Recent technological advances and applications in drug discovery. Curr. Opin. Chem. Biol. 2021, 60, 20–29. [Google Scholar] [CrossRef]
- Leung, D.; Hardouin, C.; Boger, D.L.; Cravatt, B.F. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat. Biotechnol. 2003, 21, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fang, B.; Kinose, F.; Bai, Y.; Kim, J.-Y.; Chen, Y.A.; Rix, U.; Koomen, J.M.; Haura, E.B. Target Identification in Small Cell Lung Cancer via Integrated Phenotypic Screening and Activity-Based Protein Profiling. Mol. Cancer Ther. 2016, 15, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.; Collins, I. Photoaffinity labeling in target- and binding-site identification. Future Med. Chem. 2015, 7, 159–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinsky, L.; Krom, B.P.; Meijler, M.M. Diazirine based photoaffinity labeling. Bioorg. Med. Chem. 2012, 20, 554–570. [Google Scholar] [CrossRef] [PubMed]
- Dormán, G.; Nakamura, H.; Pulsipher, A.; Prestwich, G.D. The Life of Pi Star: Exploring the Exciting and Forbidden Worlds of the Benzophenone Photophore. Chem. Rev. 2016, 116, 15284–15398. [Google Scholar] [CrossRef] [PubMed]
- Kita, M.; Yamagishi, K.; Tsuchiya, K.; Seguchi, Y.; Nakane, H.; Kigoshi, H. Development of photoaffinity derivatives of the antitumor macrolide aplyronine A, a PPI-inducer between actin and tubulin. Bioorg. Med. Chem. 2017, 25, 6322–6331. [Google Scholar] [CrossRef]
- Ge, S.-S.; Chen, B.; Wu, Y.-Y.; Long, Q.-S.; Zhao, Y.-L.; Wang, P.-Y.; Yang, S. Current advances of carbene-mediated photoaffinity labeling in medicinal chemistry. RSC Adv. 2018, 8, 29428–29454. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, K.; Ozawa, S.; Yamada, R.; Yasui, T.; Mizuno, S. Comparison of the Reactivity of Carbohydrate Photoaffinity Probes with Different Photoreactive Groups. ChemBioChem 2014, 15, 1399–1403. [Google Scholar] [CrossRef]
- Park, J.; Koh, M.; Koo, J.Y.; Lee, S.; Park, S.B. Investigation of Specific Binding Proteins to Photoaffinity Linkers for Efficient Deconvolution of Target Protein. ACS Chem. Biol. 2016, 11, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Kleiner, P.; Heydenreuter, W.; Stahl, M.; Korotkov, V.S.; Sieber, S.A. A Whole Proteome Inventory of Background Photocrosslinker Binding. Angew. Chem. Int. Ed. 2017, 56, 1396–1401. [Google Scholar] [CrossRef]
- Guo, H.; Li, Z. Developments of bioorthogonal handle-containing photo-crosslinkers for photoaffinity labeling. Med. Chem. Commun. 2017, 8, 1585–1591. [Google Scholar] [CrossRef]
- Li, Z.; Hao, P.; Li, L.; Tan, C.Y.J.; Cheng, X.; Chen, G.Y.J.; Sze, S.K.; Shen, H.-M.; Yao, S.Q. Design and Synthesis of Minimalist Terminal Alkyne-Containing Diazirine Photo-Crosslinkers and Their Incorporation into Kinase Inhibitors for Cell- and Tissue-Based Proteome Profiling. Angew. Chem. Int. Ed. 2013, 52, 8551–8556. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, D.; Li, L.; Pan, S.; Na, Z.; Tan, C.Y.J.; Yao, S.Q. “Minimalist” Cyclopropene-Containing Photo-Cross-Linkers Suitable for Live-Cell Imaging and Affinity-Based Protein Labeling. J. Am. Chem. Soc. 2014, 136, 9990–9998. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Jang, S.-Y.; Wang, D.; Liew, S.S.; Li, Z.; Lee, J.-S.; Yao, S.Q. A Suite of “Minimalist” Photo-Crosslinkers for Live-Cell Imaging and Chemical Proteomics: Case Study with BRD4 Inhibitors. Angew. Chem. Int. Ed. 2017, 56, 11816–11821. [Google Scholar] [CrossRef]
- Chen, J.; Kassenbrock, A.; Li, B.X.; Xiao, X. Discovery of a potent anti-tumor agent through regioselective mono-N-acylation of 7H-pyrrolo[3,2-f]quinazoline-1,3-diamine. Med. Chem. Commun. 2013, 4, 1275–1282. [Google Scholar] [CrossRef] [Green Version]
- Li, B.X.; Chen, J.; Chao, B.; David, L.L.; Xiao, X. Anticancer Pyrroloquinazoline LBL1 Targets Nuclear Lamins. ACS Chem. Biol. 2018, 13, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, U.; Huang, Z.; am Ende, C.W.; Butler, T.W.; Cleary, L.; Dresselhaus, E.; Evrard, E.; Fisher, E.L.; Green, M.E.; Helal, C.J.; et al. Photoaffinity Labeling and Quantitative Chemical Proteomics Identify LXRβ as the Functional Target of Enhancers of Astrocytic apoE. Cell Chem. Biol. 2020. [Google Scholar] [CrossRef]
- Park, J.; Oh, S.; Park, S.B. Discovery and Target Identification of an Antiproliferative Agent in Live Cells Using Fluorescence Difference in Two-Dimensional Gel Electrophoresis. Angew. Chem. Int. Ed. Engl. 2012, 51, 5447–5451. [Google Scholar] [CrossRef]
- Lee, K.; Ban, H.S.; Naik, R.; Hong, Y.S.; Son, S.; Kim, B.-K.; Xia, Y.; Song, K.B.; Lee, H.-S.; Won, M. Identification of Malate Dehydrogenase 2 as a Target Protein of the HIF-1 Inhibitor LW6 using Chemical Probes. Angew. Chem. Int. Ed. 2013, 52, 10286–10289. [Google Scholar] [CrossRef]
- Lee, S.; Nam, Y.; Koo, J.Y.; Lim, D.; Park, J.; Ock, J.; Kim, J.; Suk, K.; Park, S.B. A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nat. Chem. Biol. 2014, 10, 1055–1060. [Google Scholar] [CrossRef]
- Park, H.; Ha, J.; Koo, J.Y.; Park, J.; Park, S.B. Label-free target identification using in-gel fluorescence difference via thermal stability shift. Chem. Sci. 2017, 8, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A Contaminant Repository for Affinity Purification Mass Spectrometry Data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Prabhu, N.; Yu, L.Y.; Bacanu, S.; Ramos, A.D.; Nordlund, P. Horizontal Cell Biology: Monitoring Global Changes of Protein Interaction States with the Proteome-Wide Cellular Thermal Shift Assay (CETSA). Annu. Rev. Biochem. 2019, 88, 383–408. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Li, Z.; Chen, D.; Jia, L.; Guo, J.; Zhao, T.; Nordlund, P. Target identification and validation of natural products with label-free methodology: A critical review from 2005 to 2020. Pharmacol. Ther. 2020, 107690. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S.; Wang, J.; Wu, R.P.; Gomez, F.; Loo, J.A.; et al. Target identification using drug affinity responsive target stability (DARTS). Curr. Protoc. Chem. Biol. 2009, 106, 21984–21989. [Google Scholar] [CrossRef] [Green Version]
- Lomenick, B.; Olsen, R.W.; Huang, J. Identification of Direct Protein Targets of Small Molecules. ACS Chem. Biol. 2011, 6, 34–46. [Google Scholar] [CrossRef]
- Qu, Y.; Olsen, J.R.; Yuan, X.; Cheng, P.F.; Levesque, M.P.; Brokstad, K.A.; Hoffman, P.S.; Oyan, A.M.; Zhang, W.; Kalland, K.-H.; et al. Small molecule promotes β-catenin citrullination and inhibits Wnt signaling in cancer. Nat. Chem. Biol. 2018, 14, 94–101. [Google Scholar] [CrossRef]
- Dal Piaz, F.; Vera Saltos, M.B.; Franceschelli, S.; Forte, G.; Marzocco, S.; Tuccinardi, T.; Poli, G.; Nejad Ebrahimi, S.; Hamburger, M.; De Tommasi, N.; et al. Drug Affinity Responsive Target Stability (DARTS) Identifies Laurifolioside as a New Clathrin Heavy Chain Modulator. J. Nat. Prod. 2016, 79, 2681–2692. [Google Scholar] [CrossRef]
- West, G.M.; Tang, L.; Fitzgerald, M.C. Thermodynamic analysis of protein stability and ligand binding using a chemical modification- and mass spectrometry-based strategy. Anal. Chem 2008, 80, 4175–4185. [Google Scholar] [CrossRef]
- West, G.M.; Tucker, C.L.; Xu, T.; Park, S.K.; Han, X.; Yates, J.R.; Fitzgerald, M.C. Quantitative proteomics approach for identifying protein–drug interactions in complex mixtures using protein stability measurements. Proc. Natl. Acad. Sci. USA 2010, 107, 9078–9082. [Google Scholar] [CrossRef] [Green Version]
- Strickland, E.C.; Geer, M.A.; Tran, D.T.; Adhikari, J.; West, G.M.; DeArmond, P.D.; Xu, Y.; Fitzgerald, M.C. Thermodynamic analysis of protein-ligand binding interactions in complex biological mixtures using the stability of proteins from rates of oxidation. Nat. Prot. 2013, 8, 148–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, D.T.; Adhikari, J.; Fitzgerald, M.C. Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC)-Based Strategy for Proteome-Wide Thermodynamic Analysis of Protein-Ligand Binding Interactions. Mol. Cell. Proteom. 2014, 13, 1800–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geer Wallace, M.A.; Kwon, D.-Y.; Weitzel, D.H.; Lee, C.-T.; Stephenson, T.N.; Chi, J.-T.; Mook, R.A.; Dewhirst, M.W.; Hong, J.; Fitzgerald, M.C. Discovery of Manassantin A Protein Targets Using Large-Scale Protein Folding and Stability Measurements. J. Proteome Res. 2016, 15, 2688–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogburn, R.N.; Jin, L.; Meng, H.; Fitzgerald, M.C. Discovery of Tamoxifen and N-Desmethyl Tamoxifen Protein Targets in MCF-7 Cells Using Large-Scale Protein Folding and Stability Measurements. J. Proteome Res. 2017, 16, 4073–4085. [Google Scholar] [CrossRef] [PubMed]
- Molina, D.M.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundbäck, T.; Nordlund, P.; Molina, D.M. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Prot. 2014, 9, 2100. [Google Scholar] [CrossRef]
- Savitski, M.M.; Reinhard, F.B.M.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef] [Green Version]
- Dziekan, J.M.; Yu, H.; Chen, D.; Dai, L.; Wirjanata, G.; Larsson, A.; Prabhu, N.; Sobota, R.M.; Bozdech, Z.; Nordlund, P. Identifying purine nucleoside phosphorylase as the target of quinine using cellular thermal shift assay. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, M.; Liao, P.-J.; Lee, K.H.; Wong, J.; Shang, S.C.; Minami, N.; Sampetrean, O.; Saya, H.; Lingyun, D.; Prabhu, N.; et al. Dual blockade of the lipid kinase PIP4Ks and mitotic pathways leads to cancer-selective lethality. Nat. Commun. 2017, 8, 2200. [Google Scholar] [CrossRef] [Green Version]
- Becher, I.; Werner, T.; Doce, C.; Zaal, E.A.; Tögel, I.; Khan, C.A.; Rueger, A.; Muelbaier, M.; Salzer, E.; Berkers, C.R.; et al. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol. 2016, 12, 908–910. [Google Scholar] [CrossRef]
- Becher, I.; Andrés-Pons, A.; Romanov, N.; Stein, F.; Schramm, M.; Baudin, F.; Helm, D.; Kurzawa, N.; Mateus, A.; Mackmull, M.-T.; et al. Pervasive Protein Thermal Stability Variation during the Cell Cycle. Cell 2018, 173, 1495–1507.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Zhao, T.; Bisteau, X.; Sun, W.; Prabhu, N.; Lim, Y.T.; Sobota, R.M.; Kaldis, P.; Nordlund, P. Modulation of Protein-Interaction States through the Cell Cycle. Cell 2018, 173, 1481–1494.e13. [Google Scholar] [CrossRef] [PubMed]
- Ball, K.A.; Webb, K.J.; Coleman, S.J.; Cozzolino, K.A.; Jacobsen, J.; Jones, K.R.; Stowell, M.H.B.; Old, W.M. An isothermal shift assay for proteome scale drug-target identification. Commun. Biol. 2020, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, M.; Sabatier, P.; Saei, A.A.; Beusch, C.M.; Yang, Z.; Lundström, S.L.; Zubarev, R.A. Proteome Integral Solubility Alteration: A High-Throughput Proteomics Assay for Target Deconvolution. J. Proteome Res. 2019, 18, 4027–4037. [Google Scholar] [CrossRef] [PubMed]
- Chernobrovkin, A.; Marin-Vicente, C.; Visa, N.; Zubarev, R.A. Functional Identification of Target by Expression Proteomics (FITExP) reveals protein targets and highlights mechanisms of action of small molecule drugs. Sci. Rep. 2015, 5, 11176. [Google Scholar] [CrossRef]
- Saei, A.A.; Gullberg, H.; Sabatier, P.; Beusch, C.M.; Johansson, K.; Lundgren, B.; Arvidsson, P.I.; Arnér, E.S.J.; Zubarev, R.A. Comprehensive chemical proteomics for target deconvolution of the redox active drug auranofin. Redox Biol. 2020, 32, 101491. [Google Scholar] [CrossRef]
- Ruprecht, B.; Di Bernardo, J.; Wang, Z.; Mo, X.; Ursu, O.; Christopher, M.; Fernandez, R.B.; Zheng, L.; Dill, B.D.; Wang, H.; et al. A mass spectrometry-based proteome map of drug action in lung cancer cell lines. Nat. Chem. Biol. 2020, 16, 1111–1119. [Google Scholar] [CrossRef]
- Nijman, S.M.B. Functional genomics to uncover drug mechanism of action. Nat. Chem. Biol. 2015, 11, 942–948. [Google Scholar] [CrossRef]
- Kapoor, T.M.; Miller, R.M. Leveraging Chemotype-specific Resistance for Drug Target Identification and Chemical Biology. Trends Pharmacol. Sci. 2017, 38, 1100–1109. [Google Scholar] [CrossRef]
- Han, T.; Nijhawan, D. Exome Sequencing of Drug-Resistant Clones for Target Identification. In Systems Chemical Biology: Methods and Protocols; Ziegler, S., Waldmann, H., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 175–187. ISBN 978-1-4939-8891-4. [Google Scholar]
- Wacker, S.A.; Houghtaling, B.R.; Elemento, O.; Kapoor, T.M. Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat. Chem. Biol. 2012, 8, 235–237. [Google Scholar] [CrossRef] [Green Version]
- Kasap, C.; Elemento, O.; Kapoor, T.M. DrugTargetSeqR: A genomics- and CRISPR-Cas9–based method to analyze drug targets. Nat. Chem. Biol. 2014, 10, 626–628. [Google Scholar] [CrossRef] [Green Version]
- Povedano, J.M.; Liou, J.; Wei, D.; Srivatsav, A.; Kim, J.; Xie, Y.; Nijhawan, D.; McFadden, D.G. Engineering Forward Genetics into Cultured Cancer Cells for Chemical Target Identification. Cell Chem. Biol. 2019, 26, 1315–1321.e3. [Google Scholar] [CrossRef]
- Neggers, J.E.; Kwanten, B.; Dierckx, T.; Noguchi, H.; Voet, A.; Bral, L.; Minner, K.; Massant, B.; Kint, N.; Delforge, M.; et al. Target identification of small molecules using large-scale CRISPR-Cas mutagenesis scanning of essential genes. Nat. Commun. 2018, 9, 502. [Google Scholar] [CrossRef] [PubMed]
- Brammeld, J.S.; Petljak, M.; Martincorena, I.; Williams, S.P.; Alonso, L.G.; Dalmases, A.; Bellosillo, B.; Robles-Espinoza, C.D.; Price, S.; Barthorpe, S.; et al. Genome-wide chemical mutagenesis screens allow unbiased saturation of the cancer genome and identification of drug resistance mutations. Genome Res. 2017, 27, 613–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Leprohon, P.; Bigot, S.; Padmanabhan, P.K.; Mukherjee, A.; Roy, G.; Gingras, H.; Mestdagh, A.; Papadopoulou, B.; Ouellette, M. Coupling chemical mutagenesis to next generation sequencing for the identification of drug resistance mutations in Leishmania. Nat. Commun. 2019, 10, 5627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, Y.; Zyryanova, A.; Crespillo-Casado, A.; Fischer, P.M.; Harding, H.P.; Ron, D. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 2015. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic Analysis of Smoothened Inhibitor Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Kampmann, M.; Bassik, M.C.; Weissman, J.S. Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, E2317–E2326. [Google Scholar] [CrossRef] [Green Version]
- Bassik, M.C.; Kampmann, M.; Lebbink, R.J.; Wang, S.; Hein, M.Y.; Poser, I.; Weibezahn, J.; Horlbeck, M.A.; Chen, S.; Mann, M.; et al. A Systematic Mammalian Genetic Interaction Map Reveals Pathways Underlying Ricin Susceptibility. Cell 2013, 152, 909–922. [Google Scholar] [CrossRef] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Koike-Yusa, H.; Li, Y.; Tan, E.P.; Velasco-Herrera Mdel, C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014, 32, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhu, S.; Cai, C.; Yuan, P.; Li, C.; Huang, Y.; Wei, W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 2014, 509, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horlbeck, M.A.; Gilbert, L.A.; Villalta, J.E.; Adamson, B.; Pak, R.A.; Chen, Y.; Fields, A.P.; Park, C.Y.; Corn, J.E.; Kampmann, M.; et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 2016, 5, e19760. [Google Scholar] [CrossRef]
- Kampmann, M. Elucidating drug targets and mechanisms of action by genetic screens in mammalian cells. Chem. Commun. 2017, 53, 7162–7167. [Google Scholar] [CrossRef]
- Breslow, D.K.; Hoogendoorn, S.; Kopp, A.R.; Morgens, D.W.; Vu, B.K.; Kennedy, M.C.; Han, K.; Li, A.; Hess, G.T.; Bassik, M.C.; et al. A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat. Genet. 2018, 50, 460–471. [Google Scholar] [CrossRef]
- Pusapati, G.V.; Kong, J.H.; Patel, B.B.; Krishnan, A.; Sagner, A.; Kinnebrew, M.; Briscoe, J.; Aravind, L.; Rohatgi, R. CRISPR screens uncover genes that regulate target cell sensitivity to the morphogen sonic hedgehog. Dev. Cell 2018, 44, 113–129.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogendoorn, S. Small Molecules Targeting the Hedgehog Pathway: From Phenotype to Mechanistic Understanding. CHIMIA 2020, 74, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaitė, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Matheny, C.J.; Wei, M.C.; Bassik, M.C.; Donnelly, A.J.; Kampmann, M.; Iwasaki, M.; Piloto, O.; Solow-Cordero, D.E.; Bouley, D.M.; Rau, R.; et al. Next-Generation NAMPT Inhibitors Identified by Sequential High-Throughput Phenotypic Chemical and Functional Genomic Screens. Chem. Biol. 2013, 20, 1352–1363. [Google Scholar] [CrossRef] [Green Version]
- Deans, R.M.; Morgens, D.W.; Ökesli, A.; Pillay, S.; Horlbeck, M.A.; Kampmann, M.; Gilbert, L.A.; Li, A.; Mateo, R.; Smith, M.; et al. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat. Chem. Biol. 2016, 12, 361. [Google Scholar] [CrossRef] [Green Version]
- Estoppey, D.; Hewett, J.W.; Guy, C.T.; Harrington, E.; Thomas, J.R.; Schirle, M.; Cuttat, R.; Waldt, A.; Gerrits, B.; Yang, Z.; et al. Identification of a novel NAMPT inhibitor by CRISPR/Cas9 chemogenomic profiling in mammalian cells. Sci. Rep. 2017, 7, 42728. [Google Scholar] [CrossRef] [Green Version]
- Giaever, G.; Shoemaker, D.D.; Jones, T.W.; Liang, H.; Winzeler, E.A.; Astromoff, A.; Davis, R.W. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat. Genet. 1999, 21, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Estoppey, D.; Lee, C.M.; Janoschke, M.; Lee, B.H.; Wan, K.F.; Dong, H.; Mathys, P.; Filipuzzi, I.; Schuhmann, T.; Riedl, R.; et al. The Natural Product Cavinafungin Selectively Interferes with Zika and Dengue Virus Replication by Inhibition of the Host Signal Peptidase. Cell Rep. 2017, 19, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Jost, M.; Chen, Y.; Gilbert, L.A.; Horlbeck, M.A.; Krenning, L.; Menchon, G.; Rai, A.; Cho, M.Y.; Stern, J.J.; Prota, A.E.; et al. Combined CRISPRi/a-Based Chemical Genetic Screens Reveal that Rigosertib Is a Microtubule-Destabilizing Agent. Mol. Cell 2017, 68, 210–223.e6. [Google Scholar] [CrossRef] [Green Version]
- Colic, M.; Wang, G.; Zimmermann, M.; Mascall, K.; McLaughlin, M.; Bertolet, L.; Lenoir, W.F.; Moffat, J.; Angers, S.; Durocher, D.; et al. Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Med. 2019, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Pries, V.; Nöcker, C.; Khan, D.; Johnen, P.; Hong, Z.; Tripathi, A.; Keller, A.-L.; Fitz, M.; Perruccio, F.; Filipuzzi, I.; et al. Target Identification and Mechanism of Action of Picolinamide and Benzamide Chemotypes with Antifungal Properties. Cell Chem. Biol. 2018, 25, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target Identification for Small Bioactive Molecules: Finding the Needle in the Haystack. Angew. Chem. Int. Ed. 2013, 52, 2744–2792. [Google Scholar] [CrossRef] [PubMed]
- Katsila, T.; Spyroulias, G.A.; Patrinos, G.P.; Matsoukas, M.-T. Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 2016, 14, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhukar, N.S.; Khade, P.K.; Huang, L.; Gayvert, K.; Galletti, G.; Stogniew, M.; Allen, J.E.; Giannakakou, P.; Elemento, O. A Bayesian machine learning approach for drug target identification using diverse data types. Nat. Commun. 2019, 10, 5221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.-B.; Yu, Z.-J.; Liu, S.; Huang, L.-Y.; Yang, L.-L.; Lohans, C.T.; Yang, S.-Y. IFPTarget: A Customized Virtual Target Identification Method Based on Protein–Ligand Interaction Fingerprinting Analyses. J. Chem. Inf. Model. 2017, 57, 1640–1651. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.; Roy, S.; Schröder, P.; Rathmer, B.; Roos, J.; Kapoor, S.; Patil, S.; Pommerenke, C.; Maier, T.; Janning, P.; et al. Combined Proteomic and In Silico Target Identification Reveal a Role for 5-Lipoxygenase in Developmental Signaling Pathways. Cell Chem. Biol. 2018, 25, 1095–1106. [Google Scholar] [CrossRef]
- Byrne, R.; Schneider, G. In Silico Target Prediction for Small Molecules. In Systems Chemical Biology: Methods and Protocols; Ziegler, S., Waldmann, H., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 273–309. ISBN 978-1-4939-8891-4. [Google Scholar]
- Lee, A.; Lee, K.; Kim, D. Using reverse docking for target identification and its applications for drug discovery. Expert Opin. Drug Discov. 2016, 11, 707–715. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797. [Google Scholar] [CrossRef] [Green Version]
- Duran-Frigola, M.; Pauls, E.; Guitart-Pla, O.; Bertoni, M.; Alcalde, V.; Amat, D.; Juan-Blanco, T.; Aloy, P. Extending the small-molecule similarity principle to all levels of biology with the Chemical Checker. Nat. Biotechnol. 2020, 38, 1087–1096. [Google Scholar] [CrossRef]
- Keenan, A.B.; Jenkins, S.L.; Jagodnik, K.M.; Koplev, S.; He, E.; Torre, D.; Wang, Z.; Dohlman, A.B.; Silverstein, M.C.; Lachmann, A.; et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laraia, L.; Garivet, G.; Foley, D.J.; Kaiser, N.; Müller, S.; Zinken, S.; Pinkert, T.; Wilke, J.; Corkery, D.; Pahl, A.; et al. Image-Based Morphological Profiling Identifies a Lysosomotropic, Iron-Sequestering Autophagy Inhibitor. Angew. Chem. Int. Ed. 2020, 59, 5721–5729. [Google Scholar] [CrossRef] [Green Version]
- Bray, M.-A.; Singh, S.; Han, H.; Davis, C.T.; Borgeson, B.; Hartland, C.; Kost-Alimova, M.; Gustafsdottir, S.M.; Gibson, C.C.; Carpenter, A.E. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Prot. 2016, 11, 1757–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneidewind, T.; Brause, A.; Pahl, A.; Burhop, A.; Mejuch, T.; Sievers, S.; Waldmann, H.; Ziegler, S. Morphological Profiling Identifies a Common Mode of Action for Small Molecules with Different Targets. ChemBioChem 2020. [Google Scholar] [CrossRef]
- Disney, M.D. Targeting RNA with Small Molecules to Capture Opportunities at the Intersection of Chemistry, Biology, and Medicine. J. Am. Chem. Soc. 2019, 141, 6776–6790. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, H.; Blain, J.C.; Vandivier, L.E.; Chin, D.N.; Friedman, J.E.; Liu, F.; Maillet, A.; Fang, C.; Kaplan, J.B.; Li, J.; et al. PEARL-seq: A Photoaffinity Platform for the Analysis of Small Molecule-RNA Interactions. ACS Chem. Biol. 2020, 15, 2374–2381. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquer, Q.T.L.; Tsakoumagkos, I.A.; Hoogendoorn, S. From Phenotypic Hit to Chemical Probe: Chemical Biology Approaches to Elucidate Small Molecule Action in Complex Biological Systems. Molecules 2020, 25, 5702. https://doi.org/10.3390/molecules25235702
Pasquer QTL, Tsakoumagkos IA, Hoogendoorn S. From Phenotypic Hit to Chemical Probe: Chemical Biology Approaches to Elucidate Small Molecule Action in Complex Biological Systems. Molecules. 2020; 25(23):5702. https://doi.org/10.3390/molecules25235702
Chicago/Turabian StylePasquer, Quentin T. L., Ioannis A. Tsakoumagkos, and Sascha Hoogendoorn. 2020. "From Phenotypic Hit to Chemical Probe: Chemical Biology Approaches to Elucidate Small Molecule Action in Complex Biological Systems" Molecules 25, no. 23: 5702. https://doi.org/10.3390/molecules25235702
APA StylePasquer, Q. T. L., Tsakoumagkos, I. A., & Hoogendoorn, S. (2020). From Phenotypic Hit to Chemical Probe: Chemical Biology Approaches to Elucidate Small Molecule Action in Complex Biological Systems. Molecules, 25(23), 5702. https://doi.org/10.3390/molecules25235702